

Abstract
The direct C–H functionalization and arylation of benzyl ethers has been accomplished via photoredox organocatalysis. The productive merger of a thiol catalyst and a commercially available iridium photoredox catalyst in the presence of household light directly affords benzylic arylation products in good to excellent yield. The utility of this methodology was further demonstrated in the direct arylation of 2,5-dihydrofuran to form a single regioisomer.
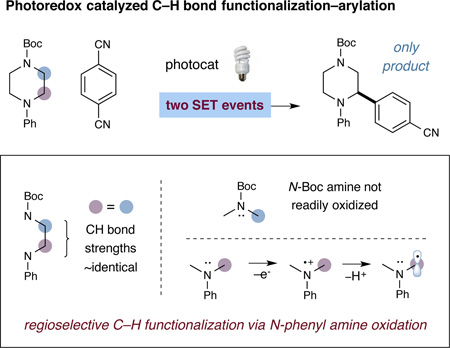
The functionalization of sp3 C–H bonds in a predictable, selective, and efficient manner has become a central challenge in modern organic chemistry.1 In this context, our laboratory recently introduced a unique activation mode that enables the direct arylation of α-methylene amines via visible light photoredox catalysis (Eq 1).2 This strategy relies on the coupling of two catalytically generated radicals: an arene radical anion formed by photocatalytic reduction of an arylnitrile, and a nucleophilic α-amino radical formed via oxidation and deprotonation of a N-phenyl amine. A remarkable feature of this activation mode is the capacity for regioselective C–H arylation adjacent to electron-rich N-phenyl amines in the presence of other moieties that have similar or weaker C–H bond strengths (including other α-amino methylene groups). As exemplified in equation 1, this oxidation potential-gated mechanism allows for predictive and selective C–H bond functionalization of α-CH2-N systems via the judicious selection of nitrogen protecting groups.3
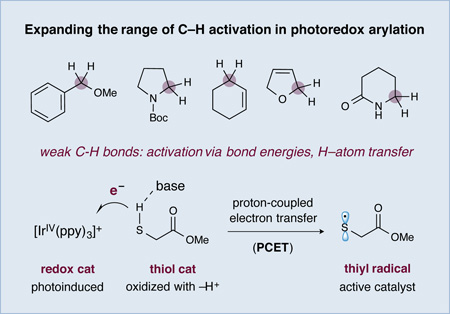
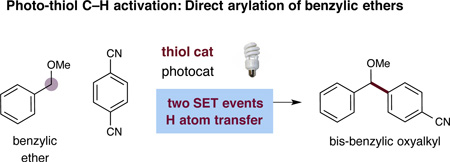
Recently, we sought to broadly expand the classes of organic molecules that will participate in photoredox mediated C–H activation. More specifically, we hoped to introduce a new photoredox-organocatalytic C–H functionalization mechanism that exploits several established physical properties (e.g. bond dissociation energies (BDEs),4 hydrogen-atom transfer (HAT) exchange constants,5 and oxidation potentials) that are predictable across a wide range of organic structure types. As shown in Figure 1, we postulated that thiol organocatalysts should undergo proton coupled electron transfer (PCET) oxidation6 in the presence of photoexcited catalysts to generate electrophilic R–S• radicals.7 These transiently formed open-shell thiyls should selectively serve to abstract H• from substrate partners that contain C–H bonds, which are both weak and hydridic based on the confluence of two known physical constants: (a) a low C–H bond dissociation energy and (b) a high HAT exchange constant.8 Moreover, the seminal studies of Roberts in the 1990s have demonstrated the remarkable utility of electrophilic thiyl systems for H• abstraction within traditional radical-based reactions.9 On this basis, we hoped to provide a C–H functionalization mechanism that is amenable to a broad range of organic subunits including benzylic, allylic, amine, or oxygen bearing methyl, methylenes or methines (within acyclic or cyclic frameworks). Furthermore, we proposed that this C–H oxidation step would be electronically balanced with a photocatalyst-mediated reduction of an accompanying aryl-cyano substrate to generate an arene radical anion (redox neutral mechanism). Coupling of the two catalytically generated organic radicals would then provide a general pathway to directly introduce aromatic and heteroaromatic rings onto a diverse range of organic sub-structures (using visible light as the driving force).10 In this communication, we describe the successful execution of these ideals and present a new synergistic catalysis approach to the direct arylation of benzylic and allylic ethers with cyano aromatics via the combination of photoredox and organocatalysis (Eq 2). As exemplified in Figure 1, bis-benzylic oxyalkyl groups are a prominent structural motif found in pharmaceutically active compounds,11 complex natural products11 and asymmetric catalysts.12 As such, we expect that this new C–H bond arylation strategy will find broad application across a variety of fields that rely on organic molecule construction.
 |
(Eq 1) |
 |
(Eq 2) |
Figure 1.

Photoredox Strategy Towards Diarylalkyl Ethers.
Detailed Design Plan
The specific mechanistic details of our proposed benzyl ether C–H arylation are outlined in Scheme 1. Irradiation of tris(2-phenylpyridinato-C2,N)iridium(III) [Ir(ppy)3] (1) by visible light (for example, a household light bulb) at room temperature produces a long-lived (1.9 µs) photoexcited state 2 (*IrIII(ppy)3). *IrIII(ppy)3 (2) is a strong reductant (E1/2IV/*III = −1.73 V versus SCE in CH3CN)13 and could undergo single-electron transfer (SET) with an electron-deficient arene, such as 1,4-dicyanobenzene (3) (E1/2red = −1.61 V versus SCE in CH3CN)14 to afford the corresponding arene radical anion (4) and oxidized photocatalyst IrIV(ppy)3 (5). We expected that the oxidation potentials of typical thiols (E1/2,red = +0.85 V versus SCE (cysteine))15 should render electron transfer to the oxidized IrIV(ppy)3 (5) (E1/2IV/III = +0.77 V vs. SCE)13 inefficient. Similarly, thiols are weakly acidic (e.g. pKa = 9.35 (methyl L-cysteinate), pKa = 8.04 (methyl 2-mercaptoacetate)),16 requiring strong bases to generate significant concentration of thiol anions. However, we anticipated that the joint action of a suitable base and electron-deficient photocatalyst 5 on the thiol catalyst 6 could facilitate efficient formation of electrophilic thiyl radical 7 via a concerted proton coupled electron transfer (PCET) event.17 Based on the BDEs of the various catalysts and substrates involved in this reaction, we hypothesized that this thiyl radical 7 (e.g. methyl 2-mercaptoacetate S–H BDE = 86.8–87.2 kcal/mol)18 should readily engage in a hydrogen atom transfer reaction with a benzyl ether substrate 8 (e.g. benzyl methyl ether αC–H BDE = 85.8 kcal/mol)19 to regenerate the thiol catalyst while forming the corresponding α-benzyl ether radical 9. At this stage we presumed that a radical–radical coupling reaction between the intermediates 9 and 4 would then represent the key bond-forming step prior to rapid elimination of cyanide to form the desired arylated benzyl ether product 10. It should be noted that in all transformations involving radical anions derived from cyano aromatics (such as 4), we have not observed a substrate homo-dimerization coupling (a step that we expect would be reversible). On this basis, we hypothesized that benzylic radical–radical anion coupling would predominate to generate the desired product. Although we postulate a PCET mechanism, we recognize that a stepwise pathway could also be operative, wherein a thiol anion will undergo electron transfer with IrIV(ppy)3 (5) to generate the thiyl activated catalyst 7. This alternative mechanism would require a thiol deprotonation step ahead of the oxidation event.
Scheme 1.

Proposed Catalytic Cycles for Benzylic C–H Arylation.
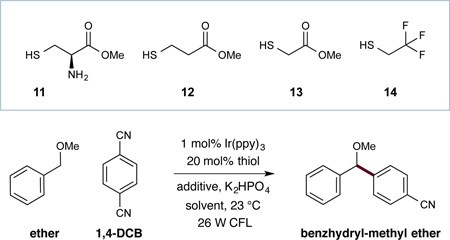
Evaluation of the proposed tandem catalysis strategy was first examined with benzyl methyl ether, K2HPO4, cysteine (11), Ir(ppy)3, a 26 W fluorescent lamp, and 1,4-dicyanobenzene as the arene coupling partner. As shown in Table 1, initial experiments revealed that the proposed C–H functionalization arylation was indeed possible (entry 1, 14% yield). Moreover, we serendipitously found in early studies that the presence of an aldehyde additive (octanal) had a beneficial effect on the reaction efficiency, presumably sequestering the cyanide anion formed during the course of the arylation step (entry 2, 32% yield). With respect to the hydrogen-atom transfer catalyst, we found methyl 2-mercaptoacetate (13) to be the most suitable, delivering the desired benzhydryl ether in 55% yield (entry 6). Next we determined that solvent selection had a significant influence on the coupling yield, with DMA proving to be the optimal medium (entry 11, 77% yield). As anticipated, control experiments established the requirement of both the organocatalyst and the photocatalyst, as no desired reaction was observed in the absence of light, Ir(ppy)3, or thiol.
Table 1.
Initial Studies Towards Benzylic C–H Arylation.
 | ||||
|---|---|---|---|---|
| Entry | Thiol catalyst | Solvent | Additive | Yielda |
| 1 | 11 | MeCN | none | 14% |
| 2 | 11 | MeCN | octanal | 32% |
| 3 | 11 | MeCN | pivaldehyde | 25% |
| 4 | 11 | MeCN | benzaldehyde | 19% |
| 5 | 12 | MeCN | octanal | 44% |
| 6 | 13 | MeCN | octanal | 55% |
| 7 | 14 | MeCN | octanal | 48% |
| 8 | 13 | DMSO | octanal | 68% |
| 9 | 13 | DMF | octanal | 72% |
| 10 | 13 | acetone | none | 16% |
| 11b | 13 | DMA | octanal | 77% |
| 12 | 13 | DMA | none | 31% |
Yield determined by IH NMR using 1-bromo-3,5-bis(trifluoromethyl)benzene as the internal standard.
Isolated yield.
With the optimized conditions in hand, we next sought to define the scope of the benzyloxy coupling partner. As revealed in Table 2, a broad array of benzyl alkyl ethers can serve as competent substrates, including cyclic analogs, such as phtalan (entry 7, 71% yield) and isochroman (entry 8, 70% yield). Notably, the use of dibenzyl ethers led to mono-arylation adducts exclusively (entry 3, 80% yield), a mechanistic selectivity that is not readily rationalized at the present time. With respect to wide spread application, it is important to note that this activation mode can also be used for the arylation of both benzyl silyl ethers (entries 10–13, 61–74% yield) and MEM-protected benzyl alcohols (entry 9, 62% yield). We also found that these mild redox conditions are compatible with a wide range of functional groups, including acetals, alkyl chlorides, alcohols, and amines (entries 9, 14–16, 62–75% yield). Perhaps most remarkable is the capacity of benzyl alcohols to undergo selective C-arylation without formation of the corresponding aldehyde or ketone products (entries 4–6, 14–16, 70–77% yield). Intriguingly, we have found that the octanal additive plays a critical role with such benzylic alcohol substrates. Specifically, 1H NMR studies of the coupling of benzyl alcohol with 1,4-dicyanobenzene clearly demonstrate the reversible formation of a hemiacetal intermediate from the substrate alcohol and octanal under our standard reaction conditions. These investigations suggest that transient acetol formation is required for selective C–H functionalization as control experiments, performed without aldehyde additive, led exclusively to benzyl alcohol oxidation in lieu of aryl coupling (e.g. benzaldehyde from benzyl alcohol). This mechanistic bifurcation as a function of acetol versus alcohol incorporation seems consistent with the capacity of benzylic alcohol radicals to undergo a rapid deprotonation-oxidation sequence that is not available to the corresponding acetol bearing radical. Importantly, we have also demonstrated the utility of this new activation mode to allow C–C bond formations at highly sterically congested centers, as highlighted with methine-bearing alcohol substrates (entries 5, 14–16). In each case, fully substituted tertiary oxy stereocenters are formed with excellent levels of efficiency (entries 5, 14–16, 70–75% yield). The broad scope of this arylation methodology was further demonstrated using a range of heteroaromatic containing ethers. Pyridines, furans, and thiophenes all undergo selective C–H arylation in high yield (entries 11–13, 61–73% yield), a notable feature with respect to medicinal agent synthesis and applications.
Table 2.
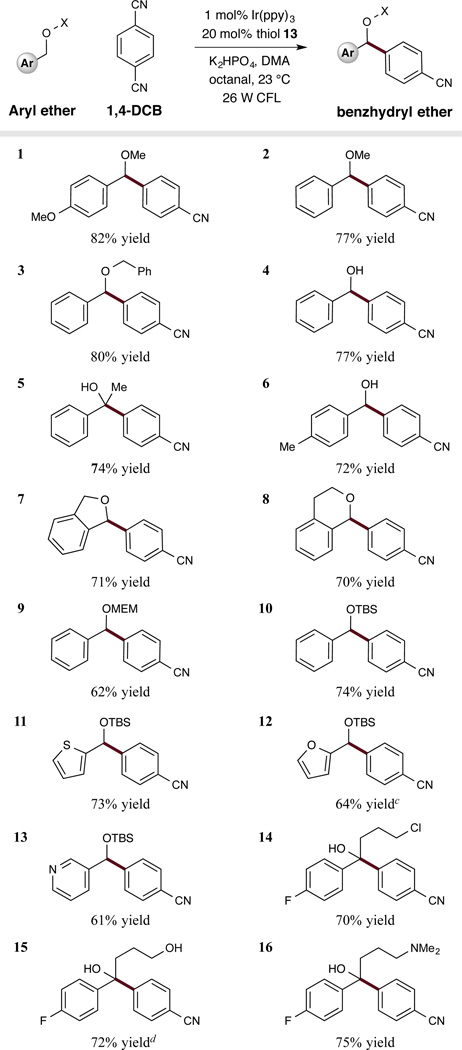 |
Yield of isolated material.
See supporting information for experimental details.
2 equiv. Na2CO3 was used as base.
2 equiv. K2HPO4 was used.
We next examined the structural diversity of the arene coupling partner in this synergistic catalysis protocol. As shown in Table 3, a range of cyanobenzenes and cyanoheteroaromatics has been found to be suitable substrates. Moreover, a variety of ortho-, meta- and para-substituted terephthalonitriles readily couple to the activated benzylic silyl ether substrate (entries 1–3, 6–7, 41–72% yield). When unsymmetrical dicyano arenes were used, mixtures of regioisomers were observed (entries 2, 3, 6). In addition, benzonitriles substituted with sulfones or esters are tolerated as radical anion coupling partners (entries 4–5, 55–71% yield). In recognizing the prevalence of heteroaromatic rings in pharmaceutical compounds, we were delighted to find that a range of substituted cyanopyridines as well as azaindole (an important indole isostere) underwent addition to the silyl benzyl ether with high efficiencies (entries 8–12; 51–86% yield).
Table 3.
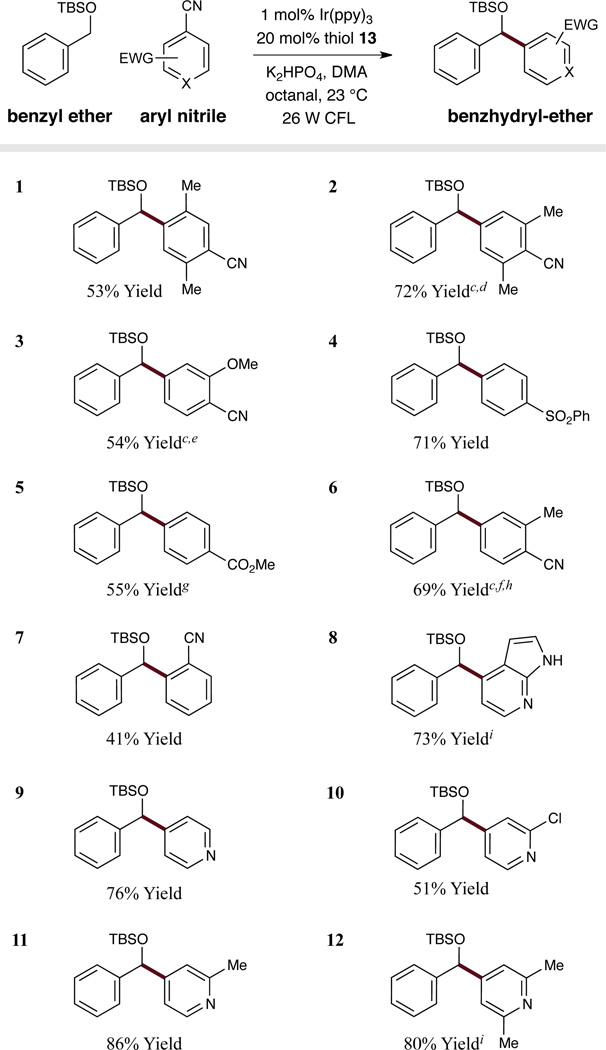 |
Yield of isolated material.
See supporting information for experimental details.
Regiomeric ratios (r.r.) determined by 1H NMR (major isomer is shown; see supporting info for minor isomer).
2:1 r.r.
2:1 r.r.
1.4:1 r.r.
3 equiv. K2HPO4 was used.
Na2CO3 was used as base.
DMA/DMSO (1:1) was used as solvent.
A defining attribute of this new C-H arylation protocol is its potential to provide direct access to a broad array of C–H arylated products. One particular challenge is the selective C–H functionalization of dihydrofuran, a ring system often found in the molecular skeletons of naturally occurring and biologically active substances.20 A major mechanistic concern, however, was the possible formation of two regioisomeric arylation products after the C–H activation step. As shown in equation 3, exposing 2,5-dihydrofuran to our optimized C–H abstraction conditions in the presence of 1,4-dicyanobenzene, resulted in the formation of an arylation adduct in excellent yield, and notably, as a single regioisomer. This regioselectivity is orthogonal to the selectivity observed in the Heck reaction of enol ethers.21
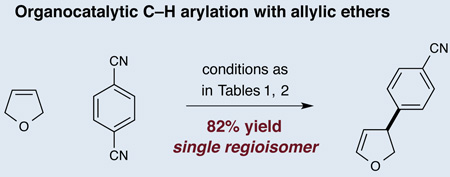 |
(Eq 3) |
In conclusion, we have developed a generic catalytic approach to the direct arylation of benzylic ether C–H bonds. This versatile method was shown to tolerate a range of functionality on both the ether and aryl components. Given the operational simplicity and mild conditions of this new C–H functionalization protocol, we anticipate it will find broad application among practitioners of organic synthesis.
Supplementary Material
Acknowledgement
Financial support was provided by the NIHGMS (R01 GM103558-01) and kind gifts from Merck, Amgen, and AbbVie. K.Q. thanks the Carlsberg Foundation for a postdoctoral fellowship.
Footnotes
Supporting Information Available. Experimental procedures and spectral data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Bergman RG. Nature. 2007;446:391. doi: 10.1038/446391a. [DOI] [PubMed] [Google Scholar]; (b) Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; (c) Labinger JA, Bercaw JE. Nature. 2002;417:507. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (d) Campos KR. Chem. Soc. Rev. 2007;36:1069. doi: 10.1039/b607547a. [DOI] [PubMed] [Google Scholar]
- 2.McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For an overview of reduction potentials of amines, amides, and carbamates, see: Steckhan E. In: Organic Electrochemistry. 4th ed. Henning L, Baizer M, editors. New York: Marcel Dekker Inc; 2001. p. 570. and references therein.
- 4.Luo Y-R. Handbook of Bond Dissociation Energies in Organic Compounds. Florida: CRC Press LLC; 2003. [Google Scholar]
- 5.Mayer JM. Acc. Chem. Res. 2011;44:36. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Mayer JM. Annu. Rev. Phys. Chem. 2004;55:363. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]; (b) Huynh MHV, Meyer TJ. Chem. Rev. 2004;107:5004. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Tyson EL, Ament MS, Yoon TP. J. Org. Chem. 2013;78:2046. doi: 10.1021/jo3020825. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeForest CA, Anseth KS. Angew. Chem. Int. Ed. 2012;51:1816. doi: 10.1002/anie.201106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For some related examples on thiyl hydrogen atom abstraction catalysis in organic synthesis, see: Feray L, Kuznetsov N, Renaud P. In: Radicals in Organic Synthesis. Renaud P, Sibi MP, editors. Vol. 2. Wiley-VCH; 2001. p. 256. and references therein.
- 9.(a) Roberts BP. Chem. Soc. Rev. 1999;28:25. [Google Scholar]; (b) Dang HS, Roberts BP. J. Chem. Soc., Perkin Trans. 1. 1998:67. [Google Scholar]; (c) Dang H-S, Roberts BP. Tetrahedron Lett. 1999;404:8929. [Google Scholar]
- 10.For some recent examples of radical-based ipso-substitution reactions of cyanoaromatics, see: Bernardi R, Caronna T, Poggi G, Vittimberga BM. J. Heterocycl. Chem. 1994;31:903. Zeng X, Cai J, Gu Y. Tetrahedron Lett. 1995;36:7275. Bernardi R, Caronna T, Dal Pio Luogo D, Morrocchi S, Poggi G, Vittimberga BM. J. Chem. Soc., Perkin Trans. 1. 1996:1593. Bernardi R, Caronna T, Morrocchi S, Ursini M. J. Heterocycl. Chem. 1996;33:1137. Tsuji M, Higashiyama K, Yamauchi T, Kubo H, Ohmiya S. Heterocycles. 2001;54:1027. Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Science. 2013;339:1593. doi: 10.1126/science.1232993. Hoshikawa T, Inoue M. Chem. Sci. 2013;4:3118.
- 11.(a) Ameen D, Snape TJ. Med. Chem. Commun. 2013;4:893. [Google Scholar]; (b) Schmidt F, Stemmler RT, Rudolph J, Bolm C. Chem. Soc. Rev. 2006;35:454. doi: 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]
- 12.Braun M. Angew. Chem. Int. Ed. 2012;51:2550. doi: 10.1002/anie.201105127. [DOI] [PubMed] [Google Scholar]
- 13.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top. Curr. Chem. 2007;281:143. [Google Scholar]
- 14.Mori Y, Sakaguchi W, Hayashi H. J. Phys. Chem. A. 2000;104:4896. [Google Scholar]
- 15.Shaidarova LG, Ziganshina S-A, Budnikov GK. J. Anal. Chem. 2003;58:640. [Google Scholar]
- 16.pKa values calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994–2013)
- 17.Tarantino KT, Liu P, Knowles RR. J. Am. Chem. Soc. 2013;135:10022. doi: 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]
- 18.Escoubet S, Gastaldi G, Vanthuyne N, Gil G, Siri D, Bertrand M. J. Org. Chem. 2006;71:7288. doi: 10.1021/jo061033l. [DOI] [PubMed] [Google Scholar]
- 19.Ochiai M, Yamane S, Hoque MM, Saito M, Miyamoto K. Chem. Commun. 2012;48:5280. doi: 10.1039/c2cc31523h. [DOI] [PubMed] [Google Scholar]
- 20.(a) Nakanishi K. Natural Products Chemistry. New York: Kodansha Ltd., Academic; 1974. [Google Scholar]; (b) Meyers AI. Heterocycles in Organic Synthesis. New York: John Wiley & Sons; 1974. [Google Scholar]; (c) Dean FM. In: Advances in Heterocyclic Chemistry. Katritzky AR, editor. Vol. 30. New York: Academic; 1982. p. 167. [Google Scholar]; (d) Dean FM, Sargent MV. In: Comprehensive Heterocyclic Chemistry. Part 3. Bird CW, Cheeseman GWH, editors. Vol. 4. New York: Pergamon; 1984. p. 531. [Google Scholar]; (e) Vernin G. The Chemistry of Heterocyclic Flavoring and Aroma Compounds. Chichester, U.K.: Ellis Horwood; 1982. [Google Scholar]; (f) Danheiser RL, Stoner EJ, Kojama H, Yamashita DS, Klade CA. J. Am. Chem. Soc. 1989;111:4407. [Google Scholar]; (g) Fraga BM. Nat. Prod. Rep. 1992;9:217. doi: 10.1039/np9920900217. [DOI] [PubMed] [Google Scholar]; (h) Merrit AT, Ley SV. Nat. Prod. Rep. 1992;9:243. doi: 10.1039/np9920900243. [DOI] [PubMed] [Google Scholar]; (i) Kilroy TG, O’Sullivan TP, Guiry PJ. Eur. J. Org. Chem. 2005;23:4929. [Google Scholar]
- 21.(a) Arai I, Daves GD., Jr J. Org. Chem. 1979;44:21. [Google Scholar]; (b) Andersson C-M, Hallberg A, Daves GD., Jr J. Org. Chem. 1987;52:3529. [Google Scholar]; (c) Shibasaki M, Boden CDJ, Kojima A. Tetrahedron. 1997;53:7371. [Google Scholar]; (d) Schmidt B. Chem. Commun. 2003:1656. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.