

Ubiquitin-related domains are present in many spliceosomal proteins, but the roles they play remain largely unknown. This study provides intriguing insights into the function of Sad1, a spliceosomal protein that contains both a Zn finger ubiquitin binding domain and a noncatalytic ubiquitin-specific protease domain. This work demonstrates that Sad1 is required for pre-mRNA splicing in vivo and for spliceosome assembly. Unexpectedly, Sad1 is unable to bind ubiquitin in vitro. By determining the crystal structure of Sad1, the authors show how the Sad1 domains indeed deviate from canonical ubiquitin binding domains.
Keywords: pre-mRNA splicing mechanism, spliceosome assembly, deubiquitinase, ubiquitin
Abstract
Sad1 is an essential splicing factor initially identified in a genetic screen in Saccharomyces cerevisiae for snRNP assembly defects. Based on sequence homology, Sad1, or USP39 in humans, is predicted to comprise two domains: a zinc finger ubiquitin binding domain (ZnF-UBP) and an inactive ubiquitin-specific protease (iUSP) domain, both of which are well conserved. The role of these domains in splicing and their interaction with ubiquitin are unknown. We first used splicing microarrays to analyze Sad1 function in vivo and found that Sad1 is critical for the splicing of nearly all yeast intron-containing genes. By using in vitro assays, we then showed that it is required for the assembly of the active spliceosome. To gain structural insights into Sad1 function, we determined the crystal structure of the full-length protein at 1.8 Å resolution. In the structure, the iUSP domain forms the characteristic ubiquitin binding pocket, though with an amino acid substitution in the active site that results in complete inactivation of the enzymatic activity of the domain. The ZnF-UBP domain of Sad1 shares high structural similarly to other ZnF-UBPs; however, Sad1's ZnF-UBP does not possess the canonical ubiquitin binding motif. Given the precedents for ZnF-UBP domains to function as activators for their neighboring USP domains, we propose that Sad1's ZnF-UBP acts in a ubiquitin-independent capacity to recruit and/or activate Sad1's iUSP domain to interact with the spliceosome.
INTRODUCTION
The removal of a pre-mRNA intron and the splicing together of its exons are catalyzed by the spliceosome, an enzyme composed of over 100 proteins that associate with small nuclear RNAs (snRNAs) to form five distinct subcomplexes, or snRNPs (small nuclear ribonucleoproteins) (Jurica and Moore 2003). The U1 snRNP binds to the 5′ splice site of the intron, and then the U2 snRNP binds in an ATP-dependent manner to the branch point of the intron. This U1/U2/pre-mRNA–bound complex is referred to as the prespliceosome. The remaining three snRNPs comprise the tri-snRNP, a super-complex composed of the U4, U5, and U6 snRNPs (for review, see Wahl et al. 2009).
The tri-snRNP is formed through the initial base-pairing of U4 and U6 snRNAs forming the U4/U6 di-snRNP, which is later joined by the U5 snRNP. Upon binding to the prespliceosome, the U4/U6 duplex is separated and the U6 snRNA base-pairs with the U2 snRNA and the 5′ splice site. The U6/U2 and U6/5′ splice-site interactions are further specified and stabilized by the binding of the nineteen complex (NTC), which contains the ubiquitin ligase Prp19 (Chan et al. 2003). This activated state of the spliceosome is competent to bind the final factors necessary to catalyze the first and second chemical steps of splicing. Upon the completion of splicing, the spliceosome disassembles and releases the mRNA product, the intron lariat, and the individual snRNPs. The snRNPs are then recycled for reassembly upon the next intron-containing mRNA.
Sad1 is required for the assembly of the U4 and U6 snRNAs into the di-snRNP particle. Named for its snRNP assembly defect, the sad1-1 temperature-sensitive (ts) yeast strain is unable to assemble newly transcribed U4 snRNAs into the di-snRNP particle at the restrictive temperature, a phenotype also observed for the known snRNP assembly factor mutants prp19-1 and prp24-1 (Lygerou et al. 1999). Inactivation of Sad1 was shown to block the splicing of several pre-mRNA substrates; however, it was unclear if Sad1 acted as a core splicing factor in yeast. Sad1 did not associate stably with any of the individual yeast snRNAs (Lygerou et al. 1999) but was detected in the penta-snRNP, a mature form of the spliceosome that contains all five snRNPs and can only be purified under low-salt conditions (Stevens et al. 2002). The human homolog of Sad1, USP39, weakly associates with the tri-snRNP snRNAs under low-salt conditions and was shown to be required for the stable association of the tri-snRNP to the prespliceosome (Makarova et al. 2001). Thus, Sad1 may be required for tri-snRNP recruitment to the prespliceosome in addition to its contributions to di-snRNP assembly. How Sad1 mediates these snRNP complex formations remains unknown.
Sad1 belongs to a group of spliceosomal proteins that contains ubiquitin-related domains (Korneta et al. 2012). Prp19 is a U-box ubiquitin ligase required for spliceosome activation and recycling (Chan et al. 2003). The tri-snRNP protein, Prp8, contains an inactive Jab1/MPN deubiquitinase domain in its C terminus that has been linked to the regulation of U4/U6 snRNA duplex separation (Bellare et al. 2006; Maeder et al. 2009). Finally, the presence of ubiquitin in the tri-snRNP has been linked to both human and yeast tri-snRNP stability (Bellare et al. 2008; Song et al. 2010). The N-terminal domain of Sad1 is homologous to a zinc finger ubiquitin binding (ZnF-UBP) domain, and the C-terminal domain is homologous to a ubiquitin-specific peptidase (USP) domain. Not all members of the ZnF-UBP domain family are competent to bind ubiquitin. For example, USP5 has two ZnF-UBP domains, one of which has been shown to bind the free C-terminal tail of ubiquitin with high affinity (Reyes-Turcu et al. 2006, 2008; Avvakumov et al. 2012). In contrast, the ZnF-UBP domain from Ubp8 does not bind ubiquitin but rather functions as a scaffold for cofactors, which then activate the USP domain of Ubp8 (Kohler et al. 2010; Samara et al. 2010, 2012). Sad1's USP domain (iUSP) is predicted to be catalytically inactive, as it is missing the essential nucleophilic cysteine in its USP catalytic triad. It is unclear whether this deviation would affect ubiquitin binding or whether Sad1's ZnF-UBP domain might be required to activate ubiquitin binding function.
To examine the mechanism of Sad1 function and its role in the regulation of the splicing reaction, we first analyzed sad1-1 inactivation with splicing microarrays and found that, like other core splicing factors, Sad1 inactivation in vivo caused a strong block to most pre-mRNA splicing events. We then produced full-length recombinant Sad1 protein and showed that it could rescue in vitro splicing activity in sad1-1–inactivated extracts. We observed that tri-snRNP joining to the prespliceosome was blocked in sad1-1 ts-inactivated extracts and that this defect could be rescued by the addition of recombinant Sad1. Finally, to gain insight into Sad1's function, we crystallized and solved the structure of full-length Sad1. We show that Sad1's ZnF-UBP is in a conformation that is incompatible with ubiquitin binding. In addition, we observed that Sad1's iUSP domain preserves many of the features of active USP domains but possesses a remodeled catalytic cleft. Together these results help us to interpret Sad1's role in splicing, and raise questions for the further exploration of its molecular function.
RESULTS
Sad1 is composed of ubiquitin-related domains
Sad1 is a member of the ZnF-UBP USP family of deubiquitinases. The ZnF-UBP domain is located at the N terminus (amino acids 29–121), and the iUSP domain is located at the C terminus (amino acids 148–448) (Fig. 1A). Although the iUSP domain has many of the residues conserved in active USPs, it and its homologs are predicted to be inactive. Whereas an active USP catalytic triad is CHN/D (Komander et al. 2009), Sad1 has DHD and its human homolog, USP39, has DSQ (Fig. 1A; Supplemental Fig. S1). Sad1's two domains are well-conserved among its homologs, with the exception of an additional N-terminal RS domain found in the human homolog (Fig. 1A; Supplemental Fig. S1). A 25-amino-acid linker connects the N and C-terminal domains of Sad1.
FIGURE 1.

Sad1 is a core splicing factor necessary for spliceosome assembly. (A) Domain topology for S. cerevisiae Sad1, the human homolog of Sad1 (USP39), and a canonical active USP domain, with the catalytic residues denoted by asterisks. The ZnF-UBP domain is colored pale orange, a short linker domain is pale green, and the iUSP domain is colored slate blue. USP39 possesses an additional N-terminal RS domain. (B) The yeast pre-mRNA splicing microarray is composed of probes that detect the pre-mRNA intron (P), the exon (T) for total, and the exon/exon junction for the mature mRNA (M). SAD1 and sad1-1 cultures were grown at 25°C to mid-log phase and were then shifted for 30 min to 37°C. Total RNA was extracted and Cy3- and Cy5-labeled cDNA produced. The cDNAs were competitively hybridized to the array, and the resulting log2-ratios for each gene were plotted after performing centroid-linkage hierarchical clustering. Shown side by side are the heat maps of plots produced by two biological replicate experiments, a and b, where increasing amounts of yellow signal for the intron probe (P) reflects intron accumulation and is indicative of a pre-mRNA splicing defect. (C) sad1-1 splicing extracts were inactivated for 10 min at 37°C, and where indicated, a 10-fold decreasing dilution series of 105 nM full-length Sad1 was added. Pre-mRNA splicing reactions were incubated at 25°C in the presence of 2 mM ATP with Cy5-labeled pre-ACT1 for 30 min and then resolved on a 6% denaturing polyacrylamide gel. (D) Splicing extracts from sad1-1 were prepared and inactivated in a manner identical to that of the in vitro splicing assay in C, but the products of the reaction were resolved by native gel electrophoresis. An RNase H–catalyzed knockdown of the U6 snRNA in a SAD1 wild-type strain is shown for comparison.
Sad1 is a general splicing factor
Sad1 appears to have a weak affinity for the spliceosome, and it was unknown whether inactivating SAD1 in vivo would have a broad, genome-wide splicing defect, as is observed upon the inactivation of essential spliceosome factors (Pleiss et al. 2007). We used pre-mRNA splicing microarrays to assess the breadth of Sad1's role in splicing after competitively hybridizing total mRNA from ts-inactivated sad1-1 cells and SAD1 cells treated under the same conditions. We cloned the sad1-1 ts allele and SAD1 into CEN/ARS vectors that can complement a SAD1 deletion in yeast. The sad1-1 ts mutation had not been mapped, and upon sequencing, we discovered that it contains two mutations, L236P and S276F. These mutations likely destabilize the protein at 37°C, as attempts to recombinantly express and purify the double-mutant Sad1-1 protein yielded insoluble protein (data not shown), whereas the wild-type Sad1 protein was well expressed and soluble. Both CEN/ARS plasmids were able to complement the SAD1 deletion at 30°C, but only the wild-type plasmid was able to complement the SAD1 deletion at 37°C (Table 1). Splicing microarray analysis revealed a strong and broad block to pre-mRNA splicing in sad1-1 after incubation for 30 min at 37°C, as can be observed by the dramatic accumulation of intron signal for many pre-mRNAs in the sad1-1 versus SAD1 strain (Fig. 1B, middle lane, labeled P for pre-mRNA, represents the intron probes). The magnitude and breadth of this splicing defect is reminiscent of the inactivation of the core splicing factors, Prp5 and Prp8 (Pleiss et al. 2007). Thus SAD1 plays a crucial role in pre-mRNA splicing.
TABLE 1.
Strains tested for complementation of sad1▵::kan by 5-FOA selection
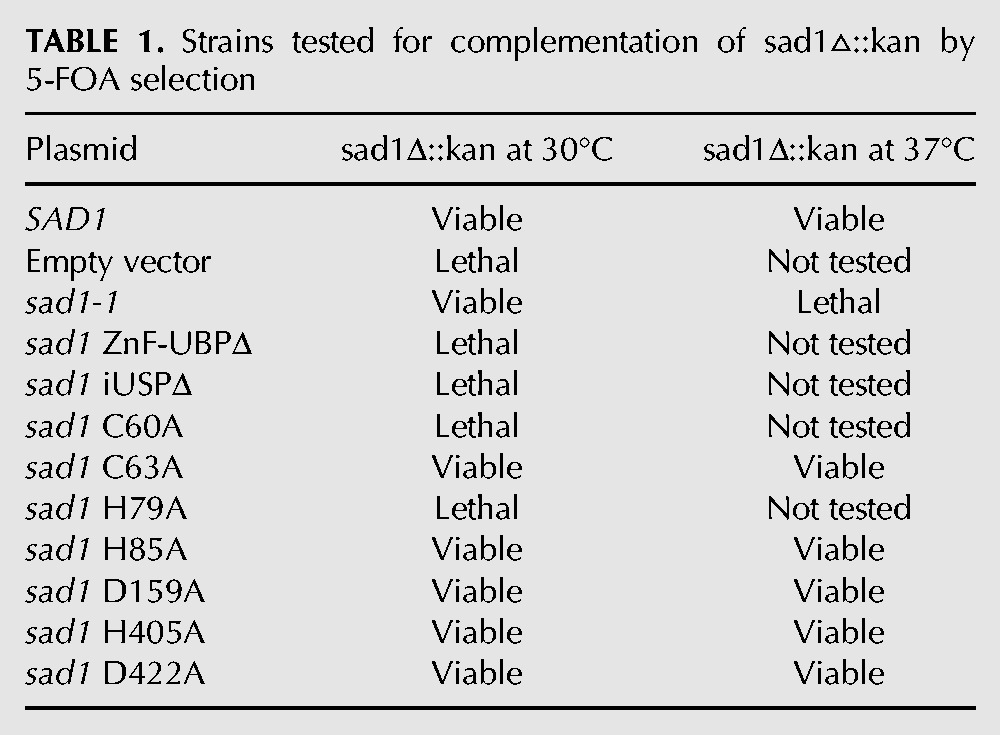
To investigate the mechanism of SAD1’s function, we used an in vitro splicing assay (Lin et al. 1985). Upon the addition of a Cy5 fluorescently labeled ACT1 pre-mRNA and ATP, the spliceosome assembles and then catalyzes two chemical steps, resulting in the removal of the intron and the ligation of the exons. The intermediates and the product of the splicing reaction are resolved by denaturing gel electrophoresis and visualized by a FluorImager. Consistent with initial observations, splicing was slightly inhibited by sad1-1 extracts grown at 30°C (Fig. 1C, cf. lanes 2 and 4; Lygerou et al. 1999), whereas splicing was entirely inhibited by heating the extracts for 10 min to 37°C (Fig. 1C, lane 3). The sad1-1 heat-inactivated lysate was unable to complete the first chemical step of splicing of the ACT1 pre-mRNA, as evidenced by the absence of splicing reaction intermediates (Fig. 1C, cf. lanes 2 and 3). In order to further determine the role of Sad1 in the splicing reaction, we purified full-length recombinant Sad1 with an N-terminal 6-His tag and found that it was able to fully rescue in vitro splicing activity (Fig. 1C).
Sad1 is required for the stable recruitment of the tri-snRNP
Since sad1-1–inactivated extracts were blocked at the first catalytic step, we sought to distinguish between a catalytic defect and a defect in the assembly of the spliceosome. Spliceosome assembly can be monitored by incubating extracts with a labeled pre-mRNA substrate and resolving the pre-mRNA bound spliceosome from its assembly intermediates by native gel electrophoresis (Cheng and Abelson 1987). As there was a low amount of ATP in both of our extracts, we saw a small amount of the prespliceosome assemble in the absence of exogenous ATP (Fig. 1D, cf. lanes 1 and 5). After the addition of ATP to 2 mM to the wild-type extracts, the prespliceosome band disappeared as the tri-snRNP joined the prespliceosome stably and a slower migrating band consisting of fully assembled spliceosomes appeared (Fig. 1D, lane 2). Depleting the U6 snRNA by anti-U6 oligo-directed RNase H digestion caused the tri-snRNP to fall apart, and as a result, even in the presence of 2 mM ATP, only the prespliceosome band is formed (Fig. 1D, lane 3; Fabrizio et al. 1989). Upon the addition of in vitro transcribed U6 snRNA, the tri-snRNP reassembled and much of the prespliceosome matured to the fully assembled spliceosome (Fig. 1D, cf. lanes 4 and 3).
To determine the role of Sad1 in spliceosome assembly, we made extracts from sad1-1 cells grown at 30°C. As with the wild-type extract, in the absence of exogenous ATP, a small amount of the prespliceosome band formed (Fig. 1D, lane 5). Unlike the wild-type extract, upon the addition of 2 mM ATP, the sad1-1 assembled spliceosome band was barely detectable and the prespliceosome band had only a slight decrease in intensity, reflecting the weakened capacity for this sad1-1 to splice at 30°C (Fig. 1D, lane 6, C, lane 2). Heat-shocking the sad1-1 extracts for 10 min at 37°C led to a large accumulation of the prespliceosome species after the addition of 2 mM ATP, consistent with the total block to splicing that we observe by in vitro splicing (Fig. 1D, lane 8, C, lane 3). This defect looked similar to the U6 snRNA knockdown in wild-type extracts (Fig. 1D, lane 3), and the addition of recombinant Sad1 to sad1-1 lysates grown at 30°C or to heat-shocked sad1-1 lysates rescued these defects (Fig. 1D, lanes 7,9). Therefore yeast Sad1 is required for the stable integration of the tri-snRNP into the prespliceosome to form the fully assembled spliceosome, similar to what was observed for USP39 (Makarova et al. 2001). Sad1 could achieve this by interacting with the prespliceosome, the tri-snRNP, or both. Additionally Sad1 could be required to maintain the stability of the tri-snRNP, as it was originally identified as a di-snRNP assembly factor (Lygerou et al. 1999). As connections between ubiquitin and the tri-snRNP had been established previously (Bellare et al. 2008; Song et al. 2010) we were intrigued by the possibility that Sad1 might somehow employ its ubiquitin-related domains to affect tri-snRNP joining to the prespliceosome.
The two ubiquitin-related domains of Sad1 do not bind ubiquitin in vitro
Sad1 is composed of two domains that have the potential to bind ubiquitin (Fig. 1A). We found that both domains are required for Sad1's function in vivo, as each domain by itself is unable to complement a SAD1 deletion (Table 1). To determine whether Sad1 binds to ubiquitin, we developed an isothermal titration calorimetry (ITC) assay to measure the interaction between Sad1 and ubiquitin. For comparison, we used the ZnF-UBP from USP5, known to bind the C-terminal tail of ubiquitin through a WRY motif that is well conserved among the ZnF-UBP domains that have been shown to bind ubiquitin (Fig. 2A; Reyes-Turcu et al. 2006; Bonnet et al. 2008; Ouyang et al. 2011). In contrast, the ZnF-UBP domains that do not bind ubiquitin have substitutions in the WRY motif (Allen and Bycroft 2007; Zhang et al. 2011), as does Sad1, which occurs as YRL (Fig. 2A).
FIGURE 2.

Sad1 does not bind ubiquitin in vitro. (A) A primary sequence alignment of ubiquitin binding and nonubiquitin binding Homo sapiens and S. cerevisiae ZnF-UBP domains. The WRY motif residues that are essential for ubiquitin binding in USP5 are annotated with asterisks; in Sad1, they appear as YRL. The residues that comprise the three zinc fingers found in ZnF-UBPs are denoted A, B, and C; Sad1 only possesses the B site zinc finger. Conservation is indicated by shades of gray, and gaps in the alignment are represented by dashes. (B) We titrated 1 mM of ubiquitin into an ITC receptor cell containing either 50 µM full-length Sad1, Sad1-iUSP, USP5 ZnF-UBP, or buffer at 25°C. The data from the USP5 ZnF-UBP titration with ubiquitin were fit to a single exponential in good agreement with one binding site and a dissociation constant of 2.3 µM. ITC curve-fitting and data analysis were performed with the MicroCal-enabled Origin software.
Using ITC, we were unable to detect an interaction between full-length Sad1 and ubiquitin. Titrating 1 mM ubiquitin into either 50 µM Sad1 or buffer produces no observable response as measured by ITC (Fig. 2A). The ZnF-UBP from USP5 produced a dissociation constant of 2.4 µM, consistent with previously published values (Fig. 2B; Reyes-Turcu et al. 2006). In addition, the iUSP domain alone of Sad1 does not bind to ubiquitin, as it, too, did not produce a measurable ITC response (Fig. 2B). The ZnF-UBP domain alone of Sad1 could not be recombinantly purified and was thus unsuitable for ITC analysis. As ITC only measures the enthalpy between two reactants, there was a possibility that Sad1 bound to ubiquitin with a purely entropic-based interaction. To address this possibility, we used an alternative method to investigate the potential binding between Sad1 and ubiquitin by performing biolayer interferometry (BLI) with immobilized ubiquitin and either USP5 ZnF-UBP or Sad1 (Supplemental Fig. S2). Consistent with the ITC results, USP5 ZnF-UBP produced a robust response with a KD similar to that previously observed and there was no specific response for Sad1.
We also used glutathione-S-transferase (GST) immunoprecipitations to investigate the possibility of an interaction between Sad1 and ubiquitin or the yeast ubiquitin-like proteins. We used the yeast pre-mRNA splicing factor Snu66, which has been shown to interact with Hub1 (Mishra et al. 2011), as a positive control. When purified N-terminal GST fusions of ubiquitin, SUMO, Nedd8, and Hub1 were incubated with either Sad1, ZnF-UBP USP5, or Snu66, the only observed interactions were between GST-ubiquitin and the ZnF-UBP of USP5 and between GST-Hub1 and Snu66 (Supplemental Fig. S3). As this absence of ubiquitin binding for Sad1 was contrary to our expectations, and given Sad1's critical role in splicing, we pursued a structural approach to gain further insights into Sad1 function.
The crystal structure of full-length SAD1
Full-length Sad1 fused to a N-terminal 6-His tag was recombinantly expressed and purified to homogeneity from Escherichia coli and was crystallized by hanging drop vapor diffusion methods. The crystal structure was solved by single anomalous diffraction using a tungsten derivative (Table 2). The structure of Sad1 was refined to 1.87 Å resolution with Rwork of 17.1%, Rfree of 20.2%, and good stereochemistry. The phased electron density maps allowed for the unambiguous placement of 414 of 448 residues of the full-length Sad1. The N-terminal 28 amino acids were disordered in the crystal and absent from the experimental map. The tips of the USP fingers subdomain were also disordered (Fig. 3A), which is a feature that has been observed in other USPs crystallized in the absence of ubiquitin (Joint Center For Structural Genomics 2004; Kohler et al. 2010; Samara et al. 2010). Additionally, a loop in the pseudo-active site of Sad1's USP domain is disordered. As mentioned, the sad1-1 ts mutations, L236P and S276F, likely destabilize the protein at 37°C. The L236P mutation introduces a proline in the middle of a central α-helix of the iUSP domain, and S276F introduces a hydrophobic residue in the solvent-exposed tips of the iUSP fingers domain, both of which would be likely to challenge the stability of the Sad1 iUSP domain.
TABLE 2.
Data collection and refinement statistics

FIGURE 3.

The crystal structure of full-length Sad1. (A) The ZnF-UBP (pale orange) and the iUSP domain (slate blue) are connected through a 25-amino-acid linker (pale green). (B) Intradomain hydrogen bonding of K199 to D43 and of K207 to D41 provides interdomain stabilization. (C) Intradomain hydrophobic packing between F206, L140, R37, and a hydrophobic patch on the ZnF-UBP domain further specifies the orientation of the two domains.
The orientation of Sad1's ZnF-UBP domain to its iUSP domain is specified by a 25-amino-acid linker composed of a short helix and two short anti-parallel β-strands (Fig. 3A). The linker sequence, amino acids 122–147, is not well conserved among Sad1 homologs. The length of Sad1's linker is only three amino acids shorter in the Schizosaccharomyces pombe and human homologs, suggesting some conservation in their interdomain orientations (Supplemental Fig. S1). The linker is structurally distinct from the linker observed in the structure of Ubp8, another ZnF-UBP USP in the PDB, which is longer and composed of multiple short α-helices (Kohler et al. 2010; Samara et al. 2010). Sad1 K199 from the iUSP domain makes hydrogen bonds to D43 from the ZnF-UBP domain. Sad1 K207 from the iUSP domain connects the two domains with a hydrogen bond to D41 from the ZnF-UBP domain (Fig. 3B). Lastly, interdomain contacts are further specified through hydrophobic residues that are within van der Waals distances of each other. F206 from the iUSP domain and L140 from the linker domain interact with a hydrophobic patch from the ZnF-UBP domain, which is composed of the aliphatic part of the conserved R37 side-chain and the side-chains of L40 and I117 residues (Fig. 3C).
The ZnF-UBP of Sad1
The ZnF-UBP domain from Sad1 shares the same (∼95-amino-acid) mixed α–β core structure found in other ZnF-UBP proteins, possessing a five-stranded anti-parallel β-sheet, flanked by two α-helices. Table 3 lists the RMSDs for these ZnF-UBPs aligned to Sad1's ZnF-UBP. The first α-helix, αA, is connected to the end of the second β-strand through a loop, Loop L2A (Reyes-Turcu et al. 2006), that displays varied orientations in the different ZnF-UBP structures (Fig. 4A). The second α-helix, αB, is held closely to the opposite side of the β-sheet wall through a network of hydrophobic residues.
TABLE 3.
ZnF-UBP domains from the PDB with the highest structural similarity as determined by the DALI server for the ZnF-UBP domain of Sad1 (amino acids 29–124)
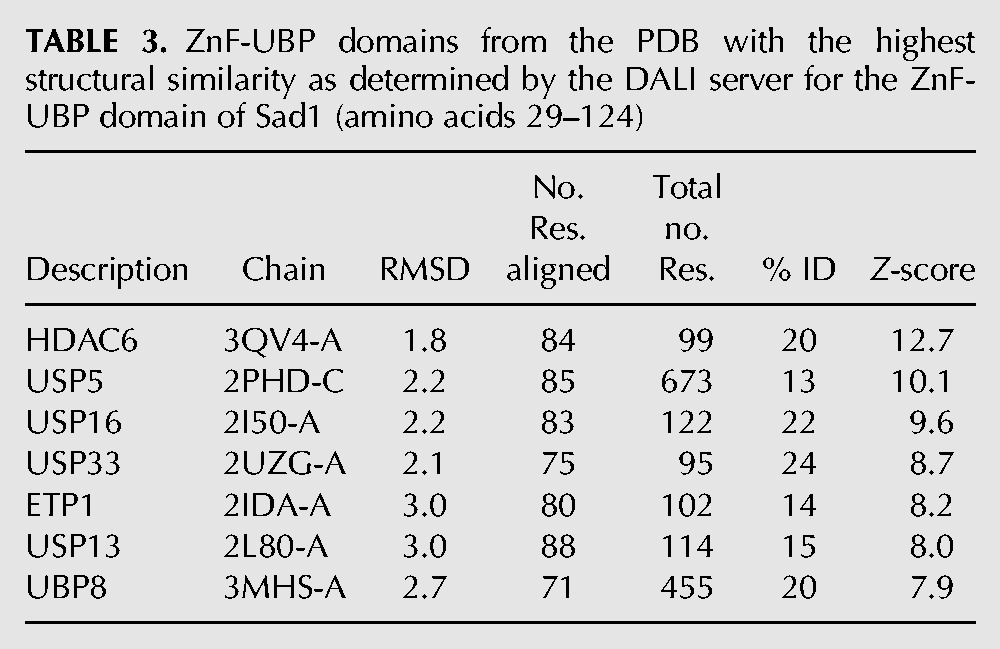
FIGURE 4.
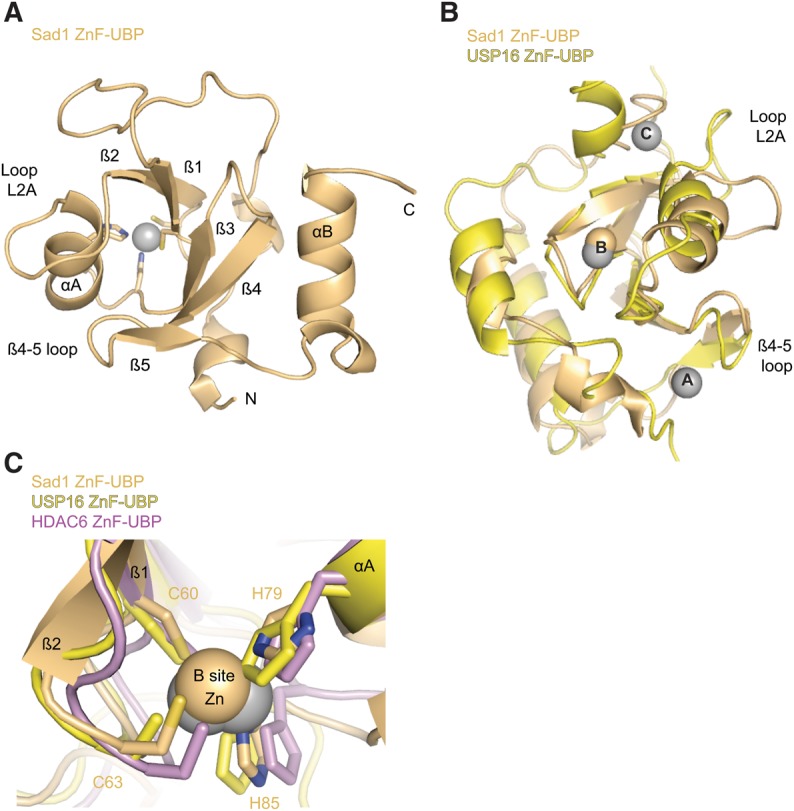
The ZnF-UBP domain of Sad1 binds one Zn atom. (A) Sad1's ZnF-UBP domain (pale orange) with secondary structure elements labeled, including loop L2A and the β4-5 loop. This view is from a 50° rotation about the y-axis of the full-length molecule in Figure 3B with the iUSP domain removed for clarity. (B) A structural alignment produced with Pymol of Sad1's ZnF-UBP to the ZnF-UBP domain from H. sapiens USP16 (yellow; PDB ID 2I50). The three zinc atoms of USP16, A through C, appear as gray spheres, and the B-site zinc atom from Sad1 appears as a pale orange sphere. (C) Structural alignment of the B-site zincs from SAD1 ZnF-UBP, H. sapiens HDAC6 ZnF-UBP (violet, PDB ID 3C5K), and USP16 ZnF-UBP. The residues that comprise the CCHH zinc finger of Sad1 are labeled.
ZnF-UBPs can bind up to three zinc atoms, as is the case with the ZnF-UBP from USP16 (Fig. 4B; Pai et al. 2007). Unlike USP16, Sad1's ZnF-UBP only binds a single zinc atom at the B site through a CCHH zinc finger, which serves to connect the first and second β-strand to the end of the first α-helix (Fig. 4B,C). Sad1's B-site CCHH zinc-binding finger is conserved among all of Sad1's splicing homologs and aligns well with the B-site CCHH zinc fingers from the ZnF-UBP domains of USP16 and HDAC6 (Fig. 4C; Supplemental Fig. S1).
The B-site CCHH finger may be generally important for the stability of ZnF-UBP domains. Alanine substitutions at any of the CCHH zinc-binding residues of Sad1's B site results in aggregated protein as determined by size-exclusion chromatography (data not shown), and the Sad1 ACHH and the CCHA substitutions were lethal in vivo (Table 1). Every ZnF-UBP from human and yeast contains a B-site CCHH zinc finger, with the sole exception being USP5, which possesses residues that are engaged in compensatory interactions (Supplemental Fig. S4; Reyes-Turcu et al. 2006; Bonnet et al. 2008).
Sad1 does not possess a canonical ubiquitin binding site
Comparison of Sad1's ZnF-UBP primary sequence with those ZnF-UBPs that bind ubiquitin provided little insight into why ubiquitin binding was not observed for Sad1 (Fig. 2A). Inspection of the available ubiquitin-bound ZnF-UBP crystal structures in the PDB shows that the ubiquitin binding site lies between the L2A loop and the loop that connects the fourth and fifth β-strands, the β4-5 loop (Reyes-Turcu et al. 2006; Dong et al. 2008). Although overall RMSDs for Sad1's ZnF-UBP with other ZnF-UBPs in the PDB are low (Table 3), a structural alignment of Sad1's ZnF-UBP with the ubiquitin binding ZnF-UBPs from USP5 and HDAC6 reveals notable differences in the conformations of their L2A and β4-5 loops. In Sad1, these loops are positioned closer to one another relative to the USP5 and HDAC6 ZnF-UBP structures (Fig. 5A). Additionally, the closed conformation of Sad1 does not align well with any of the ZnF-UBP structures available in the PDB (Fig. 5B).
FIGURE 5.

The L2A and β4-5 loops of Sad1's ZnF-UBP domain are contracted relative to other ZnF-UBPs. (A) Structural alignment of Sad1's ZnF-UBP domain (pale orange) to the ZnF-UBP domains from H. sapiens HDAC6 (violet; PDB ID 3C5K) and H. sapiens USP5 (cyan; PDB ID 2G43). The zinc atoms of HDAC6 and USP5 are depicted as gray spheres, and the zinc atom from Sad1 is depicted as an orange sphere. Structural alignments were performed in Pymol. (B) Structural alignment of Sad1's ZnF-UBP main-chain (pale orange) to all other ZnF-UBP domain main-chains available in the PDB (gray; 2G45, 2G43, 3IHP, 3C5K, 3PHD, 3GV4, 2UZG, 2I50, 2L80, 2IDA, 3M99, 3MHH, and 3MHS). Sad1 is an outlier with respect to the distance between its L2A and β4-5 loops. (C) Structural alignment of Sad1's ZnF-UBP domain to the apo ZnF-UBP domain from USP5 (PDB ID 2G43). The ubiquitin interacting residue R221 of USP5 is pointed toward solvent. In contrast, the corresponding residue in Sad1, R70, is held close to the surface through two hydrogen bonds to Y58 and N102. Hydrogen bonds were determined by Pymol with a 3.1 Å cut-off. (D) Structural alignment of Sad1's ZnF-UBP to the ZnF-UBP domain from USP5 bound to ubiquitin (raspberry; PDB ID 2G45), shows that the ubiquitin tail as it binds to USP5 would clash with the unique placement of Sad1 R70. (E) Surface representations of the ZnF-UBP domains from USP5 and Sad1, either with or without ubiquitin. The structural alignment from 4D was used to model Sad1 with ubiquitin, and USP5 was removed for clarity. The deep depression present on the surface of USP5's ZnF-UBP domain is entirely filled by the unique placement of Sad1 R70. (F) Structural alignment of the A site zinc finger of HDAC6 to the corresponding regions of USP5 and Sad1. The residues of USP5 that are engaged in compensatory hydrogen bond interactions are labeled. (G) Primary sequence alignment of USP5 homologs aligned with the T-coffee server. The H. sapiens USP5 residues that engage in compensatory A-site zinc finger hydrogen bond interactions are highlighted. Shown below the alignment for reference are the equivalent residues in Sad1.
As a result of the contraction between the L2A and β4-5 loops in Sad1, homologous residues that would be used for ubiquitin recognition in USP5 are in a conformation that blocks ubiquitin binding by Sad1. Ubiquitin binding by USP5's ZnF-UBP is dependent upon the presence of a group of residues that are conserved in those ZnF-UBPs competent to bind ubiquitin (Fig. 2A; Reyes-Turcu et al. 2006). R221 from USP5's loop L2A forms two hydrogen bonds to the main-chain carbonyl oxygen of ubiquitin G75 (Fig. 5D). Sad1's ZnF-UBP also has an arginine at this position, Sad1 R70 (Fig. 2A). However, instead of pointing toward solvent to make itself available for interaction with ubiquitin, the crystal structure of Sad1 reveals that R70 packs against the surface of Sad1's ZnF-UBP (Fig. 5C,D). This conformation of Sad1 R70 is stabilized by hydrogen bonds to Y58 from the first β-strand and to N102 from the β4-5 loop (Fig. 5C). The homologous residue of Sad1 Y58 corresponds to W209 of USP5, which interacts with ubiquitin and is conserved among ZnF-UBPs that bind ubiquitin (Fig. 2A). Although the positions of the planar rings of Sad1 Y58 and USP5 W209 align well, Sad1 Y58 introduces a hydroxyl group that functions as a hydrogen bond acceptor for the terminal amine of Sad1 R70 (Fig. 5C). Sad1 R70 is further stabilized through a second hydrogen bond to N102 (Fig. 5C). Sad1 N102 corresponds to D264 in USP5, which in USP5's ZnF-UBP points toward the solvent to interact with the terminal amines of ubiquitin R72 and R74 (Fig. 5C,D; Reyes-Turcu et al. 2006). Aligning Sad1 to the crystal structure of USP5 bound to ubiquitin shows that, as a result of the unique stabilization of Sad1's R70, the R70 arginine side-chain lies directly in the path of where ubiquitin would be expected to bind (Fig. 5D).
The more contracted conformation of Sad1's L2A and β4-5 loops and R70's blockage of the ubiquitin binding path also results in an altered ZnF-UBP surface. Whereas the USP5 ZnF-UBP ubiquitin binding site appears as a prominent cavity on the surface of both the apo and the ubiquitin bound structures (Fig. 5E), the same area on Sad1, which corresponds to the space between the L2A and β4-5 loops, is entirely filled by the contraction of these loops and the placement of Sad1's R70 side-chain (Fig. 5E).
The absence of the A and C zinc binding sites in Sad1 allows for the observed contraction between the L2A loop and the β4-5 loop
Unlike Sad1, those ZnF-UBPs that are competent to bind ubiquitin have open structures stabilized by the presence of A and C zinc binding sites. In HDAC6, the A zinc binds the β4-5 loop and the C zinc binds to the L2A loop, constraining the positions of these loops and leading to the observed open conformation (Fig. 5A). The ZnF-UBP from USP5 also has a more open conformation relative to Sad1. USP5 has the C-site zinc finger, and although it is missing the A-site zinc finger, it appears to compensate for this absence by constraining the β4-5 loop with intraloop hydrogen bonds between E263, S260, and S177 (Fig. 5F). The importance of these bonds is supported by the observation that the only deviation from these well-conserved residues is with the Saccharomyces cerevisiae and S. pombe homologs of USP5, which contain a CHCC zinc finger (Fig. 5G). Shown below the alignment is the corresponding sequence in Sad1, which with the crystal structure shows that Sad1 possesses no such mechanism to constrain the position of its β4-5 loop. With the absence of the A and C zinc fingers in Sad1 and its homologs, a more closed conformation between the L2A and β4-5 loops is likely to be a general feature of the splicing-specific ZnF-UBPs, making ubiquitin binding unlikely.
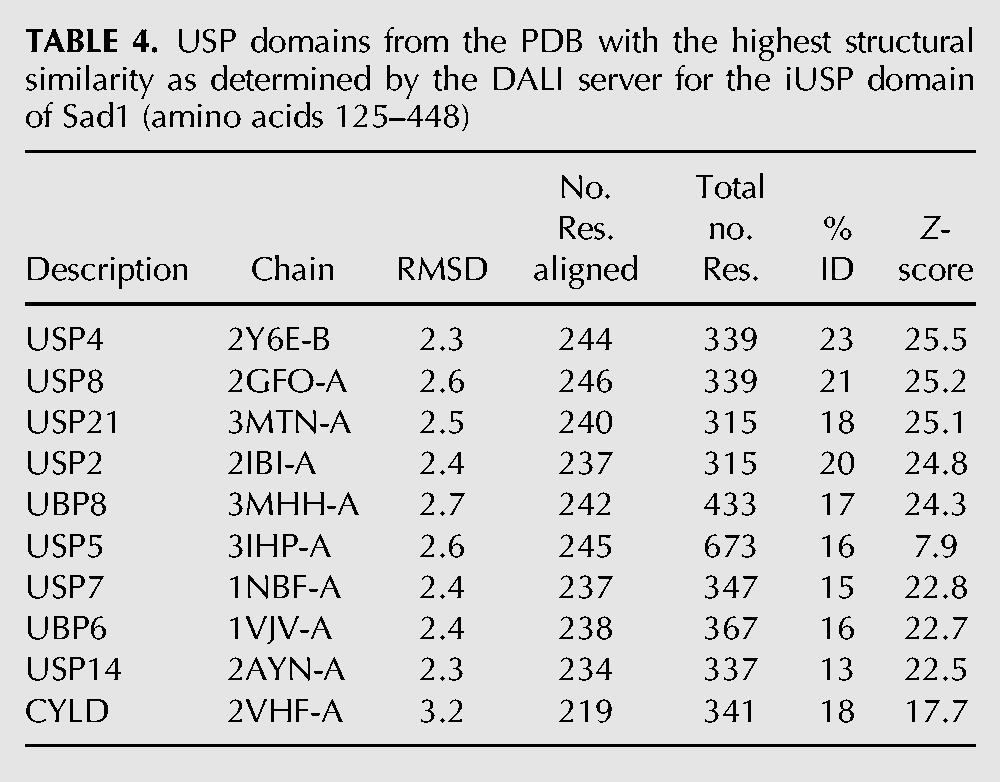
The iUSP domain from Sad1
The iUSP domain of Sad1 possesses the finger, thumb, and palm subdomains that are characteristic of USPs, and a Dali structural alignment search with the iUSP domain of Sad1 shows a high degree of structural similarity to other USP structures (Fig. 6A; Table 4; Hasegawa and Holm 2009; Komander et al. 2009). To analyze the structure of Sad1's iUSP domain, we used the USP domain from USP2 for comparison, as it has been well characterized biochemically and has been crystallized with ubiquitin, allowing us to model ubiquitin onto Sad1 (Renatus et al. 2006).
FIGURE 6.

The iUSP domain of Sad1 aligns well with known USP structures. (A) The finger, palm, and thumb subdomains of Sad1's iUSP domain (slate blue) are circled and the blocking loops indicated. (B) Structural alignment of the Sad1 iUSP domain to the USP domain from H. sapiens USP2 (lime) bound to ubiquitin (PDB ID 2HD5), where ubiquitin was removed afterward for clarity. The structural alignment was produced in Pymol. (C) Transparent surface representation of USP2 bound to ubiquitin (raspberry). The cup-like surface, which binds to the globular fold of ubiquitin, and the catalytic cleft, which binds to the C-terminal tail of ubiquitin, are highlighted. (D) A hypothetical model of the transparent surface representation of Sad1's iUSP domain after a structural alignment of Sad's iUSP domain to USP2 bound to ubiquitin. USP2 was removed to allow modeling of Sad1 iUSP's capacity to accommodate ubiquitin in its partial cup-like surface. (E) A zoomed image of the catalytic cleft from USP2 bound to ubiquitin aligned to Sad1's iUSP domain. The two blocking loops, the QDE loop of USP2 and the homologous PDL loop of Sad1, are labeled to highlight their different conformations. (F) A zoomed image of the catalytic triad from USP2 aligned to the pseudo-catalytic triad of Sad1's iUSP domain shows a high degree of structural similarity.
TABLE 4.
USP domains from the PDB with the highest structural similarity as determined by the DALI server for the iUSP domain of Sad1 (amino acids 125–448)
A structural alignment of Sad1's iUSP domain to USP2 bound to ubiquitin shows similarities in the palm and thumb subdomains but also reveals the extent to which Sad1's iUSP fingers could not be built due to the high degree of disorder in this region (Fig. 6B). As can be observed from the structural alignment, USP2 binds one zinc atom in the tips of its fingers, the presence of which has been implicated in ubiquitin recognition (Renatus et al. 2006). However, zinc binding in the fingertips is not a prerequisite for ubiquitin binding, as USP14 and USP7 are active USPs that are missing this zinc binding site in their fingertips; instead, these USPs compensate for this absence through the formation of a zinc ribbon–like fold (Hu et al. 2002, 2005). It is unclear how Sad1's fingertips are structured as it is missing the zinc binding residues identified in USP2, and the high degree of disorder in this region makes it impossible to determine if they adopt the compensatory zinc ribbon–like fold.
The overall fold and dimensions of Sad1's iUSP domain and catalytic core are similar to other USPs; however, the consensus motif for the catalytic triad of active USP domains, CHD/N, is changed (Komander et al. 2009). Instead of the essential nucleophilic cysteine, Sad1 and its homologs contain an aspartate, which in Sad1 is D159. Sad1 D159 participates in a hydrogen bond network with Sad1 H405 and D422, the conformations of which closely resemble that of the USP2 active catalytic triad, composed of C33, H314, and N331 (Fig. 6F). However, consistent with the hypothesis that Sad1 is catalytically inactive, Sad1 H405 and D422 are not well conserved, and single alanine mutations of the residues in the pseudo-catalytic triad are tolerated in vivo and in vitro, suggesting that they are dispensable for Sad1 function (Table 1).
The USP domain of USP2 recognizes ubiquitin through two distinct sets of interactions. A large cup-like surface formed between the finger, palm, and thumb subdomains recognizes the core of ubiquitin, residues 1–71, and a deep catalytic cleft formed between the palm and thumb subdomains interacts extensively with the C-terminal tail of ubiquitin, residues 71–76 (Fig. 6C; Renatus et al. 2006). Sad1's iUSP domain contains a similar cup-like feature that appears large enough to accommodate the core domain of ubiquitin in a hypothetical model of Sad1 bound to ubiquitin (Fig. 6D); however, the cup is partially disordered in the fingers region, complicating the interpretation of Sad1's putative ubiquitin binding surface.
The structure of Sad1's iUSP domain reveals the presence of a cleft that is reminiscent of the catalytic cleft seen in USP2, but it is missing critical elements. The catalytic cleft of USP2 appears as a deep groove that binds to the last five amino acids of the ubiquitin tail (Fig. 6C; Renatus et al. 2006). This groove is composed of the “Cys Box,” the “His Box,” the “QDE Box,” and two loops, Blocking Loop 1 (BL1) and Blocking Loop 2 (BL2) (Hu et al. 2005; Renatus et al. 2006). Sad1 is missing the catalytic cysteine from the Cys Box, but has the histidine and aspartate residues from the His box (Fig. 6F). The QDE box contacts the C terminus of ubiquitin in the active site of USP2, but in Sad1, the homologous sequence appears as PDL and is in an alternate orientation (Fig. 6E). Finally, BL1 and BL2, which abut the active site in USP2, are larger, and BL2 appears more flexible in Sad1 compared with USP2, as it was unable to be resolved in the crystal structure due to its high degree of disorder (Fig. 6E).
In USPs, BL1 and BL2 are connected to the palm subdomain in the catalytic cleft and must move to accommodate the docking of the C-terminal ubiquitin tail (Hu et al. 2005; Luna-Vargas et al. 2011). As is found with other USPs crystallized in the absence of ubiquitin, Sad1's BL1 is in the closed conformation, yet BL1 possesses an extra helical region that points toward the ubiquitin tail and is larger than the USP BL1s in the PDB (Fig. 6E). The BL2s from all USPs crystallized to date are short, well-ordered loops. In Sad1, BL2 not only is five amino acids longer than those of all other USPs but also is disordered, suggesting an increased degree of flexibility (Fig. 6E). Thus, the increased bulk of BL1 and BL2 may prevent Sad1 from creating an open pocket for ubiquitin binding, which may account for the observed inability to recognize ubiquitin in vitro.
DISCUSSION
Sad1 protein is a general splicing factor
We have examined the structure and function of the essential splicing factor Sad1, which is composed of two conserved and essential domains, a zinc finger ubiquitin binding domain and an inactive USP domain. Using pre-mRNA splicing microarrays (Fig. 1B), we show that Sad1 is a general splicing factor. Inactivation of SAD1 in vivo causes a rapid pre-mRNA splicing defect that is both broad and severe, and this phenotype is most likely due to a tri-snRNP joining defect, as inactivation of SAD1 in vitro causes an immediate block to spliceosome assembly at the prespliceosome stage (Fig. 1D). Recombinant Sad1 was able to complement the spliceosome assembly defect and restore in vitro splicing (Fig. 1C). We determined a 1.8 Å crystal structure of full-length Sad1 (Fig. 3A) and found that although the two domains of Sad1 are broadly similar to the ubiquitin signaling families from which they are derived, they deviate sharply in the regions that would be predicted to interact with ubiquitin, which may explain the inability to observe ubiquitin binding to purified Sad1 in vitro.
Sad1 protein does not bind ubiquitin in vitro
Sad1 lacks two of the zinc-binding sites found in ZnF-UBPs. Taken together with the dramatic rearrangement of the ubiquitin binding pocket, it is clear that the ZnF-UBP domain of Sad1 does not have the high affinity ubiquitin binding site seen in other members of this family (Figs. 2A, 5D). There are three other structures of ZnF-UBP domains that do not bind ubiquitin, and sequence alignments of all ZnF-UBPs in humans and yeast indicate that there may be more (Fig. 2A; Bonnet et al. 2008). Thus, as has been suggested previously, there are two classes of ZnF-UBPs: those that can bind ubiquitin and those, like Sad1, that cannot (Bonnet et al. 2008). Based upon the structural studies presented here and the high degree of conservation in the residues that dictates Sad1's unique L2A and β4-5 loop rearrangements, it is expected that the ZnF-UBP domain from all of Sad1's splicing homologs will similarly not bind ubiquitin (Supplemental Fig. S1).
While it is clear from the crystal structure of Sad1 why the ZnF-UBP domain is unable to bind ubiquitin, it is less obvious why Sad1's iUSP domain does not bind ubiquitin. Sad1's iUSP structure resembles other USP structures; it contains the prominent, cup-like surface that contributes to the binding of the core domain of ubiquitin in other USPs. However, the catalytic cleft also contributes to ubiquitin binding in active USPs, and this region, although present in Sad1, is dramatically remodeled. The pseudo-active site formed by Sad1's catalytic cleft lacks the essential catalytic USP residues and contains active site loops that are longer and more disordered than those that have been seen in other USP structures. Such changes may explain the inability to observe ubiquitin binding to the USP domain of Sad1 in vitro and may point to an unidentified role for these features in vivo.
The ZnF-UBP domain as an activator for the neighboring USP domain
ZnF-UBPs domains have been shown to act as ubiquitin-dependent activators for the deubiquitinase activity of their neighboring USP domains. For example, the deubiquitinase activity of USP5 is stimulated when the free C-terminal tail of ubiquitin binds to its ZnF-UPB domain, most likely through contributions to substrate recruitment (Reyes-Turcu et al. 2006, 2008; Avvakumov et al. 2012).
The ZnF-UBP domain has also been shown to contribute to USP activity in a ubiquitin-independent manner. Instead of binding ubiquitin, the ZnF-UBP from Ubp8 functions as an assembly lobe to recruit cofactors, which then stimulate the catalytic activity of its neighboring USP domain (Kohler et al. 2010; Samara et al. 2010, 2012). Analogous to our current examination of Sad1, initial studies of Ubp8 showed that purified full-length Ubp8 did not bind ubiquitin nor did it possess in vitro deubiquitinase activity (Lee et al. 2005). It was only upon the purification of Ubp8 from cell lysates and the identification of its interacting partners that in vitro deubiquitinase activity could be observed (Ingvarsdottir et al. 2005; Lee et al. 2005). Finally Ubp8 crystallized in the presence of these factors revealed that they assembled upon the ZnF-UBP domain, which then allowed them to interact with the fingers and catalytic cleft of Ubp8's USP domain to activate USP activity (Kohler et al. 2010; Samara et al. 2010, 2012). Consistent with its ubiquitin-independent function, the ZnF-UBP domain from Ubp8 is missing many of the residues important for ubiquitin recognition (Fig. 2A).
In this work, we show that Sad1's ZnF-UBP is of the class that does not bind ubiquitin and possesses an iUSP domain that closely resembles the USP fold (Table 4). Given UBP8's dependence upon its nonubiquitin binding ZnF-UBP domain for activity, it is plausible that Sad1's function similarly depends upon activation through cofactor binding to its upstream ZnF-UBP domain. This could result in the activation of a ubiquitin receptor-like function in its neighboring iUSP domain given its high similarity to known USP structures. Additionally, the ZnF-UBP of Sad1 may be essential for its recruitment to the spliceosome, as the cofactors that decorate the ZnF-UBP of Ubp8 for activation of the USP domain are also essential for its recruitment to the SAGA complex (Ingvarsdottir et al. 2005; Lee et al. 2005; Kohler et al. 2006).
If Sad1's ZnF-UBP does act as a scaffold, then these interactions occur in the pre-mRNA splicing lysate during in vitro splicing and assembly. When these cofactors are identified, we predict that Sad1's iUSP domain will become competent to interact with ubiquitin. An alternative to this model is that Sad1's iUSP domain may not need activation to bind ubiquitin but rather may require the recognition of both ubiquitin and the spliceosomal protein to which it is conjugated. Finally, Sad1's iUSP domain may not bind ubiquitin and instead may serve an entirely different function in the splicing reaction. With the resolution of which proteins directly interact with Sad1, which will be a challenge given Sad1's low affinity for splicing factors, we will distinguish between these potential mechanisms. The work presented here provides critical insight into the function of Sad1 by demonstrating Sad1's role in spliceosome assembly as a general splicing factor and by providing a structural perspective on the role of its essential and well-conserved ubiquitin-related domains.
MATERIALS AND METHODS
Strain construction and 5-fluoroorotic acid selection
The sad1-1 allele was cloned from the original sad1-1 isolate (Lygerou et al. 1999) along with 800 bp upstream of the start site and 900 bp downstream from the stop codon into a pRS313 HIS3 CEN/ARS plasmid. The same primers were used to clone a wild-type copy of SAD1 from S. cerevisiae S288C into both a pRS316 URA3 CEN/ARS plasmid and a pRS313 HIS3 CEN/ARS plasmid. The sequences were identical with the exception of T707C and C827T, resulting in the amino acid changes L236P and S276F, respectively. Subsequent mutants and truncations of SAD1 were produced by standard PCR mutagenesis of the SAD1 pRS313 plasmid. To produce a deletion strain of SAD1, the SAD1 gene was deleted using PCR-based homologous recombination in a diploid S288C strain using the KanMX marker (Longtine et al. 1998). After selection for kanamycin resistance, an individual colony was transformed with the wild-type SAD1 pRS316 plasmid, and after selection in the absence of uracil and the presence of kanamycin, the transformants were allowed to sporulate. Tetrads were dissected and grown on complete media, and then replica were plated to media lacking uracil and containing kanamycin for the selection of haploid cells harboring both the KanMX marked SAD1 deletion and the wild-type SAD1 pRS316 plasmid, producing the SAD1 pRS316/sad1::kan strain.
5-Fluoroorotic acid (5-FOA) screening was performed by transforming the SAD1 pRS316/sad1::kan strain with the wild-type SAD1 pRS313 plasmid, an empty pRS313 plasmid, or a pRS313 plasmid containing a mutation or truncation of SAD1. Selection was performed in the absence of histidine and in the presence of uracil to allow for the passive loss of the wild-type SAD1 pRS316 plasmid. Individual colonies were patched, then replica were plated to 5-FOA–containing plates. Growth indicated the ability to lose the wild-type SAD1 pRS316 plasmid and, therefore, the viability of the genetic manipulation. Transformants that survived on 5-FOA were further screened for temperature sensitivity by testing for growth at 30°C and 37°C.
Protein purification
Full-length Sad1, amino acids 1–448, was cloned from S. cerevisiae S288C into the pET-28b plasmid at the Bam HI and XhoI sites, which fuses a 6-His tag to the N terminus. BL21 E. coli cells were transformed and induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside at an optical density of 0.8 at 600 nm, and the cultures were allowed to grow overnight at 18°C. Cells were flash frozen and resuspended in 50 mM Hepes (pH 7.9), 500 mM NaCl, 20 mM Imidazole, 1 mM tris (2-chloroethyl) phosphate (TCEP). After sonication, the soluble lysate was incubated with Ni-NTA; washed with 50 mM Hepes (pH 7.9), 150 mM NaCl, 20 mM imidazole, 1 mM TCEP; and eluted with wash buffer supplemented with imidazole to 300 mM. The eluate was concentrated and applied to a S200 Superdex size-exclusion column equilibrated with 20 mM Hepes (pH 7.9), 50 mM KCl, and 1 mM TCEP. The peak fractions were pooled, concentrated to 50 μM Sad1, and then flash frozen. This protein was used for crystallization and for use in in vitro splicing and assembly assays.
Crystallization
The initial screen to identify crystallization conditions for full-length Sad1 was conducted by the Hauptman-Woodward Institute with 50 μM 6-His-Sad1. Under oil hits were optimized by vapor-diffusion hanging drop methods, and the optimal crystal conditions were determined to be 1:1 50 μM 6-His Sad1with 13% PEG 4000, 0.1 M sodium acetate, and 0.1 M magnesium acetate (pH 5.5) at 25°C and were fully grown within 48 h. Tungsten-derivative Sad1 crystals were produced by soaking native crystals for 2 h in mother liquor supplemented with 10 mM sodium tungstate. Optimal cryogenic conditions were achieved by soaking crystals in a drop of mother liquor supplemented with PEG 400 to 25%.
Structure solution
Data were collected at 100 K at the Advanced Light Source and processed using the program suite Elves (Holton and Alber 2004). Phases were determined from a single-wavelength anomalous dispersion experiment. A single tungsten site was found in the HYSS module of Autosol in Phenix (Adams et al. 2010), which was used to calculate phases with the program Phaser and then to generate an electron density map with Resolve for Autobuild. Three hundred seventy-five of 414 residues were placed by Autobuild, and then 39 residues were placed manually into the density. The final model was refined in Phenix to an R/Rfree of 17.1/20.2 (Adams et al. 2010). Coordinates and structure factors were deposited in the Protein Data Bank with accession code 4MSX.
Pre-mRNA splicing arrays
The following is adapted from previously designed S. cerevisiae pre-mRNA microarray studies (Pleiss et al. 2007). Briefly, 100 mL of SAD1 pRS313/sad1▵::kan and sad1-1 pRS313/sad1▵::kan cells were grown to mid-log phase at 25°C and then split to two 50 mL cultures, which were either kept at 25°C or shifted to 37°C for 30 min until cells were collected and frozen. RNA was extracted and split for dN9 cDNA synthesis in the presence of either Cy5- or Cy3-labeled dUTP. Cy3/Cy5-labeled SAD1 and sad1-1 cDNAs were competitively hybridized to pre-mRNA microarrays composed of probes that were designed to hybridize to sequence elements in the intron, the exon, and the junction of every yeast intron-containing gene. Microarray fluorescence was measured at 532 and 635 nm with an Axon Instruments GenePix 4000B, and images were processed with Axon Instruments GenePix Pro, version 5.1. Six technical replicate spots per array and dye flipped replicate arrays were combined and normalized as log2 transformed ratios that were then averaged and subjected to centroid-linkage hierarchical clustering using Cluster 3.0 and displayed graphically to produce heat maps with Java Treeview.
In vitro splicing and assembly
Splicing extracts were prepared as follows from SAD1 pRS313/sad1▵::kan and sad1-1 pRS313/sad1▵::kan grown at 30°C to an optical density of 2 at 600 nm. Cells were resuspended in 0.4× volumes AGK buffer (20 mM Hepes at pH 7.9, 200 mM KCl, 2.5 mM MgCl2, protease inhibitors) and frozen. Cells were ball-milled five times at 10 Hz for 3 min, and thawed lysates were centrifuged at 50,000g for 30 min to remove membranes and centrifuged again at 125,000g for 80 min to remove ribosomes. Finally, lysates were buffer exchanged into buffer D (20 mM Hepes at pH 7.9, 10% glycerol, 50 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT) using a desalting column.
In vitro splicing reactions were prepared with 8 µL of splicing extract, which was added to a reaction containing 3% PEG 8000, 60 mM potassium phosphate (pH 7.0), 2.5 mM magnesium chloride, and 10 units of RNasin. The ACT1 pre-mRNA, transcribed with Cy5-labeled UTP (as 5% of the UTP), was incubated with sad1-1 splicing extracts that were either inactivated completely with an incubation at 37°C or not. Full-length Sad1 was supplemented to the reactions in a 10-fold serial dilution starting from 105 nM. After the addition of ATP to 2 mM, the reactions were incubated for 30 min at 25°C and stopped with the addition of SDS to 2.5%, 1 mM EDTA. Ten units of Proteinase K were added, and the reactions were incubated for 10 min at 60°C and then loaded onto a 6% polyacrylamide, 6 M urea, TBE gel.
Using a P32-labeled ACT1 pre-mRNA, in vitro assembly reactions were prepared in an identical manner to that of the in vitro splicing reactions with the exception that the reactions were stopped with the addition of 2× Native buffer (60 mM KPO4, 3 mM MgCl2, 3% PEG 8000, 8% glycerol, 10 mg/mL heparin) and were then resolved on a 26-cm-long, 0.5-mm-thick, 4% 80:1 polyacrylamide gel that was run at 450 V for 4.5 h in TAE.
Isothermal titration calorimetry
To express the iUSP domain of Sad1 for ITC, residues 124–448 were cloned and purified in a similar manner to full-length Sad1, but to aid in solubility, the Hepes (pH 7.9) buffer was replaced with Tris-Cl (pH 8.0) and the size-exclusion buffer was 20 mM Tris-Cl (pH 8.0), 150 mM NaCl, and 1 mM TCEP. Full-length Sad1 was also purified in this manner for the ITC experiments and showed no difference in potency in the in vitro splicing or assembly add-back assays (data not shown). The plasmid to express the ZnF-UBP domain from USP5 was a gift from Dr. Hong-Yu Hu, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China, and the protocol followed for purifying the protein did not vary from the published one (Zhang et al. 2011). Ubiquitin was purchased from Boston Biochemicals.
We dizlyzed 50 μM of Sad1, 50 μM of the iUSP domain from Sad1, 50 μM of the ZnF-UBP domain from USP5, and 1 mM ubiquitin extensively against 25 mM Tris (pH 7.4), 150 mM NaCl, and 0.5 mM TCEP. The receptor cell was filled with 50 μM Sad1, 50 μM Sad1-iUSP, or 50 μM USP5- ZnF-UBP. We used 1 mM ubiquitin as the titrant. Experiments were carried out on a Microcal VPITC at 25°C.
GST-UBL immunoprecipitations
Hub1, SUMO, ubiquitin, and Rub1 were cloned from S. cerevisiae S288C into the pGEX 6-P-1 vector, producing N-terminal fusions with GST. Proteins were purified in a similar manner to the 6-His tagged proteins above. We used 20 mM Tris-Cl (pH 8.0), 150 mM NaCland 1 mM TCEP as the resuspension and wash buffer, which was then supplemented with 20 mM glutathione for elution. Eluates were concentrated and dialyzed extensively against 20 mM Tris-Cl (pH 8.0), 150 mM NaCl, and 0.5 mM TCEP. Full-length Snu66 was cloned from S. cerevisiae S288C into pET28b, placing a 6-His tag on its N terminus. 6-His Snu66 was purified in a manner identical to the Sad1 iUSP domain.
Six nanomoles of each of the GST-UBLs was incubated separately in 500 μL of wash buffer with 12.5 μL magnetic glutathione beads for 1 h at 4°C to allow for binding, and then beads were washed with 20 mM Tris-Cl (pH 8.0), 150 mM NaCl and 1 mM TCEP. Five hundred microliters of 50 μM Sad1, 50 μM Snu66, or 50 μM USP5-ZnF-UB was added to the beads and incubated for an additional hour at 4°C before washing and eluting in 50 μL wash buffer supplemented with 20 mM glutathione. Eluates were analyzed on 8% SDS-polyacrylamide gel electrophoresis.
Biolayer interferometry
Ubiquitin labeled with a single biotin on its N terminus was purchased from Thermo Scientific. Sad1 and USP5 were prepared in a manner identical to that used for the ITC experiments. BLI was measured at 25°C with an Octet384 (ForteBio). Biotin ubiquitin was immobilized on streptavidin biosensors at 10 μg/mL in assay buffer (25 mM Tris-Cl at pH 8.0, 150 mM NaCl, and 1 mM TCEP) for 5 min. Probes were then washed with assay buffer for 1 min, regeneration buffer (0.1% SDS, 10 mM glycine-HCl at pH 2.5) for 30 sec, and assay buffer again for 1 min. All binding measurements were carried out at the indicated concentrations of either USP5 ZnF-UBP or Sad1 for the indicated amounts of time. A single biosensor was used to measure ubiquitin binding at each concentration. BLI responses to buffer alone and responses to streptavidin coated tips without immobilized ubiquitin were subtracted from each curve using the ForteBio analysis suite. Data for processed association binding curves were analyzed with GraphPad Prism 6.0 (GraphPad), where they were fit using the equation for a one-phase association. Saturation binding values from the plateaus of the one-phase associations (Req) were plotted as a function of titrant concentration and fit with a single-site saturation binding model to obtain KDs when possible.
DATA DEPOSITION
The coordinates and the structure factors have been deposited in the Protein Data Bank with accession code 4MSX.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Andre Schonichen and Erica Moehle for their technical contributions. We thank Sarah Ledoux and other members of the Guthrie laboratory for their invaluable contributions to the text and helpful discussions. The USP5 ZnF-UBP expression plasmid was a kind gift from Dr. Hong-Yu Hu. We particularly thank Robert Stroud and Jeffery Cox for their input and support. The staff at BL8.3.1 at ALS, especially James Holton, provided invaluable technical support and advice. This work was supported by NIH grant GM21119 to C.G. and a predoctoral fellowship from the National Science Foundation to H.H. C.G. is an American Cancer Society Research Professor of Molecular Genetics. O.S.R. is supported by NIH K08AI091656. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract no. DE-AC02-05CH11231.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66(Pt 2): 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Bycroft M 2007. The solution structure of the ZnF UBP domain of USP33/VDU1. Protein Sci 16: 2072–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov GV, Walker JR, Xue S, Allali-Hassani A, Asinas A, Nair UB, Fang X, Zuo X, Wang YX, Wilkinson KD, et al. 2012. Two ZnF-UBP domains in isopeptidase T (USP5). Biochemistry 51: 1188–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellare P, Kutach A, Rines A, Guthrie C, Sontheimer E 2006. Ubiquitin binding by a variant Jab1/MPN domain in the essential pre-mRNA splicing factor Prp8p. RNA 12: 292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellare P, Small EC, Huang X, Wohlschlegel JA, Staley JP, Sontheimer EJ 2008. A role for ubiquitin in the spliceosome assembly pathway. Nat Struct Mol Biol 15: 444–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Romier C, Tora L, Devys D 2008. Zinc-finger UBPs: regulators of deubiquitylation. Trends Biochem Sci 33: 369–375 [DOI] [PubMed] [Google Scholar]
- Chan S, Kao D, Tsai W, Cheng S 2003. The Prp19p-associated complex in spliceosome activation. Science 302: 279–282 [DOI] [PubMed] [Google Scholar]
- Cheng S, Abelson J 1987. Spliceosome assembly in yeast. Genes Dev 1: 1014–1027 [DOI] [PubMed] [Google Scholar]
- Dong A, Ravichandran M, Schuetz A, Loppnau P, Li Y, MacKenzie F, Kozieradzki I, Edwards AM, Arrowsmith CH, Weigelt J, et al. 2008. Crystal structure of human HDAC6 zinc finger domain. http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=3C5K
- Fabrizio P, McPheeters D, Abelson J 1989. In vitro assembly of yeast U6 snRNP: a functional assay. Genes Dev 3: 2137–2150 [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Holm L 2009. Advances and pitfalls of protein structural alignment. Curr Opin Struct Biol 19: 341–348 [DOI] [PubMed] [Google Scholar]
- Holton J, Alber T 2004. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci 101: 1537–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Li P, Li M, Li W, Yao T, Wu J, Gu W, Cohen R, Shi Y 2002. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111: 1041–1054 [DOI] [PubMed] [Google Scholar]
- Hu M, Li P, Song L, Jeffrey P, Chenova T, Wilkinson K, Cohen R, Shi Y 2005. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J 24: 3747–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsdottir K, Krogan N, Emre N, Wyce A, Thompson N, Emili A, Hughes T, Greenblatt J, Berger S 2005. H2B ubiquitin protease Ubp8 and Sgf11 constitute a discrete functional module within the Saccharomyces cerevisiae SAGA complex. Mol Cell Biol 25: 1162–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joint Center For Structural Genomics. 2004. Crystal structure of ubiquitin carboxyl-terminal hydrolase 6 (yfr010w) from Saccharomyces cerevisiae at 1.74 Å resolution. http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=1vjv
- Jurica MS, Moore MJ 2003. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell 12: 5–14 [DOI] [PubMed] [Google Scholar]
- Kohler A, Pascual-Garcia P, Llopis A, Zapater M, Posas F, Hurt E, Rodriguez-Navarro S 2006. The mRNA export factor Sus1 is involved in Spt/Ada/Gcn5 acetyltransferase-mediated H2B deubiquitinylation through its interaction with Ubp8 and Sgf11. Mol Biol Cell 17: 4228–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler A, Zimmerman E, Schneider M, Hurt E, Zheng N 2010. Structural basis for assembly and activation of the heterotetrameric SAGA histone H2B deubiquitinase module. Cell 141: 606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Clague M, Urbé S 2009. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 10: 550–563 [DOI] [PubMed] [Google Scholar]
- Korneta I, Magnus M, Bujnicki JM 2012. Structural bioinformatics of the human spliceosomal proteome. Nucleic Acids Res 40: 7046–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Florens L, Swanson S, Washburn M, Workman J 2005. The deubiquitylation activity of Ubp8 is dependent upon Sgf11 and its association with the SAGA complex. Mol Cell Biol 25: 1173–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Newman AJ, Cheng SC, Abelson J 1985. Yeast mRNA splicing in vitro. J Biol Chem 260: 14780–14792 [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Luna-Vargas MP, Faesen AC, van Dijk WJ, Rape M, Fish A, Sixma TK 2011. Ubiquitin-specific protease 4 is inhibited by its ubiquitin-like domain. EMBO Rep 12: 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lygerou Z, Christophides G, Séraphin B 1999. A novel genetic screen for snRNP assembly factors in yeast identifies a conserved protein, Sad1p, also required for pre-mRNA splicing. Mol Cell Biol 19: 2008–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder C, Kutach A, Guthrie C 2009. ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat Struct Mol Biol 16: 42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova O, Makarov E, Lührmann R 2001. The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP are essential for the assembly of mature spliceosomes. EMBO J 20: 2553–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Ammon T, Popowicz G, Krajewski M, Nagel R, Ares M, Holak TA, Jentsch S 2011. Role of the ubiquitin-like protein Hub1 in splice-site usage and alternative splicing. Nature 474: 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang H, Ali Y, Ravichandran M, Dong A, Qiu W, MacKenzie F, Dhe-Paganon S, Arrowsmith C, Zhai R 2011. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem 287: 2317–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai M, Tzeng S, Kovacs J, Keaton M, Li S, Yao T, Zhou P 2007. Solution structure of the Ubp-M BUZ domain, a highly specific protein module that recognizes the C-terminal tail of free ubiquitin. J Mol Biol 370: 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss J, Whitworth G, Bergkessel M, Guthrie C 2007. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol 5: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renatus M, Parrado S, D'Arcy A, Eidhoff U, Gerhartz B, Hassiepen U, Pierrat B, Riedl R, Vinzenz D, Worpenberg S, et al. 2006. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure 14: 1293–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu F, Horton J, Mullally J, Heroux A, Cheng X, Wilkinson K 2006. The ubiquitin binding domain ZnF UBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell 124: 1197–1208 [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu F, Shanks J, Komander D, Wilkinson K 2008. Recognition of polyubiquitin isoforms by the multiple ubiquitin binding modules of isopeptidase T. J Biol Chem 283: 19581–19592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samara N, Datta A, Berndsen C, Zhang X, Yao T, Cohen R, Wolberger C 2010. Structural insights into the assembly and function of the SAGA deubiquitinating module. Science 328: 1025–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samara N, Ringel A, Wolberger C 2012. A role for intersubunit interactions in maintaining SAGA deubiquitinating module structure and activity. Structure 20: 1414–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E, Werner S, Neubauer J, Stegmeier F, Aspden J, Rio D, Harper J, Elledge S, Kirschner M, Rape M 2010. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev 24: 1434–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SW, Ryan DE, Ge HY, Moore RE, Young MK, Lee TD, Abelson J 2002. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol Cell 9: 31–44 [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhou C, Zhou Z, Song A, Hu H 2011. Domain analysis reveals that a deubiquitinating enzyme USP13 performs non-activating catalysis for Lys63-linked polyubiquitin. PLoS One 6: e29362. [DOI] [PMC free article] [PubMed] [Google Scholar]