

Abstract
Carotenoid cleavage oxygenases (CCOs, also referred to as carotenoid cleavage dioxygenases (CCDs) in the literature) are a new class of non-heme iron-type enzymes that oxidatively cleave double bonds in the conjugated carbon chain of carotenoids. The oxidative cleavage mechanism of these enzymes is not clear and both monooxygenase and dioxygenase mechanisms have been proposed for different carotenoid cleavage enzymes. CCOs have been described from plants, animals, fungi and cyanobacteria but little is known about their distribution and activities in bacteria other than cyanobacteria. We surveyed bacterial genome sequences for CCO homologs and report the characterization of CCO homologs identified in Novosphingobium aromaticivorans DSM 12444 (NOV1 and NOV2) and in Bradyrhizobium sp. (BRA-J and BRA-S). In vitro and in vivo assays with carotenoid and stilbene compounds were used to investigate cleavage activities of the recombinant enzymes. The NOV enzymes cleaved the interphenyl α-β double bond of stilbenes with an oxygen functional group at the 4’ carbon (e.g. resveratrol, piceatannol, and rhaponticin) to the corresponding aldehyde products. Carotenoids and apocarotenoids were not substrates for these enzymes. The two homologous enzymes from Bradyrhizobium sp. did not possess carotenoid or stilbene cleavage oxygenase activities, but showed activity with farnesol. To investigate whether oxidative cleavage of stilbenes proceeds via a monooxygenase or dioxygenase reaction, oxygen labeling studies were conducted with NOV2. Our labeling studies show that double-bond cleavage of stilbenes occurs via a monooxygenase reaction mechanism.
Keywords: carotenoids, enzymes, microbiology, phytohormones, oxygen
Introduction
Mono- and dioxygenases play important roles in the oxidative modification and cleavage of metabolic compounds. Recently, a new non-heme iron family of oxygenases has been described that catalyze cleavage of double bonds in the conjugated carbon chain of carotenoids to produce apocarotenoids.[1,2] Carotenoid cleavage products have important biological functions as signal molecules, hormones, and attractants for pollinators (reviewed in [1]) and are also of considerable interest for medical and agricultural applications (reviewed in [1-3]).
Carotenoid cleavage oxygenases (CCOs, also referred to as carotenoid cleavage dioxygenases (CCDs) in the literature, see below) have now been identified in all taxa. [4-9] In higher plants, cleavage enzymes have been identified that produce signaling molecules to regulate growth and development, influence fruit color, and affect aroma (reviewed in [8]). Carotenoid oxygenases also play important metabolic and signaling roles in metazoans. [7,9] The symmetric cleavage of β,β-carotene to retinal via carotenoid oxygenase activity, for example, was an important medical discovery (reviewed in [3]). The function of the oxygenase enzymes in microorganisms is far less clear, although recent studies on cloning and characterization of CCOs from cyanobacteria begin to address these questions. [4, 10, 11]
Cloning and characterization of a number of mostly plant derived CCOs showed that these enzymes exhibit different cleavage site and substrate specificities. [6, 12-18] However, very little is known about the mechanism by which these enzymes catalyze oxidative cleavage of double bonds to form two aldehyde cleavage products. Currently only one crystal structure is available for an apocarotenoid specific CCO from the cyanobacteria Synechocystis sp. PCC6803. [19] The structure shows that the enzyme contains a Fe2+ coordinated to four His residues in the active site, which is embedded in a seven-bladed β-propeller chain arrangement topped by a dome comprised of six large loops. However, whether this enzyme catalyzes oxidative cleavage via a mono- or dioxygenase mechanism cannot be deduced from the structure. Labeling studies from plants producing abscisic acid suggested a dioxygenase mechanism. [20] These data were supported by labeling studies examining the production of the aroma compound β-ionone by Arabidopsis thaliana CCD1 (AtCCD1).[21] However, researchers studying vitamin A biosynthesis have suggested at different times with different enzyme examples both a dioxygenase mechanism and a monooxygenase-like mechanism through a postulated epoxy intermediate.[22] Consequently, we chose to use the term CCO (for carotenoid cleavage oxygenase) to describe this class of enzymes.
We have recently begun to characterize putative CCO homologs identified in genome sequences of cyanobacterial strains in order to gain a better understanding of their functions in photosynthetic bacteria.[4] Our analysis of bacterial genome sequences for new members of the CCO family also identified several putative CCO homologs in carotenogenic and noncarotenogenic bacteria, indicating that at least some of these enzymes probably cleave substrates other than carotenoids. In the early 1990's enzymes that cleave the interphenyl α,β double bond of trans-stilbenes have been described from the soil bacterium Sphinogomonas paucimobilis TMY1009 (four isoforms SPA1-4).[23,24] These enzymes are believed to have a catabolic function by cleaving stilbene-type compounds derived from lignin degradation and have therefore been named lignostilbene-alpha,beta-dioxygenases (LSD, EC 1.13.11.43); although dioxygen incorporation has never been experimentally established for these enzymes. The identification of CCOs several years later showed that the Sphingomonas enzymes are CCO homologs that are most closely related to 9-cis-epoxycarotenoid dioxygenases (NCEDs) that generate the precursor of the plant hormone abscisic acid.[16,25]
In this study we survey bacterial genomes for other CCO homologs and describe the characterization of two CCO paralogs NOV1 and NOV2 identified in the non-carotenogenic Novosphingobium aromaticivorans DSM12444 and two CCO homologs from the non-carotenogenic Bradyrhizobium japonicum USDA110 and carotenogenic Bradyrhizobium sp. BTAi1 (BRA-J and BRA-S respectively). In addition, isotopic oxygen labeling experiments show that NOV2 is a monooxygenase, which is in contrast to a recent study suggesting a dioxygenase mechanism for the Arabidopsis thaliana CCD1 enzyme.[21]
Results and Discussion
Bacterial CCO homologs
We previously surveyed cyanobacterial genome sequences for putative CCO enzymes and characterized cleavage activities of several recombinant enzymes.[4] Like in plants, we expected to find in cyanobacteria mostly CCO enzymes that cleave (apo)carotenoids as based on their presumed function in general carotenoid breakdown and synthesis of apocarotenoids for light-sensing (retinal, rhodopsin) and/or other signaling functions. In fact, cloned putative cyanobacterial CCOs cleaved (apo)carotenoids with different selectivities and cleavage specificities (9, 10, 9’10’; 15,15’; apo-9,10), although several of these CCO sequences, e.g. from Nostoc punctiforme and Nostoc sp. PCC7120, were annotated as lignostilbene α, β dioxygenases.[4] A BLAST analysis of published bacterial genome sequences with sequences of experimentally characterized CCOs identifies a number of CCO homologs in bacteria with and without annotated carotenoid pathways in their genomes (Figure 1 and Supplementary Table 1). The absence of carotenoid biosynthetic pathways in some bacteria suggests alternative catalytic activities other than carotenoid cleavage. We selected four sequences from carotenogenic and non-carotenogenic proteobacteria for further characterization: two paralogs from Novosphingobium aromaticivorans DSM12444 (NOV1 and NOV2), one each from Bradyrhizobium japonicum USDA110 (BRA-J) and Bradyrhizobium sp. BTAi1 (BRA-S). Novosphingobium does not have characterized carotenoid biosynthetic pathways, although there are several carotenoid associated genes in the genome according to the KEGG database.[26] Novosphingobium is well known for its ability to degrade phenolic structures.[27] Bradyrhizobium strains are nitrogen-fixing symbionts of legumes.[28] While B. japonicum USDA110 is non-photosynthetic and does not synthesize carotenoids, B. sp. BTAi1 is photosynthetic and therefore makes carotenoids.
Figure 1.

Phylogenetic tree [average distance by percent identity] from NOV1 and NOV2 amino acid sequences with additional representatives from sequenced microbial genomes (BLAST hits with high identity) and other characterized carotenoid oxygenase family representatives. Lignostilbene oxygenase activity: NOV1 (accession no. YP_496081), NOV2 (accession no. YP_498079) (this study), Sphingomonas paucimobilis isoform 1 (SPA1, accession no. AAC60447) [23] and isoform 3 (SPA3, accession no. AAB35856 ).[24] Apocarotenoid cleavage activity: Synechocystis PCC6803 (SYC2, accession no. S76169),[10] Nostoc sp. PCC7120 9,10 (NSC3, accession no. ZP_00112423).[4] 15, 15’ carotenoid cleavage activity: Synechococcus elongates PCC 7942 (SYO, accession no. ZP_00351210) (unpublished), Nostoc sp. PCC7120 (NSC2, accession no. accession no. AE2341),[4] mouse 15,15’-dioxygenase (MmBCO1, accession no. Q9JJS6).[41] Unknown activity: Bradyrhizobium japonicum USDA110 (BRA-J, accession no. NP_772430) (this study), Bradyrhizobium sp. Btai1 (BRA-S, accession no. ZP_008636652) (this study), Synechocystis PCC6803 (SYC1, accession no. S76206).[10] 9,10-9’10’ carotenoid cleavage activity: mouse 9,10-9’10’-dioxygenase (MmBCO2, accession no. Q99NF1),[9] Zea mays (ZmCCD1, ABF8565B), Phaseolus vulgaris (PvCCD1, Q94IR2) [6], Arabidopsis thaliana (AtCCD1, accession no. NP_191911.1) [6], Lycopersicon esculentum (LeCCD1, accession no. AAT68187),[17] Petunia × hybrida (PhCCD1, accession no. AAT68189),[18] Nostoc sp. PCC7120 (NSC1, accession no. BAB73063).[4] Isomerase activity: mouse RPE protein (MmRPE65, accession no. Q91ZQ5);[42] 9-cis-epoxycarotenoid 11,12 cleavage activity: Zea mays (VP14, accession no. AAB621811.1),[16] Arabidopsis thaliana (AtNCED1, accession no. AAN17413). Additional enzymes and accession numbers for putative oxygenases can be found in Supplemental Table 1.
The phylogenetic tree in Figure 1 shows that BRA protein sequences are most closely related to putative CCO homologs in Ralstonia and Rhodopseudomonas, while NOV sequences cluster with two known LSDs from Sphingomonas paucimobilis SPA1 (accession number AAC60447) and SPA3 (accession number AAB35856).[29] All four putative CCO sequences (NOV and BRA) are more closely related to cyanobacterial enzymes than to plant CCOs. Alignments show that NOV1 and the SPA1 and SPA3 proteins from Sphingomonas paucimobilis share the highest degree of amino acid sequence identity (55% and 56% respectively) between genera (Supplemental Figure 1). This is considerably different than the similarity between SPA enzymes and NOV2, which are 37-38%. The SPA enzymes are more alike one another (68% identity) than are the NOV enzymes (36%). The two BRA proteins share 32-37% identity to both the SPA enzymes and the NOV enzymes (with 80% similarity between BRA-J and BRA-S).
Survey of cleavage activities in carotenoid or stilbene synthesizing E. coli strains
A CCO enzyme from Synechocystis has been shown to be membrane associated;[11] membrane association is one contributing factor to the difficulty of developing optimal in vitro assay conditions for CCO enzymes (other difficulties are described in [30]). As a result, in vivo detection of carotenoid cleavage activity through co-expression of the CCO enzyme in question with carotenoid biosynthetic pathways has become the standard approach to identifying active enzymes. To determine the cleavage activity of NOV and BRA enzymes, genes were amplified from genomic DNA and cloned into the constitutive E. coli expression vector pUCmod.[31] For a survey of stilbene or carotenoid cleavage activities, genes were expressed in recombinant E. coli producing β-carotene, zeaxanthin, torulene or different stilbene compounds.
In vivo cleavage of carotenoid structures produced in E. coli was investigated essentially as described previously for the characterization of cyanobacterial CCOs.[4] Briefly, CCO homologs on pUCmod were co-expressed with a carotenoid pathway expressed from pACmod. Based on previous results showing that the bicyclic carotenoid β-carotene is a substrate for many CCOs [6, 9, 22], β-carotene produced by genes encoded on plasmid pAC-crtE-crtB-crtI14-crtY [31] was chosen as the model carotenoid for this study. Two additional carotenoids with other structural features were also tested: torulene as a monocyclic carotenoid with one β-ionone end group and a linear end, and zeaxanthin as a bicyclic carotenoid with hydroxylated β-ionone end-groups. Cleavage of carotenoids in E. coli destroys the chromophore, causing a loss of cell color (also referred to as bleaching) that can be visually detected when compared to control cells. However, none of the tested four enzymes caused bleaching of the cell color of carotenoid producing E. coli suggesting that carotenoids are not likely a substrate of NOV and BRA enzymes.
We applied the same in vivo experimental approach to test the activity of NOV and BRA enzymes against stilbene substrates. In previous research, we created recombinant E. coli cells that co-express stilbene synthase (STS) and 4-coumaroyl CoA-ligase (4CL), enabling the synthesis of stilbene compounds from phenylpropionic acid precursor compounds fed to the recombinant cells.[32] Biotranformation of phenylpropionic acid precursors coumaric acid, cinnamic acid, or caffeic acid results in the synthesis of resveratrol (3,5,4’-trihydroxy-trans-stilbene), pinosylvin (5-(2-phenylvinyl)-1,3-benzenediol), and piceatannol (3,3’.4,5-tetrahydroxy-trans-stilbene), respectively (Figure 2). E. coli cells expressing the stilbene pathway were co-transformed with NOV and BRA genes and enzymatic cleavage of resveratrol, pinosylvin and piceatannol produced by the recombinant cultures was investigated. In addition, E. coli cells co-expressing only 4CL and putative CCO enzymes were fed phenylpropionic acid precursors and the resulting compounds analyzed to rule out cleavage of CoA-activated phenylpropionic acids by NOV and BRA enzymes.
Figure 2.
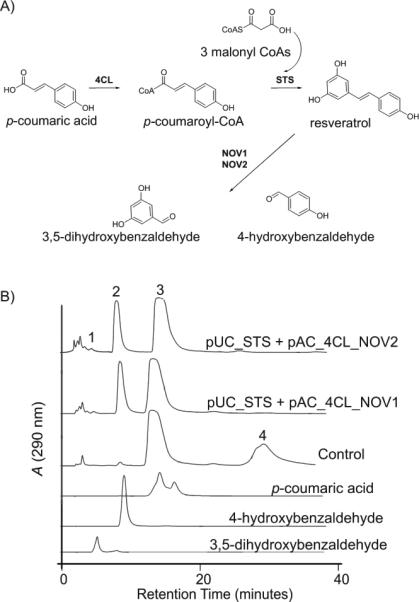
In vivo cleavage of resveratrol by NOV1 and NOV2. A) Engineered pathway in E. coli for resveratrol biosynthesis from fed coumaric acid and cleavage of synthesized resveratrol in E. coli by co-expressed NOV enzymes. Enzymes shown are 4-coumaroyl:CoA ligase (4CL; EC 6.2.1.12), stilbene synthase (STS; EC 2.3.1.95), and NOV oxygenases (NOV1, NOV2). B) HPLC analysis of extracts from coumaric acid fed recombinant E. coli cultures co-expressing stilbene biosynthetic genes and NOV1 (pUC_STS + pAC_4CL+NOV1) or NOV2 (pUC_STS + pAC_4CL+NOV2). The control culture contained only stilbene biosynthesis genes (pUC_STS + pAC_4CL). Shown are HPLC traces of culture extracts and of authentic standard compounds. Peaks are (1) 3,5-dihydroxybenzaldehyde (m/z 137.0), (2) 4-hydroxybenzaldehyde (m/z 121.0), (3) p-coumaric acid (m/z 163.2), (4) resveratrol (m/z 227.1). Control cultures convert coumaric acid to resveratrol. Addition of the oxygenase enzymes NOV1 and NOV1 results in a decrease in resveratrol and the appearance of 4-hydroxybenzaldeyde and 3,5-dihydroxybenaldehyde. The products were confirmed by standards and mass spectral analysis. 3,5-dihydroxybenzaldehyde is not extracted from the medium in stoichiometric amounts.
NOV1 and NOV2 both efficiently cleaved resveratrol resulting in complete degradation of resveratrol by recombinant E. coli strains after 16 hrs of cultivation and in the accumulation of two new products (Figure 2). The two new compounds were structurally identified as 4-hydroxybenzaldehyde and 3,5-dihydroxybenzaldehyde by comparison of retention time and mass spectra with those from authentic compounds. The dihydroxy product, 3,5-dihydroxybenzaldehyde, was not extracted from the medium in stoichiometric amounts suggesting it was further degraded by E. coli enzymes or formed Schiff's base adducts. Piceatannol, the stilbene compound produced from caffeic acid, was also cleaved by NOV1 and NOV2 into the corresponding aldehyde products 3,4-dihydroxybenzaldehyde and 3,5-dihydroxybenzaldehyde (data not shown). However, since conversion of fed caffeic acid to the corresponding stilbene piceatannol by the recombinant E. coli cells is much slower compared to the production of resveratrol from 4-coumaric acid [32], the amounts of piceatannol cleavage products detected in the cultures was correspondingly lower. Cleavage of pinosylvin, the stilbene product from cinnamic acid, by NOV1 or NOV2 was not observed. Both NOV1 and NOV2 were specific for stilbene cleavage and no cleavage products of fed phenylpropionic acids or CoA activated phenylpropionic acids were detected in culture extracts (data not shown).
Surprisingly, no stilbene cleavage products were detected in recombinant E. coli cultures expressing BRA-J or BRA-S despite their sequence similarity to the NOV and SPA enzymes. Previously characterized cyanobacterial CCOs such as NSC1, NSC2, and SYC2 [4, 10] were also expressed in stilbene producing E. coli cells and found to not cleave stilbenes.
Characterization of in vitro cleavage activities
Assays with purified protein and/or whole cell protein extracts were conducted to confirm the cleavage results obtained in recombinant E. coli and to test additional substrates. NOV and BRA genes were overexpressed from a pET expression vector in a recombinant E. coli strain that also expressed the GroEL and GroES chaperones to aid in the production of soluble protein. Overexpressed histidine-tagged proteins were purified by immobilized metal affinity chromatography and used in in vitro assays. However, as with other reports from this family of enzymes ([21,30]), purified enzymes were far less active than enzymes in protein extracts from whole cell lysates (losing more than 75% of their activity during purification). As a consequence, protein extracts from whole cell lysates are frequently used in assays with CCO enzymes [21]. We used both purified proteins and protein extracts from whole cell lysates for in vitro assays with NOV and BRA enzymes and observed similar cleavage specificities for both preparations; but ~ 5-fold lower cleavage rates were obtained with purified proteins. A series of cofactors and reducing agents tested with the enzyme (NAD, FAD, NADH, FADH, ascorbate, excess Fe2+, glutathione, dithiothreitol) did not improve enzyme activity. Protein extracts from whole cell lysates were prepared from E. coli cells overexpressing NOV and BRA genes from the constitutive expression vector pUCmod (Supplementary Figure 2).
A series of stilbene substrates (rhapontigenin, resveratrol, rhaponticin, piceatannol and pinosylvin) with different hydroxyl and methoxy functional groups were tested in in vitro assays with NOV and BRA enzymes (Figure 3). NOV1 and NOV2 cleaved stilbene compounds that have a hydroxy or methoxy group at the 4’ position at the central double bond. As observed in the in vivo cleavage survey, pinosylvin which carries no substitution at the 4’ position, was not a substrate for NOV enzymes. Resveratrol was the preferred substrate for NOV1 and NOV2, followed by piceatannol and the 4’ methoxy group bearing stilbenes rhapontigenin and rhaponticin. In vitro assays with equal amounts of protein lysates resulted in complete cleavage of 1 mM (68.5 μg) resveratrol in 20 min, while complete cleavage of 1 mM (73.3 μg) piceatannol required 60 min of incubation (Figure 3). Cleavage of 1 mM (77.4 μg) rhapontigenin and its glucosylated derivative rhaponticin by NOV1 and NOV2 was much slower and only 20% of the substrates were cleaved after 60 min (data not shown).
Figure 3.
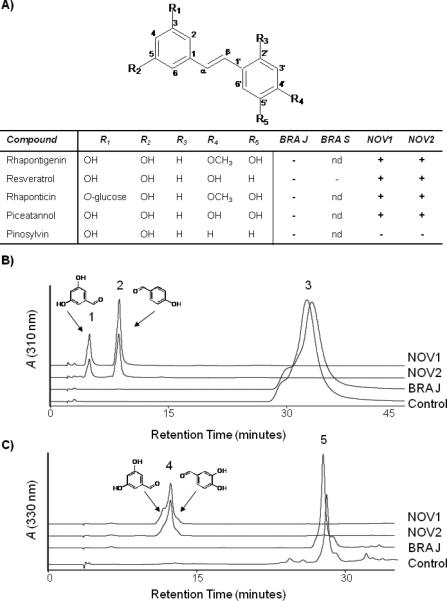
In vitro cleavage of stilbene compounds. A) Stilbene compounds with different functional groups were chosen as substrates for in vitro assays with NOV1, NOV2, BRA-J, and BRA-S protein lysates. Cleavage of the substrates is indicated by a + in the table based on product identification by HPLC and LC-MS. None of the enzymes cleaved pinosylvin. B) HPLC analysis of in vitro assays with resveratrol as a substrate. Synthesis of 3,5-dihydroxybenzaldehyde (1) and 4-hydroxybenzaldehyde cleavage products (2) from 1 mM resveratrol (3) with NOV1 and NOV2 protein extracts. No residual resveratrol was detected by HPLC or LCMS. C) HPLC trace of in vitro assay with piceatannol as a substrate. Synthesis of 3,5-dihydroxybenzaldehyde and 3,4-dihydroxybenzaldehyde cleavage products (4) from 1 mM piceatannol (5) with NOV1 and NOV2 enzymes protein extracts. Small amounts of residual piceatannol could be detected for NOV2. BRA-J protein extracts did not produce cleavage products with either resveratrol or piceatannol. Protein extracts from E. coli served as a control.
To confirm the in vivo results showing that none of the four tested enzymes cleaved carotenoids, NOV and BRA enzymes were tested against the apocarotenoid substrate β-apo-8’-carotenal. β-apo-8’-carotenal was chosen as a substrate instead of β,β-carotene because of its greater solubility, resulting in higher activities of carotenoid cleaving enzymes with apocarotenoid substrates compared to full-length carotenoids.[21] Moreover, some carotenoid cleavage enzymes are specific for apocarotenoids.[10] However, no cleavage products of β-apo-8’-carotenal were detected in in vitro assays with NOV and BRA enzymes.
BRA-J and BRA-S enzymes were also assayed with several stilbene compounds (Figure 3). However, no cleavage activity of stilbene compounds was detected confirming the results obtained with stilbene producing E. coli cells. Bradyrhizobium strain USDA 110 has been shown to produce the plant phytohormone abscisic acid via an unknown pathway.[50] We confirmed abscisic acid biosynthesis for strain B. japonicum USDA 110 and Bradyrhizobium sp. BTAi1 (Supplemental Figure 4). Because these Bradyrhizobium strains produce either no carotenoids at all (USDA 110) or no epoxy-carotenoids (BTAi1), abscisic acid biosynthesis in Bradyrhizobium must occur via a different route than the plant pathway, which involves oxidative cleavage of epoxy-carotenoids by a CCO (NCEDs).[16] In filamentous fungi, abscisic acid is synthesized from farnesol via a partially described pathway. The recent identification of an abscisic acid gene cluster in Botrytis cinerea suggests the involvement of several oxidative steps in the conversion of farnesol to abscisic acid.[34] Therefore we tested whether the BRA CCO homologs have activity against farnesol. GC-MS analysis of in vitro assays with farnesol as substrate showed conversion of farnesol into a new compound by BRA-J and BRA-S, but not by the NOV enzymes or in the control reaction (Supplemental Figure 5). The parent ion detected for this new product is m/z 290, which is consistent with it being a methanol adduct [M + 32] of a farnesol derivative containing two additional oxygen groups m/z 258 (M+). However, the structure of this compound can not be deduced from the MS data alone.
Oxygen labeling studies with NOV2
Cleavage of the central double bond of stilbenes catalyzed by CCO homologs from Novosphingobium identified in this study and Sphingomonas LSDs reported previously [35], is similar to the carotenoid cleavage reaction observed with NCED enzymes in the production of abscisic acid [16] and the central double bond cleavage observed by CCOs responsible for the production of retinal from β-carotene.[36] The enzyme mechanism using molecular oxygen and ferrous iron is thought to be similar among the different types of carotenoid or stilbene cleaving oxygenases.[25] Incorporation of one or two molecules of oxygen from atmospheric oxygen during catalysis by these enzymes is still controversial and both mono- and dioxygenase mechanisms have been suggested for carotenoid cleaving oxygenases (Scheme 1).[20-22] Poor activities of CCOs in in vitro assays and cleavage of water insoluble substrates may largely be responsible for the lack of rigorous mechanistic studies of this new class of non-heme iron oxygenases. Despite a few reports of characterized purified recombinant CCOs ([10, 36]), many studies rely on protein lysates.[21] However, compared to previously studied CCOs [4, 21], cleavage reactions with NOV enzymes were fast and stilbene substrates are much more soluble than carotenoids and can be analyzed by GC-MS. Both properties allow short assay times followed by a quick analysis of reaction products, thereby minimizing unspecific label exchange during oxygen labeling studies. We therefore sought to perform oxygen labeling studies with NOV2 to determine whether this class of oxygenases uses a mono- or dioxygenase mechanism.
Scheme 1.
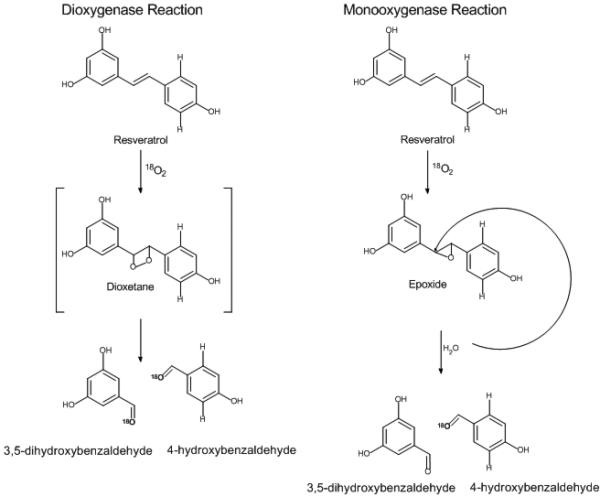
Possible mechanisms for the oxidative cleavage of resveratrol. Shown are two proposed mechanisms for oxidative cleavage with CCO enzymes. A dioxygenase mechanism results in both aldehyde cleavage products labeled with isotopic 18O when the reactions are performed in an 18O atmosphere. The monooxygenase mechanism results in a single isotopic 18O labeled cleavage product when reactions are performed in an 18O atmosphere.
First, resveratrol cleavage by NOV2 was assayed in an atmosphere of labeled oxygen 18O2. The reaction was stopped after 15 min and labeled cleavage products analyzed by GC-MS. Only one of the expected cleavage products, 4-hydroxybenzaldehyde, was found to carry significant amount of the 18O label (over 69% labeled) (Figure 4). In contrast, the 3,5-dihydroxybenzaldehyde product did not contain an equivalent amount of the heavy oxygen label, suggesting that its aldehyde oxygen must come from unlabeled water in the reaction.
Figure 4.

GC-MS analysis of oxygen labeled cleavage products of resveratrol synthesized by NOV2. A) Silylated esters of cleavage products 4-hydroxybenzaldehyde (1) and 3,5-dihydroxybenzaldehyde (2). B) Incorporation of molecular oxygen into 4-hydroxybenzaldehyde. Mass spectra of 4-hydroxybenzaldehyde (left spectrum) and 3,5-dihydroxybenzaldehyde (right spectrum) in 18O2-atmosphere, showing labeled 4-hydroxybenzaldehyde (m/z 196) and unlabeled 3,5-dihydroxybenzaldehyde (m/z 282). C) Mass spectral data from incorporation of oxygen from water H218O. Incorporation of label into 3,5-dihydroxybenzaldehyde (m/z 284), but 4-hydroxybenzaldehyde remains unlabeled (m/z 194). Note that the main fragments in panels 1B and 1C (m/z 181 and m/z 179) result from the loss of a [–CH3] group from 4-hydroxybenzaldehyde.
To confirm the oxygen labeling results, a second assay was performed with labeled H218O. As expected, a reverse labeling pattern of the cleavage products was observed (Table 1). Now, the 3,5-dihydroxybenzaldehyde carried the 18O label (7.1 (m/z 282) : 92.9 (m/z 284)) and the 4-hydroxybenzaldehyde cleavage product was predominately unlabeled (93.3 (m/z 194): 6.7 (m/z 196)) (Figure 4). The ratio is determined by the ratio of the extracted ion chromatograms [M+H] ratio of heavy to total 4-hydroxybenzaldehyde (i.e. 196/194+196) as in Schmidt et al. 2006 [21]. The ratio of labeled to unlabeled aldehyde cleavage products were higher in this H218O labeling experiment compared to the corresponding atmospheric oxygen labeling experiment. One reason for the apparent imbalance may be due to nonenzymatic oxygen exchange of the hydroxyl groups in the labeled water leading to an over representation of the labeled 3,5-dihydroxybenzaldehyde product. A time course (1, 5, 15, 30 minute) was performed to monitor oxygen exchange during the enzyme assay and in control reactions containing authentic 4-hydroxybenzaldehye and 3,5-dihydroxybenzaldehyde without NOV2 protein (Supplemental Figure 3 and Supplemental Table 2). Exchange of oxygen label in the enzyme assays does occur more rapidly with the di-hydroxy cleavage product, 3,5-dihydroxylbenzaldehyde, than with the mono-substituted 4-hydroxybenzaldehyde (Supplemental Figure 3). After 15 minutes the labeling pattern in the enzyme reaction clearly showed heavy label mainly on one product, 3,5-dihydroxylbenzaldehyde and only a small fraction of the 4-hydroxybenzaldehyde was labeled. Control reactions show that some non-enzymatic label exchange occurs predominantly with the di-hydroxy cleavage product. Together these results suggest that NOV2 cleaves resveratrol via a monooxygenase mechanism and may have a stereo-preference for oxygen attack.
Table 1.
Summary of labeling pattern from isotope experiments
| Product Mass | 18O2 | Product Mass | H218O | |
|---|---|---|---|---|
| 3,5-d ihydroxybenzaldehyde | m/z 282 | Unlabeled ~65% | m/z 284 | Labeled ~92% |
| 4-hydroxybenzaldehyde | m/z 196 | Labeled ~69% | m/z 194 | Unlabeled ~90% |
Conclusion
In this work we describe the cloning and partial characterization of four bacterial enzymes belonging to a recently described class of non-heme iron oxygenases that so far mostly includes carotenoid cleaving enzymes from plants, mammals and cyanobacteria. Two enzymes (SPA1 and SPA3) from Sphingomonas paucimobilis, previously known to cleave substrates other than carotenoids, have been named lignostilbene dioxygenases (LSDs) (for reviews see [3]). This study expands the number of non-carotenoid cleaving family members with the finding that two enzymes from Novosphingomonas aromaticivorans DSM12444 are stilbene cleaving oxygenases. We show in vivo and in vitro that the enzymes NOV1 and NOV2 cleave the central double bond of trans-stilbene derivatives but do not cleave bicyclic, monocyclic, or hydroxylated model carotenoid substrates. In contrast, the putative CCOs from Bradyrhizobium japonicum USDA110 and Bradyrhizobium sp. BTAi1 were not active against stilbenes or carotenoids but showed activity with farnesol. In the course of these studies, we also tested known carotenoid cleavage enzymes for their ability to cleave stilbenes and we did not find stilbene cleavage activity with enzymes from the cyanobacteria Nostoc punctiforme, Nostoc sp. PCC7120, or Synechocystis sp. PCC6803.[4, 10, 11]
NOV1 and NOV2 are, to our knowledge, the second reported examples of stilbene cleaving oxygenases. Studies of LSD isoforms from Sphingomonas paucimobilis differ from this report in a few significant respects.[23, 24, 29, 35, 37] In our assays, the recombinant enzymes were tested in vivo and in vitro against natural substrates from plants such as resveratrol and piceatannol. Previous reports identified the 4’hydroxyl group of stilbenes as a key structural feature for cleavage.[35] We found that the recombinant Novosphingobium enzymes cleaved compounds with 4’ hydroxyl groups or 4’methoxy functional groups in vitro suggesting that an oxygen functional group (not specifically a hydroxyl) in that position may be important for substrate binding. NOV1 and NOV2 did not cleave pinosylvin, a substrate lacking oxygen on the 4’ carbon. The NOV enzymes in this study displayed similar substrate preferences (cleaving resveratrol better than the other substrates) as opposed to the Sphingomonas enzymes which all had different substrate specificities.[29] Finally, we performed labeling studies to determine the oxygenase cleavage mechanism of these enzymes.
The current controversy over the oxygenase mechanism of this family of non-heme iron enzymes stems from contradictory findings from previous labeling studies and a lack of rigorous biophysical studies as the result of the difficulties associated with assays using purified carotenoid cleavage enzymes.[21,22] The stilbene cleavage reaction has several advantages over carotenoid cleavage, making it a good candidate for mechanistic studies. First, the reaction, when carried out with protein extracts from whole cell lysates, is fast (complete resveratrol cleavage under 20 minutes with 5 μg of protein). Second, the stilbene substrate resveratrol is more soluble in aqueous systems than the lipophilic carotenoids and the cleavage products can be easily worked up in organic solvents limiting water exchange. Third, stilbene cleavage products can be readily identified by GC-MS after derivatization, limiting the amount of oxygen exchange of the carbonyl group. In contrast, the aqueous and acidic HPLC conditions used for the analysis of carotenoid cleavage products in previous labeling studies lead to oxygen exchange rendering the interpretation of results difficult.[20,21]
Assays performed in an 18O2 environment with NOV2 and resveratrol as substrate resulted in predominant labeling (~ 69%) of one product, 4-hydroxybenzaldehdye. In the converse experiment with H218O in an unlabeled O2 environment, the aldehyde group of the other cleavage product, 3,5-dihydroxybenzaldehyde, is almost completely labeled. Control experiments with authentic aldehyde cleavage products and no enzyme added (Supplemental Table 2 and Supplemental Figure 3) show that some unspecific label exchange is observed with the more reactive 3,5-dihydroxybenzaldehedye product and H218O. Despite the observed label exchange with the 3,5-dihydroxybenzaldehyde product, the data from these assays indicate that the recombinant NOV2 stilbene oxygenase uses a monooxygenase reaction mechanism and that the atmospheric oxygen is preferentially added to the 4-hydroxybenzaldehyde cleavage product.
Our results contradict findings from an abscisic acid labeling study in plants as well as a recent in vitro oxygen labeling study conducted with the recombinant CCO from Arabidopsis (AtCCD1) which suggest a dioxygenase mechanism for this enzyme family. [20, 21] The abscisic acid study has been criticized for examining only one cleavage product and prematurely describing the reaction as a dioxygenase mechanism; we examined both cleavage products and found regio-selectivity, which could explain the abundant label found on the one cleavage product analyzed in the abscisic acid study. In vitro labeling studies with a 15,15’ carotenoid cleavage oxygenase from chicken also suggest a monooxygenase mechanism, which is similar to our findings.[22] However, the Leuenberger et al. study used a coupled enzyme reaction in which the formed aldehyde cleavage products are converted in situ to the corresponding less reactive alcohol products in order to reduce label exchange and facilitate GS-MS analysis of derivatized alcohols. It is possible that different members of this enzyme family catalyze similar reactions by a different oxygenase mechanism, which may even vary depending on the cleaved substrate. Rieske oxygenase family members, for example, have been shown to have mono- or di-oxygenase activity based on different substrates and enzymes. [43] Examinations of Rieske-type oxygenases show that subtle changes in the active site can alter the enzyme mechanism.[43] Perturbations in the active site may create an environment advantageous for a monoxygenase-like mechanism rather than a dioxygenase mechanism or vice versa.
Exact mechanistic details have not been determined for this new class of non-heme iron oxygenases because of the poor activity of in vitro assays. Assays frequently use a reductant such as ascorbate, DTT, TCEP, or excess Fe2+ to preserve the ferrous iron, but no other cofactors, iron-sulfur proteins or reductases have been identified as required to balance the electron flow. It has been suggested that all the electrons in the product can come exclusively from the substrate and oxygen.[2] Unfortunately, there is only one crystal structure of a carotenoid cleavage enzyme (Synechocystis sp. PCC6803) [19] available and the stilbene oxygenases from Novosphingobium model poorly onto the solved structure (Supplemental Figure 6). The β-strands forming the propellers are conserved but there is large variation in the amino acid residues forming the dome and entrance loop. Descriptions of the Synechocystis protein structure state that ring structures will not fit through the active site tunnel. [29] Cleavage of stilbene structures, however, requires positioning of at least one phenol ring in the active site of the NOV enzyme, illustrating that there may be important differences in structure and function among the members of this enzyme family. NOV2 residues surrounding the tunnel entrance loops (residues Leu239-Lsy243 and Phe106-Pro110) are structurally different in the model than in the Synechocystis structure. More labeling and mechanistic studies along with structural investigations are required to begin to understand catalysis of these enzymes.
This study also showed that analysis of sequence information is not sufficient to predict carotenoid or stilbene activity and substrate specificity need to be determined empirically for all new examples of these oxygenases. The two Bradyrhizobium enzymes share a similar degree of sequence identity to Sphingomonas enzymes as the NOV2 enzyme does (~35%). However, the two enzymes from Bradyrhizobium did not cleave stilbenes or carotenoids and instead showed activity with farnesol. Although the mass fragmentation pattern of the farnesol reaction product does not allow structural assignment, the fragment at m/z 259 [M+] arising from the loss of methanol from the methanol adduct parent (m/z 290) suggests the addition of two oxygen groups to farnesol (and likely bond rearrangement in order to arrive at m/z 258) rather than oxidative cleavage of farnesol by BRA enzymes which would result in products with lower molecular weight and shorter retention times. Farnesol has been identified as the precursor of abscisic acid in filamentous fungi.[34] It is postulated that conversion of farnesol to abscisic acid involves several oxidative steps.[34] Knockout studies in Botrytis cinerea have identified two P450 monooxygenases that likely catalyze two of the postulated oxidation reactions.[38] Biosynthesis of the phytohormone abscisic acid in the Bradyrhizobium must also occur through a different route than the epoxy-carotenoid cleavage pathway in plants as these bacteria are either non-carotenogenic or do not produce epoxy-carotenoids. Additional studies involving the creation of gene knockouts will be necessary to investigate whether BRA-J and BRA-S are involved in abscisic acid biosynthesis in the plant symbiont Bradyrhizobium. Interestingly, a BLAST search of the Botrytis cinerea genome sequence (Broad Institute) with the BRA protein sequences identifies two putative CCO homologs. Deletion of these putative CCO genes could establish whether one of them catalyzes yet unknown steps in abscisic acid biosynthesis in this ascomycete.
The activity of the BRA enzymes indicates that there may be many new activities to be discovered for other putative microbial CCO homologs (Figure 1). Of equal interest are investigations aimed at identifying the biological functions of these enzymes in bacteria and fungi. Carotenoid cleavage in plants and mammals has functions that extend beyond pigment degradation and synthesis of visual pigments as more and more roles of carotenoid cleavage compounds in signaling are being discovered.[8] It can be assumed that the bacterial and fungal representatives of the CCO family have similarly diverse functions beyond simple degradation.
Experimental Section
Chemicals and materials
Caffeic acid, ferulic acid, piceatannol, rhaponticin, β-apo-8’-carotenal and bistrimethylsilytrifluoroacetamide (BSTFA) were purchased from Sigma Aldrich (St. Louis, MO). 4-coumaric acid was purchased from ICN (Aurora, OH) and resveratrol was from Calbiochem (San Diego, CA). The 95% heavy water H218O was from Cambridge Isotope Laboratories (Andover, MA). All solvents were of HPLC grade and purchased through Fisher Scientific (Pittsburgh, PA). HPLC grade water was purchased from Malllinckrodt Chemicals (Phillipsburg, NJ). Vent DNA polymerase, T4 DNA ligase and restriction enzymes were from New England Biolabs (NEB, Boston, MA). Restriction buffers were SuRE/Cut buffers from Roche (Indianapolis, IN).
Gene cloning
Homology searches were performed using NCBI BLAST software based on the Sphingomonas paucimobilis lignostilbene oxygenase proteins (SPA1 (isoform I) AAC60447; SPA3 (isoform III) AAB35856 [24, 29] and previously characterized cyanobacterial CCO's [4, 10]. CCO homologs were identified in published complete genome sequences obtained from NCBI and Joint Genome Institute (Figure 1 and Supplementary Table 1). Putative CCO homologs from Novosphingobium aromaticivorans DSM 12444 (NOV1 (YP_496081); NOV2 (YP_498079)), Bradyrhizobium japonicum USDA110 (BRA-J (NP_772430) and Bradyrhizobium sp. BTAi (BRA-S) were selected for cloning and functional characterization. The putative CCO genes were amplified from genomic DNA by PCR with Vent polymerase using gene specific primers with added restriction sites. NOV1 and NOV2 were cloned into the BglII and NotI sites of the constitutive expression vector pUCmod [31] to give pUCmod-NOV1 and pUCmod-NOV2. BRA-J and BRA-S were cloned into the NdeI and XhoI sites of pUCmod yielding pUCmod-BRA-J and pUCmod-BRA-S.
For expression of higher protein amounts for protein purification of NOV1 and NOV2, genes were subcloned into the NdeI and XhoI sites of the inducible expression vector pET28b+ (Invitrogen, Carlsbad, CA) giving plasmids pET-NOV1 and pET-NOV2. The stop codon was eliminated from the sequences for in frame fusion with a C-terminal 6X histidine tag encoded on the pET28b+ vector to facilitate protein purification. BRA-J and BRA-S were subcloned in a similar fashion into the inducible expression vector pET24b+ (Invitrogen, Carlsbad, CA) to yield plasmids pET-BRA-J and pET-BRA-S.
For co-expression of CCO homologs NOV1, NOV2, BRA-J and BRA-S (in the following collectively referred to as CCO's) with stilbene biosynthetic genes in E. coli, the entire CCO expression cassettes (including the constitutive lac-promoter and gene coding region as described in [31]) in pUCmod-CCO were amplified by PCR using sequence specific primers with added restriction sites and subcloned into the XbaI of plasmid pAC-4CL [32] that contains the gene for 4-coumaroyl ligase (4CL) from Arabidopsis thaliana under the control of a constitutive lac-promoter. The resulting plasmid was called pAC-4CL-CCO.
All cloning and DNA manipulation were carried out in E. coli strain JM109 by following standard techniques described elsewhere [57]. Cloned gene sequences were verified by sequencing.
Culture conditions and strains
Unless otherwise indicated, E. coli cultures were grown in Luria-Bertani (LB) medium supplemented with appropriate antibiotics ampicillin (100 μg ml−1) and chloramphenicol (50 μg ml−1) at 30°C. E. coli strains JM109 and BL21(DE3) were used for gene expression from pUCmod and pET-plasmids, respectively.
E. coli strain BW27784 [40] was used for in vivo analysis of stilbene cleavage by CCO homologs. A modified M9 medium containing yeast extract (1.25 g/L), glycerol (0.5% v/v) and appropriate antibiotics was used for stilbene biosynthesis as described previously.[32]
Protein expression and purification
Both pET-CCO and pUCmod-CCO plasmids were used for protein overexpression. For expression of genes from pUCmod-CCO, recombinant E. coli JM109 overnight cultures (4 ml) were used to inoculate 1:100 LB medium (400 ml) containing the appropriate antibiotics. Cultures were grown for 16 hrs at 30°C and cells were harvested by centrifugation and stored at −20 °C until used. Cells collected from the culture (50 ml) were lysed with BugBuster® protein extraction reagent (2 ml) (Novagen, Madison, WI ). Cell debris was cleared by centrifugation (14,000 rpm, 5 minutes, 4°C) and the cleared protein extract used in in vitro activity assays. Protein levels were estimated by SDS gel electrophoresis and concentrations adjusted so that comparable levels were added to assays.
The pET-CCO plasmids were used for the overexpression of CCO proteins for protein purification. As previously observed with other CCO's [4, 21], these proteins are prone to inclusion body formation when expressed at high levels (e.g. from the strong T7-promoter present in pET plasmids). To facilitate the expression of soluble proteins, pET-CCO plasmids were transformed into E. coli BL21(DE3) harboring plasmid pGRO7 (Takara, Madison, Wisconsin) that expresses the GroEL and GroES chaperones (see also [4]). E. coli BL21 co-transformed with putative CCO homolog and groES-groEL were grown overnight at 30°C in LB media (4 mL). This culture was used to inoculate (1:100) LB (400 mL) and chaperone expression was induced with arabinose (0.5 mg/mL). The cells were grown at 30°C until an OD600 of 0.6. Then the cultures were cooled on ice and induced with isopropyl-β-D-thiogalactopyranoside (IPTG, 1 m M) before cultivation was continued overnight at 18°C and cells were harvested by centrifugation and stored at −20 °C until used. Cells collected from culture (50 ml) were lysed with BugBuster® protein extraction reagent (2 ml) (Novagen, Madison, WI). Cell debris was cleared by centrifugation (14,000 rpm, 5 minutes, 4°C). Aliquots of the cleared protein extract were saved for in vitro assays and the remainder used to purify CCO proteins by metal affinity chromatography. Soluble protein was loaded onto a Talon Resin immobilized metal affinity chromatography (IMAC) (Invitrogen, Carlsbad, CA) column and eluted in 50 mM phosphate buffer pH 7.2 with imidazole (300 mM) following three washing steps. The CCO proteins eluted in a fraction (4 mL) and was concentrated using an Amicon ultracentrifuge concentrator with a 10 kDa molecular weight cut off. The Amicon concentrator was used to desalt the protein by exchanging the buffer 4 times against phosphate buffer (50 mM pH 7.2). The protein was subjected to iron center reconstitution by incubation with FeSO4 (100 mM) under argon gas for 30 minutes to insure incorporation of Fe2+ into the active site. Protein concentrations were determined using Bradford reagent (BioRad, Hercules, CA).
In vivo analysis of carotenoid cleavage activity
To investigate in vivo carotenoid cleavage, CCO enzymes on pUCmod were co-expressed with β, β-carotene biosynthetic genes expressed from pAC-crtE-crtB-crtI14-crtY [31], pAC-crtE-crtB-crtI14-crtY-crtX, pAC-crtE-crtB-crtI14-crtY2 in E. coli JM109 as described previously for cyanobacterial CCO.[4] Briefly, single colonies of E. coli JM109 transformants harboring the carotenoid and CCO plasmids to be tested were grown overnight and for 48 hrs in LB media (50 ml) supplemented with ampicillin and chloramphenicol at 30°C. The color intensities of the resulting cell pellets from six replicate cultures were then compared by visible inspection to control cells harboring the corresponding carotenoid plasmid and empty pUCmod plasmid.
In vivo analysis of stilbene cleavage activity
CCO enzymes on plasmid pAC-4CL-CCO also containing 4-coumaroyl CoA-ligase 4CL were co-expressed with stilbene synthase (STS) from Arachis hypogaea (peanut) on the constitutive expression vector pUC-STS. The construction of stilbene and flavonoid biosynthetic pathways has been described previously [31,32].
To investigate in vivo stilbene cleavage by CCO's, single colonies of E. coli BW27784 transformants harboring plasmids pUC-STS and pAC-4CL-CCO or only pAC-4CL-CCO (control for cleavage of CoA-activated phenylpropionic acids) were grown overnight in modified M9 medium (4 ml) at 30°C and used to inoculate 1:100 modified M9 medium (50 ml). Cultures were grown to an OD of 0.1 at 30°C when phenyl propionic acid precursor compounds (1 mM, 200 μl of 4-coumaric acid, caffeic acid or ferulic acid in DMSO) were added to the cultures to initiate their biotransformation into stilbene compounds by the recombinant E. coli pathway (STS and 4CL). Following an additional 16 hrs of cultivation at 30°C, the culture supernatant was extracted and analyzed for product formation essentially as described in Watts et al. 2006 [32]. In brief, culture (1 ml) was centrifuged at maximum speed to pellet cells. The media was decanted to a fresh 1.5 ml microfuge tube and the pH was adjusted by addition of 1N hydrochloric acid (50 μl). Then the media was extracted twice with ethyl acetate (500 μl) and the extracts were combined and dried under nitrogen. The dried residue was resuspended in methanol (100 μL) and all samples were stored at –20°C prior to HPLC and LC-MS analysis (see below).
In vitro assays
Assays were performed with purified proteins (100-250 μg) or protein extracts (50 μL) in 300 μl reactions containing phosphate buffer (50 mM, pH 7.2), NaCl (300 mM), sodium ascorbate (10 mM) and FeSO4 (0.5 mM). After 5 min of equilibration, stilbene substrates (1 mM from 1M resveratrol, piceatannol or rhapotinigenin dissolved in DMSO) or carotenoid substrate (0.27 mM from 2 mM β-apo-8’-carotenal dissolved in 1% Tween 40 [4]). In vitro reactions were carried out at 30°C for the prescribed amount of time (5 min to 12 hrs) before being stopped with HCL (1N, 50 μL) and extracted three times with ethyl acetate (500 μl). β-apo-8’-carotenal assays were extracted with diethyl ether. The organic fractions were combined, dried under nitrogen and stored until HPLC or GC analysis.
Isotope labeling
For labeling experiments with H218O, the protein extracts were freeze-dried to remove all water and residues were resuspended in of H218O (100 μl). No buffer, NaCl or FeSO4 was added to avoid contamination with unlabeled H2O. Reactions were started by adding resveratrol (1 mM in DMSO). After 15 min of incubation at 30°C, assays were extracted two times with ethyl acetate (500 μl), dried under nitrogen and immediately derivatized to silyl ethers for GC-MS analysis as described below.
Labeling experiments with 18O2 were performed in screw-capped glass vessels (2 ml) with a gas-tight Teflon septum using reaction conditions described above for standard assays with resveratrol. The vials were flushed three times with 18O2 and protein extract was added with an airtight Hamilton syringe. The reaction mixture was allowed to equilibrate for 5 minutes before resveratrol (1 mM ) was injected into the vial. The reaction was stirred for 15 minutes before being extracted two times with ethyl acetate (500 μl). Samples were dried and immediately derivatized to silyl ethers for GC-MS analysis as described below.
HPLC and LC/MS analysis
HPLC analysis was performed using an Agilent 1100 HP system with a quaternary pump and a photodiode array detector (Palo Alto, CA). Several HPLC conditions were utilized to analyze possible β-apo-8’-carotenal cleavage products as described in Marasco et al. [4]. Briefly, cleavage of β-apo-8’-carotenal at the 15,15’ position to retinal was analyzed by applying sample (50 μL) to an Adsorbosil C-18 column (4.6 × 250 mm, 5 μm; Alltech, Deerfield, IL). The gradient program was modified from Ruch et al. [10] using a solvent system of MeOH : tert-butylmethyl ether : water (120:4:40, v/v/v) (B) and MeOH : tert-butylmethyl ether (500:500 v/v) (A). The gradient conditions were solvent B (100%) to solvent B (43%) over 45 min, solvent B (43%) to solvent B (0%) for 11 min, solvent B (0%) for 14 min with a flow rate of 1 mL min−1. Dialdehyde cleavage products were determined by applying sample (100 μL) to a Zorbax RX-C18 column (4.6 × 250 mm, 5 μm; Agilent Technologies, Palo Alto, CA). The solvent system was MeOH : water (70:30, v/v) with 0.1% ammonium acetate (B) and MeOH (A). The gradient conditions were solvent B (100%) to solvent B (0%) over 16 min, solvent B (0%) until 26 min, and then return to A (100%) with a flow rate of 0.8 mL min−1.
Stilbene cleavage products were detected using conditions modified from HPLC methods described by Watts et al. [32] for the analysis of stilbene compounds. 20 μL of sample was applied to a reverse phase Eclipse XDB-C8 column (4.6 × 150 mm, 5 μm; Alltech, Deerfield, IL) and analyzed with an isocratic program using a solvent system of water:trifluoroacetic acid (99.9:0.1, v/v) (A) and methanol:trifluoroacetic acid (99.9:0.1, v/v) (B) in a ratio of 73:27 with a flow rate of 0.8 ml min−1. To achieve better resolution of piceatannol cleavage products, a gradient program was used with a flow rate of 0.5 mL/min and the following conditions: from 0-10 min 75:25 A:B, followed by a gradient from 75:25 A:B to 50:50 A:B in 15 minutes, followed by 5 min 50:50 A:B. Stilbenes and cleavage compounds were identified by comparisons of retention times and UV/Vis spectra of standard compounds (resveratrol, piceatannol, rhaptonin, ferulic acid, coumaric acid, caffeic acid, 4-hydroxybenzaldehyde, 3,5-dihydroxybenzladehyde, 3,4-dihydroxybenzaldehyde) and mass spectrometry. For quantification of products, standard curves were constructed by plotting peak areas of known quantities of standards.
Mass fragmentation spectra were monitored in a mass range of m/z 50–500 on a LCQ mass spectrophotometer equipped with electrospray chemical ionization interface (Thermo Finnigan, USA). Mass fragmentation spectra of standard compounds and the extracted compounds were monitored with a negative electron spray ionization (ESI) interface. Negative ion values for standard compounds were as follows: 4-coumaric acid (m/z 163.1), caffeic acid (m/z 179.1), ferulic ac i d (m/z 193.1), resveratrol (m/z 227.1), piceatannol (m/z 243.1), 4-hydroxybenzaldehyde (m/z 121.0), 3,5-dihydroxybenzaldhyde (m/z 137.0). The chromatography conditions were identical to the HPLC conditions described above excluding the addition of trifluoroacetic acid.
GC-MS analysis
For GC-MS analysis, dried samples were derivatized to silyl ethers by addition of BSTFA (50 μl) (N,O-Bis(trimethylsilyl)acetamide) reagent. GC-MS analyses were performed with a HP6890 Series GC coupled to a HP5973 mass selective detector. GC conditions consisted of an HP-5 column (30 m by 0.25 mm ID by 1.5 μm coated with 5% phenyl methyl silicone) and a split injector (1:20) set to a temperature of 250 °C. The temperature started at 60 °C and increased to 280 °C at 8°C/minute intervals with a helium flow rate of 1 ml/min. The EI-MS ionization voltage was 70 eV (electron impact ionization) and the ion source and interface temperature were both 250 °C. Mass spectra were scanned in a range of m/z 40-500 at 1 sec intervals.
Supplementary Material
Acknowledgements
This work was supported by the National Science Foundation (#0332478). E. Marasco is supported by the National Institute of General Medical Sciences / National Institutes of Health (NIGMS/NIH) Biotechnology Training Grant (#T32 GM08347). We thank Tom Markis (Lipscomb Laboratory, University of Minnesota) for his help in the labeling experiments, and the Wilmot laboratory (University of Minnesota) for their generous gift of labeled water.
The abbreviations used are
- CCO
carotenoid cleavage oxygenase
- NCED
9-cis-epoxydioxygenase
- LSD
lignostilbene-alpha,beta-dioxygenases
- STS
stilbene synthase
- 4CL
4-coumaroyl ligase
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.a Bouvier F, Isner JC, Dogbo O, Camara B. Trends Plant Sci. 2005;10:187. doi: 10.1016/j.tplants.2005.02.007. [DOI] [PubMed] [Google Scholar]; b Moise AR, von Lintig J, Palczewski K. Trends Plant Sci. 2005;10:178. doi: 10.1016/j.tplants.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Kloer DP, Schulz GE. Cell. Mol. Life Sci. 2006;63:2291. doi: 10.1007/s00018-006-6176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Giuliano G, Al-Babili S, von Lintig J. Trends Plant Sci. 2003;8:145. doi: 10.1016/S1360-1385(03)00053-0. [DOI] [PubMed] [Google Scholar]; b Wyss A. J. Nutr. 2004;134:246S. doi: 10.1093/jn/134.1.246S. [DOI] [PubMed] [Google Scholar]
- 4.Marasco EK, Vay K.-l., Schmidt-Dannert C. J. Biol. Chem. 2006;281:31583. doi: 10.1074/jbc.M606299200. [DOI] [PubMed] [Google Scholar]
- 5.a Prado-Cabrero A, Scherzinger D, Avalos J, Al-Babili S. Eukaryotic Cell. 2007;6:650. doi: 10.1128/EC.00392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Prado-Cabrero A, Estrada AF, Al-Babili S, Avalos J. Molecular Microbiology. 2007;64:448. doi: 10.1111/j.1365-2958.2007.05665.x. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz SH, Qin XQ, Zeevaart JAD. J. Biol. Chem. 2001;276:25208. doi: 10.1074/jbc.M102146200. [DOI] [PubMed] [Google Scholar]
- 7.von Lintig J, Vogt K. J. Biol. Chem. 2000;275:11915. doi: 10.1074/jbc.275.16.11915. [DOI] [PubMed] [Google Scholar]
- 8.Auldridge ME, McCarty DR, Klee HJ. Curr. Opin. Plant Biol. 2006;9:315. doi: 10.1016/j.pbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 9.a Ho CC, de Moura FF, Kim SH, Clifford AJ. Am. J. Clin. Nutr. 2007;85:770. doi: 10.1093/ajcn/85.3.770. [DOI] [PubMed] [Google Scholar]; b Morales A, Rosas A, Gonzalez A, Antaramian A, Varela-Echavarria A, Shimada A, Mora O. Intern. J. Vit. Nutr. Res. 2006;76:9. doi: 10.1024/0300-9831.76.1.9. [DOI] [PubMed] [Google Scholar]; c Paik J, During A, Harrison EH, Mendelsohn CL, Lai K, Blaner WS. J. Biol. Chem. 2001;276:32160. doi: 10.1074/jbc.M010086200. [DOI] [PubMed] [Google Scholar]; d Redmond TM, Gentleman S, Duncan T, Yu S, Wiggert B, Gantt E, Cunningham FX. J. Biol. Chem. 2001;276:6560. doi: 10.1074/jbc.M009030200. [DOI] [PubMed] [Google Scholar]; e von Lintig J, Vogt K. J. Nutr. 2004;134:251S. doi: 10.1093/jn/134.1.251S. [DOI] [PubMed] [Google Scholar]; f von Lintig J, Wyss A. Arch. Biochem. Biophys. 2001;385:47. doi: 10.1006/abbi.2000.2096. [DOI] [PubMed] [Google Scholar]
- 10.Ruch S, Beyer P, Ernst H, Al-Babili S. Mol. Microbiol. 2005;55:1015. doi: 10.1111/j.1365-2958.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- 11.Scherzinger D, Ruch S, Kloer DP, Wilde A, Al-Babili S. Biochem. J. 2006;398:361. doi: 10.1042/BJ20060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auldridge ME, Block A, Vogel JT, Dabney-Smith C, Mila I, Bouzayen M, Magallanes-Lundback M, DellaPenna D, McCarty DR, Klee HJ. Plant J. 2006;45:982. doi: 10.1111/j.1365-313X.2006.02666.x. [DOI] [PubMed] [Google Scholar]
- 13.Bouvier F, Dogbo O, Camara B. Science. 2003;300:2089. doi: 10.1126/science.1085162. [DOI] [PubMed] [Google Scholar]
- 14.Bouvier F, Suire C, Mutterer J, Camara B. Plant Cell. 2003;15:47. doi: 10.1105/tpc.006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohmiya A, Kishimoto S, Aida R, Yoshioka S, Sumitomo K. Plant Physiol. 2006;142:1193. doi: 10.1104/pp.106.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a Schwartz SH, Tan BC, Gage DA, Zeevaart JAD, McCarty DR. Science. 1997;276:1872. doi: 10.1126/science.276.5320.1872. [DOI] [PubMed] [Google Scholar]; b Schwartz SH, Tan BC, McCarty DR, Welch W, Zeevaart JAD. Biochim. Biophys. Acta. 2003;1619:9. doi: 10.1016/s0304-4165(02)00422-1. [DOI] [PubMed] [Google Scholar]; c Tan BC, Joseph LM, Deng WT, Liu LJ, Li QB, Cline K, McCarty DR. Plant J. 2003;35:44. doi: 10.1046/j.1365-313x.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 17.Simkin AJ, Schwartz SH, Auldridge M, Taylor MG, Klee HJ. Plant J. 2004;40:882. doi: 10.1111/j.1365-313X.2004.02263.x. [DOI] [PubMed] [Google Scholar]
- 18.Simkin AJ, Underwood BA, Auldridge M, Loucas HM, Shibuya K, Schmelz E, Clark DG, Klee HJ. Plant Physiol. 2004;136:3504. doi: 10.1104/pp.104.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kloer DP, Ruch S, Al-Babili S, Beyer P, Schulz GE. Science. 2005;308:267. doi: 10.1126/science.1108965. [DOI] [PubMed] [Google Scholar]
- 20.a Zeevaart JAD, Heath TG, Gage DA. Plant Physiol. 1989;91:1594. doi: 10.1104/pp.91.4.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Creelman RA, Zeevaart JAD. Plant Physiol. 1984;75:166. doi: 10.1104/pp.75.1.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt H, Kurtzer R, Eisenreich W, Schwab W. J. Biol. Chem. 2006;281:9845. doi: 10.1074/jbc.M511668200. [DOI] [PubMed] [Google Scholar]
- 22.a Leuenberger MG, Engeloch-Jarret C, Woggon WD. Angew. Chem. Int. Ed. Engl. 2001;40:2614. doi: 10.1002/1521-3773(20010716)40:14<2613::AID-ANIE2613>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; b Woggon WD. Pure Appl. Chem. 2002;74:1397. [Google Scholar]
- 23.Kamoda S, Saburi Y. Biosci. Biotechnol. Biochem. 1993;57:931. doi: 10.1271/bbb.57.931. [DOI] [PubMed] [Google Scholar]
- 24.Kamoda S, Saburi Y. Biosci. Biotechnol. Biochem. 1995;59:1866. doi: 10.1271/bbb.59.1866. [DOI] [PubMed] [Google Scholar]
- 25.Han SY, Inoue H, Terada T, Kamoda S, Saburi Y, Sekimata K, Saito T, Kobayashi M, Shinozaki K, Yoshida S, Asami T. Bioorg. Med. Chem. Lett. 2002;12:1139. doi: 10.1016/s0960-894x(02)00126-9. [DOI] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. Nucl. Acids Res. 2006;34:D354. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a Demaneche S, Meyer C, Micoud J, Louwagie M, Willison JC, Jouanneau Y. Appl. Environ. Microbiol. 2004;70:6714. doi: 10.1128/AEM.70.11.6714-6725.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shi T, Fredrickson JK, Balkwill DL. J. Ind. Microbiol. Biotech. 2001;26:283. doi: 10.1038/sj.jim.7000130. [DOI] [PubMed] [Google Scholar]
- 28.Giraud E, Moulin L, Vallenet D, Barbe V, Cytryn E, Avarre J-C, Jaubert M, Simon D, Cartieaux F, Prin Y, Bena G, Hannibal L, Fardoux J, Kojadinovic M, Vuillet L, Lajus A, Cruveiller S, Rouy Z, Mangenot S, Segurens B, Dossat C, Franck WL, Chang W-S, Saunders E, Bruce D, Richardson P, Normand P, Dreyfus B, Pignol D, Stacey G, Emerich D, Vermeglio A, Medigue C, Sadowsky M. Science. 2007;316:1307. doi: 10.1126/science.1139548. [DOI] [PubMed] [Google Scholar]
- 29.a Kamoda S, Saburi Y. Biosci. Biotechnol. Biochem. 1993;57:926. doi: 10.1271/bbb.57.926. [DOI] [PubMed] [Google Scholar]; b Kamoda S, Terada T, Saburi Y. Biosci. Biotechnol. Biochem. 1997;61:1575. doi: 10.1271/bbb.61.1575. [DOI] [PubMed] [Google Scholar]
- 30.a Schilling M, Patett F, Schwab W, Schrader J. Appl Microbiol Biotechnol. 2007;75:829. doi: 10.1007/s00253-007-0878-z. [DOI] [PubMed] [Google Scholar]; b Mathieu S, Bigey F, Procureur J, Terrier N, Gunata Z. Biotechnology Letters. 2007;29:837. doi: 10.1007/s10529-007-9315-8. [DOI] [PubMed] [Google Scholar]
- 31.a Schmidt-Dannert C, Umeno D, Arnold FH. Nat. Biotechnol. 2000;18:750. doi: 10.1038/77319. [DOI] [PubMed] [Google Scholar]; b Lee PC, Momen AZR, Mijts BN, Schmidt-Dannert C. Chem. Biol. 2003;10:453. doi: 10.1016/s1074-5521(03)00103-0. [DOI] [PubMed] [Google Scholar]
- 32.Watts KT, Lee PC, Schmidt-Dannert C. BMC Biotechnology. 2006:6. doi: 10.1186/1472-6750-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boiero L, Perrig D, Masciarelli O, Penna C, Cassán F, Luna V. Appl. Microbiol. Biotechnol. 2007;74:874. doi: 10.1007/s00253-006-0731-9. [DOI] [PubMed] [Google Scholar]
- 34.Siewers V, Kokkelink L, Smedsgaard J, Tudzynski P. Appl Environ Microbiol. 2006;72:4619. doi: 10.1128/AEM.02919-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamoda S, Terada T, Saburi Y. Biosci. Biotechnol. Biochem. 2003;67:1394. doi: 10.1271/bbb.67.1394. [DOI] [PubMed] [Google Scholar]
- 36.Lindqvist A, Andersson S. J. Biol. Chem. 2002;277:23942. doi: 10.1074/jbc.M202756200. [DOI] [PubMed] [Google Scholar]
- 37.Kamoda S, Terada T, Saburi Y. Biosci. Biotechnol. Biochem. 2005;69:635. doi: 10.1271/bbb.69.635. [DOI] [PubMed] [Google Scholar]
- 38.Siewers V, Smedsgaard J, Tudzynski P. Appl Environ Microbiol. 2004;70:3868. doi: 10.1128/AEM.70.7.3868-3876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambrook J. Molecular Cloning - A Laboratory Manual, Vol. Three ed. Vol. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 40.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD. Microbiology. 2001;147:3241. doi: 10.1099/00221287-147-12-3241. [DOI] [PubMed] [Google Scholar]
- 41.Wyss A, Wirtz GM, Woggon WD, Brugger R, Wyss M, Friedlein A, Riss G, Bachmann H, Hunziker W. Biochem. J. 2001;354:521. doi: 10.1042/0264-6021:3540521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boulanger A, Liu SY, Yu S, Redmond T. Molecular Vision. 2001;7:283. [PubMed] [Google Scholar]
- 43.Wackett LP. Mechanism and applications of Rieske non-heme iron dioxygenases. Vol. 31. Enzyme Microb. Technol; 2002. pp. 577–587. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.