

A new method and data analysis pipeline, called Mod-seq, for assaying the structure of RNAs by high-throughput sequencing is described. It can be utilized both in vivo and in vitro, with any small molecule that modifies RNA and consequently impedes reverse transcriptase. As proof-of-principle, we used dimethyl sulfate (DMS) to probe the in vivo structure of total cellular RNAs in Saccharomyces cerevisiae. While this method is applicable to RNAs of any length, its high-throughput nature makes Mod-seq ideal for studying long RNAs and complex RNA mixtures.
Keywords: RNA-binding protein, RNA structure, high-throughput sequencing
Abstract
The functions of RNA molecules are intimately linked to their ability to fold into complex secondary and tertiary structures. Thus, understanding how these molecules fold is essential to determining how they function. Current methods for investigating RNA structure often use small molecules, enzymes, or ions that cleave or modify the RNA in a solvent-accessible manner. While these methods have been invaluable to understanding RNA structure, they can be fairly labor intensive and often focus on short regions of single RNAs. Here we present a new method (Mod-seq) and data analysis pipeline (Mod-seeker) for assaying the structure of RNAs by high-throughput sequencing. This technique can be utilized both in vivo and in vitro, with any small molecule that modifies RNA and consequently impedes reverse transcriptase. As proof-of-principle, we used dimethyl sulfate (DMS) to probe the in vivo structure of total cellular RNAs in Saccharomyces cerevisiae. Mod-seq analysis simultaneously revealed secondary structural information for all four ribosomal RNAs and 32 additional noncoding RNAs. We further show that Mod-seq can be used to detect structural changes in 5.8S and 25S rRNAs in the absence of ribosomal protein L26, correctly identifying its binding site on the ribosome. While this method is applicable to RNAs of any length, its high-throughput nature makes Mod-seq ideal for studying long RNAs and complex RNA mixtures.
INTRODUCTION
RNA is an extremely versatile biomolecule. The last several decades have revealed that RNA is not only a mediator of the genetic code between DNA and protein, but also functions in a wide variety of cellular processes, including transcription, RNA splicing and editing, mRNA turnover, protein synthesis, and cell signaling. The folding of RNA into complex secondary and tertiary structures serves many functions, such as creating binding sites for regulatory proteins and small molecules (Cruz and Westhof 2009). Consequently, determining RNA structures can provide molecular insights into their functions in a diverse array of cellular processes.
Classical approaches to probing RNA structure rely on small molecules, ions, or enzymes to cleave or modify the RNA, often in a manner dependent upon solvent accessibility (Peattie and Gilbert 1980; Brunel and Romby 2000; Weeks 2010). Cleavages or modifications can then be detected using a radioactively labeled primer, reverse transcription, and PAGE analysis (Stern et al. 1988). While these approaches have been invaluable to the study of RNA secondary and tertiary structure, they are often labor-intensive and time-consuming. The resolution of primer extension and SDS-PAGE analysis only allows interrogation of ∼150 nucleotides per radiolabeled primer, and only a handful of RNAs can be investigated at a time.
A relatively new approach to assaying RNA structure, selective 2′-hydroxyl acylation and primer extension (SHAPE) measures nucleotide flexibility, providing information on both base-pairing and tertiary interactions (Merino et al. 2005). Combining SHAPE with fluorescently labeled oligonucleotides and capillary electrophoresis (SHAPE-CE) has allowed rapid interrogation of much longer RNAs, including E. coli and yeast ribosomal RNAs (rRNA) (Deigan et al. 2009; Leshin et al. 2011) and the HIV-I RNA genome (Wilkinson et al. 2008; Watts et al. 2009). The improvements offered by SHAPE-CE, in combination with automated sequencing and data analysis, allow ∼600–700 nucleotides to be assayed per fluorescently labeled oligonucleotide.
While SHAPE-CE has greatly improved the speed and accuracy of RNA structure probing data, there are still limitations to this approach. For example, analysis of the analog data obtained by capillary electrophoresis is complicated. Data must be corrected for background, mobility shifts due to the different primers, and signal decay of longer read lengths. Then, the sequencing traces must be aligned, and the area under each nucleotide peak calculated. Analysis of SHAPE-CE data requires complicated transformation and quantification of analog fluorescence data into relative reactivity values, including modeling probabilities of reverse transcriptase drop-off. Second, because this approach uses oligonucleotides complementary to an RNA of interest, structure probing of long RNAs requires the use of more than one custom oligonucleotide. Finally, the low-throughput nature of capillary electrophoresis restricts SHAPE-CE analysis to a small number of RNAs that can be assayed at a time.
In an effort to characterize the structures of multiple RNAs in a single experiment, recent approaches have begun utilizing high-throughput sequencing to assay modifications or cleavages in RNA structure probing experiments. FragSeq and parallel analysis of RNA structure (PARS) rely on structure-specific nucleases that cleave either single-stranded or double-stranded RNAs (Kertesz et al. 2010; Underwood et al. 2010). SHAPE-Seq combines SHAPE chemistry with deep-sequencing technology to simultaneously assay nucleotide flexibility of multiple RNAs (Lucks et al. 2011). While providing higher resolution structural information, SHAPE-Seq is limited to the analysis of relatively short RNAs (∼300 nt) transcribed in vitro, as RT reactions are initiated at the extreme 3′ end of the molecule. SHAPE-Seq also requires complex models of RT drop-off, as all reverse transcription reactions are initiated from common primer binding sites. While a significant step forward, the major caveat of these techniques is that they are limited to in vitro analysis of denatured and refolded RNA (Westhof and Romby 2010). For example, in vivo nuclease expression would be highly impractical, difficult to control, and is known to cause cell death (Leland and Raines 2001).
Here we present a new strategy and analysis pipeline for assaying the in vivo structure of long RNAs by high-throughput sequencing—Mod-seq. This approach is applicable both in vivo and in vitro and can be utilized with any small molecule that modifies RNA and blocks reverse transcription. We probed the secondary structure of yeast total RNA using dimethyl sulfate (DMS), and identified significant sites of modification simultaneously on all four ribosomal RNAs and 32 additional noncoding RNAs in vivo. Our approach produced DMS modification patterns consistent with the published structure of the yeast ribosome (Ben-Shem et al. 2011), highlighting the power of this technique to investigate the structure of especially long RNAs. We further assayed the structural changes that occur in 80S yeast ribosomes in a previously characterized ribosomal protein L26 deletion mutant (rpl26Δ) (Babiano et al. 2012). By combining in vivo DMS probing with deep sequencing, we detected known structural changes in 5.8S and 25S rRNAs and footprinted the L26 binding site. We believe that Mod-seq will greatly facilitate structural studies of long RNAs and complex RNA mixtures.
RESULTS
We sought a high-throughput method to probe long RNA structures in vivo to advance research in RNA biology. While DMS probing is a well established, powerful approach to interrogate RNA structural characteristics in living cells, identifying the sites of DMS modification using traditional electrophoresis approaches is tedious and relatively slow. We developed a new method to map RNA chemical modification using high-throughput sequencing (Mod-seq) (Fig. 1A). Mod-seq introduces several features that allow high-throughput probing of long RNAs. Chemically treated RNA is randomly fragmented and an adapter oligonucleotide is ligated to 3′ ends of RNA fragments. This 3′ adapter serves as a universal primer-binding site for reverse transcription, allowing modification mapping on RNAs of any length. A second adapter oligonucleotide is ligated to 5′ ends of RNA fragments to differentiate RT stops due to chemical modification from those resulting from run-off of random RNA fragment ends. RNA fragments containing the 5′ adapter are enriched and converted to cDNA via reverse transcription using a primer containing a 5′ phosphorylated end. Finally, cDNA products are circularized and PCR amplified to produce barcoded libraries for multiplexed high-throughput sequencing. High-throughput sequence reads that initiate with the 5′ adapter indicate RT drop off at random fragment ends, while reads without the 5′ adapter result from RT release at a modified nucleotide.
FIGURE 1.

Mapping DMS modification sites with high-throughput sequencing. (A) RNA from DMS-treated cells is randomly fragmented, ligated to specific 5′ (blue) and 3′ (green) adapter oligonucleotides, and reverse transcribed. cDNA is circularized and products containing 5′ adapter sequences are reduced via subtractive hybridization. Remaining cDNA products are PCR amplified to introduce Illumina library sequences. Each library is given a unique 6-nt barcode and pooled for high-throughput sequencing. Reads containing the 5′ adapter sequence are removed from further analysis. (B) Genome browser view (IGV) showing the number of 5′ read ends from DMS treated (+, purple) and untreated (−, blue) samples. The peak in the “no DMS” sample corresponds to a known site of endogenous rRNA methylation. The difference between these plots is shown in red, after shifting peaks 1 nt 5′ to represent actual sites of chemical modification. (C) Enlarged genome browser view of structure probing on the 5.8S rRNA. Reverse-transcriptase blocks due to DMS modification are mostly A and C nucleotides, and map to known single-stranded regions in 5.8S rRNA.
We developed a data analysis pipeline, “Mod-seeker,” to identify RT termination sites that are significantly enriched in RNA from DMS-treated cells. For each RNA species (e.g., 25S, 18S, and 5.8S rRNA), the fractions of RT stops at each nucleotide from treated and untreated samples are compared to estimate the odds ratio of enrichment at each site using Cochran-Mantel-Haenszel tests. This approach takes into consideration experimental noise by using data from replicate experiments. Significant sites of modification are identified using a minimum 1.5-fold enrichment and 5% false discovery rate. To facilitate use of our approach, a python implementation of Mod-seeker is included in the Supplemental Files. This pipeline takes high-throughput sequence data, a target sequence for alignment, and gene annotations as input, and returns sites of significant modification for each annotated gene.
As proof-of-principle, we used Mod-seq to identify sites of in vivo DMS modification of yeast ribosomal RNA. We first performed a titration experiment to determine optimal DMS treatment conditions (Supplemental Fig. S1). Mod-seq libraries were then prepared using RNA from cells treated with 100 mM DMS for 2 min, and control cells not treated with DMS. Libraries were sequenced to produce an average of 5.7 million reads from each of two replicate experiments (Supplemental Table S1). We identified 614 nucleotides from 35 highly abundant noncoding RNAs (rRNA, tRNA, and snoRNA) with significantly more chemical modification after DMS treatment (Supplemental Table S2). The fold-enrichment values for sites on scR1 and rRNAs are depicted in Figure 2A and Supplemental Figure S3, respectively. Sites with more than 1.5-fold enrichment are localized to regions of single-stranded RNA, and were consistent with traditional primer extension analysis (Supplemental Fig. S4). DMS-specific RT stops were enriched at A residues, consistent with the known base-specificity of DMS (1.8-fold enrichment compared with genomic A frequency, Fisher's Exact Test, P = 2.1 × 10−8) (Fig. 2B). In addition, several robust RT stops were present in negative control experiments. These DMS-independent stops occurred at known endogenously modified rRNA nucleotides (Fig. 1B). We conclude that Mod-seq can successfully map chemically modified nucleotides on large RNAs from living cells.
FIGURE 2.

Sites of yeast in vivo DMS modification identified by Mod-seq. (A) Example of Mod-seq sites on the 522-nt-long SRP RNA (SCR1). Nucleotides with significantly enriched sites of DMS modification are shown with colored bars depicting relative modification strength (fold enrichment). (B) Nucleotide composition of all significant modification sites identified. Bar graph shows the number of significant modifications at each of the four RNA nucleotide bases. Bars are colored to represent the distribution of fold enrichment for each nucleotide. Sites with high fold enrichment are almost exclusively As and Cs, while significant modification of Gs have smaller enrichment values.
DMS methylation of adenine N1 and cytosine N3 leads to termination of reverse transcription by disrupting base-pairing with incoming nucleotides. Guanine N7 is also methylated by DMS, which is not on the base-pairing face. While m7G methylation does not affect reverse transcription, several studies have reported rare increases in reverse transcription termination at G residues upon treatment with DMS (Wells et al. 2000). This has previously been interpreted as resulting from rare methylation at position N1, which may occur on enol-tautomers of guanosine (Moazed and Noller 1986). Surprisingly, roughly one-quarter of significant modifications mapped to G residues (Fig. 2B). However, G modifications have a weaker effect on reverse transcription termination. Ninety percent of sites in the highest quintile of DMS-dependent fold enrichment are A or C residues, with few Gs. In contrast, G residues make up roughly half of the sites in the lower two quintiles. Modified U residues were rarely identified, comprising only 1% of all significant sites.
We next determined the efficacy of using Mod-seq to map the binding site of an RNA-binding protein (RBP) in vivo. We used ribosomal protein L26 as a test case, as deletion of the RPL26 gene has little effect on growth in rich media (Babiano et al. 2012) and its binding site on the ribosome is well known from the crystal structure model (Ben-Shem et al. 2011). Mod-seq was performed in duplicate experiments on an RPL26 deletion strain. We identified 58 nucleotides with significantly more modification in rpl26Δ than in wild-type yeast (Fig. 3; Supplemental Table S3). Viewed on the rRNA secondary structure, L26-sensitive sites of DMS probing are concentrated on the base-pairing interface of 5.8S and 25S rRNAs (Supplemental Fig. S5). These sites form a cluster of nucleotides around the L26 protein in the yeast ribosome crystal structure (Fig. 3). Thus, we conclude that Mod-seq is an effective method to map specific binding sites of RBPs.
FIGURE 3.

Footprinting the binding site of an RNA binding protein. (A) Mod-seq probing results in the L26 deletion mutant and wild-type cells. (B) Views of the X-ray-crystal structure model of the yeast 80S ribosome generated with Pymol (PDB: 3U5F, 3U5G, 3U5H, 3U5I). The large (left) and small (right) subunits are shown in different shades of gray (upper). Ribosomal protein L26 is shown in a transparent surface rendering. Nucleotides with increased modification by DMS in the absence of L26 protein in vivo are shown in colors representing their fold-enrichment values. Magnified views showing residues with the largest differences in DMS sensitivity (lower; red and orange nucleotides). These nucleotides cluster around the site of L26 binding.
DISCUSSION
Chemical probing is a powerful tool for investigating RNA structure and RNA–protein interactions in vivo. However, traditional methods to identify modified nucleotides are time consuming, tedious, and have relatively low-throughput. Here, we demonstrate Mod-seq, a new method that overcomes these limitations. Mod-seq differs from prior approaches in that it can be used both in vivo or in vitro, and can be applied to RNAs of any length. Random fragmentation of cellular RNA ensures that reverse transcription will initiate at random positions, alleviating the need for complex RT stop probability modeling. However, random fragmentation introduces a problem—differentiating RT stops due to chemical modification from those resulting from run-off of random fragment ends. We solved this problem by ligating 5′ adapters to our RNA before reverse transcription. High-throughput sequence reads that initiate with the 5′ adapter indicate RT drop off at random fragment ends, while reads without the adapter result from RT release at a modified nucleotide.
We used Mod-seq to map significant sites of DMS modification simultaneously on 36 noncoding RNAs in vivo. Identified modification sites were consistent with known secondary structure models from these RNAs. Mod-seq sites were qualitatively similar to traditional primer extension for both 5.8S and 25S rRNAs. However, quantitative comparisons were much more consistent for the longer 25S rRNA (Supplemental Fig. S4). The better performance on long RNAs is likely due to less fragmentation noise, as 5.8S RNA is as short as the selected fragment size. Finally, we show that comparing Mod-seq results in the presence and absence of an RNA-binding protein can reveal its binding site and resulting structural changes in its absence.
We also present a python implementation of the Mod-seeker analysis pipeline. Mod-seeker can be run on Macintosh or Linux computers, with any number of replicate samples. This pipeline uses simple settings files in which researchers can choose their own False Discovery Rate (FDR) and fold-enrichment thresholds, and can set specific 5′ and 3′ adapter sequences used during library preparation. Analysis of the data in this manuscript was completed in under 1 h on a Macbook Pro laptop. For larger projects, Mod-seeker can simultaneously analyze data from multiple files using multiple CPU threads. This flexible analysis package should facilitate the use of Mod-seq.
While most significant DMS-dependent sites of modification were located at A and C residues, a number were observed at Gs and Us. Reverse transcriptase is known to add untemplated nucleotides after dissociation from RNA templates (Ingolia et al. 2011), which could contribute substantially to the appearance of significant G- and U-sites. Indeed, 20% and 17% of significant G- and U-sites are 1 or 2 nt 5′ of significant A- or C-sites, respectively. However, some G- and U-sites likely represent legitimate enrichment of DMS-modification for the following reasons. First, false-positive sites should be equally likely to occur at U or G residues, yet we see very few DMS-dependent termination sites at Us. Second, some of the rRNA nucleotides with increased modification in the rpl26Δ strain that are footprinted to the L26 binding site are Gs, and were previously observed when assayed with radiolabeled primer extension (Babiano et al. 2012). Finally, methylation of guanosine N7 (m7G) is known to occur more rapidly than that of As and Cs (Lawley and Brookes 1963).
Several factors could make Mod-seq more sensitive to m7G, as compared with traditional approaches using primer extension and gel electrophoresis. For example, m7G could cause a subtle increase in the probability of RT termination only detectable with deep sequencing. Another possibility is that this modification may increase the probability of RNA fragmentation or impede ligation of 5′ linkers by RNA ligase 1. DMS-dependent sites of m7G can be identified by depurination and strand cleavage by treatment with sodium borohydride and aniline (Wells et al. 2000). Like borohydride, the Zn catalyst used in RNA fragmentation acts as a reducing agent. Thus, it is possible that fragmentation leads to depurination of m7G residues, which in turn would increase the rate of premature termination by reverse transcriptase.
We envision many applications for Mod-seq. As this method is suitable for RNA of any length, selective removal of rRNA before library preparation would allow genome-wide probing of mRNA structure in vivo. For more targeted studies, RNA of interest can be biochemically isolated prior to sequencing library preparation. This strategy would be applicable to analyses of structural changes during assembly of large RNP complexes such as the ribosome and the spliceosome. Precise footprinting of protein-binding sites, as demonstrated here with ribosomal protein L26, is also possible. Combinations of the above would allow transcriptome-wide mapping of RBP binding sites. Furthermore, applying Mod-seq with SHAPE reagents, either in vitro or in vivo (Spitale et al. 2012), would provide a powerful sequence-independent approach to interrogating RNA structure. In the absence of DMS, we observed RT termination sites at nucleotides with known endogenous modifications (e.g., m1A, at position 645 in 25S rRNA), suggesting that Mod-seq could be used to identify naturally occurring modifications. Finally, Mod-seq could be used to investigate structures and protein interactions of lncRNAs, some of which are longer than rRNA.
Transcriptome-wide applications would require very deep sequencing coverage. For example, traditional RNA-seq analysis usually requires 20 to 30 million reads, and a 1-kb transcript could be deemed “expressed” (RPKM = 1) with as few as 20 aligned reads (one read every 50 nt). By comparison, the nucleotide resolution required to identify sites of DMS modification necessitates much deeper coverage, with roughly one read per nucleotide. Indeed, while this manuscript was in revision, two studies were published using similar approaches to probe the secondary structure of the Arabidopsis thaliana and S. cerevisiae transcriptomes (Ding et al. 2013; Rouskin et al. 2013). Although the investigators of these studies did not test each modification site for statistical significance over background, they report structural information for thousands of transcripts using just a few lanes of Illumina Hiseq data. Thus transcriptome-wide studies using these approaches should be accessible to labs of any size.
MATERIALS AND METHODS
Growth of yeast strains, DMS probing, and RNA extraction
All strains were grown at 30°C in liquid YEPD media (2% dextrose, 2% peptone, and 1% yeast extract). DMS probing was performed as previously described with the following modifications (Wells et al. 2000): 10-mL cultures of WT (BY4741; MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Euroscarf) and rpl26Δ (JWY9670; MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 RPL26A::kanMX4 RPL26B::kanMK4) (Babiano et al. 2012) strains were grown to mid-logarithmic phase, at 3–5 × 107 cells/mL. A total of 400 μL of freshly diluted DMS (1:4 vol/vol in 95% ethanol) (Sigma-Aldrich) was added to the cells to a final concentration of 100 mM. Cells were incubated with shaking at 30°C for 2 min. The DMS reactions were quenched by placing the cultures on ice and adding 5 mL of ice-cold 0.6 M β-mercaptoethanol and water-saturated isoamyl alcohol. Cells were pelleted by centrifugation at 5000g for 5 min. Cell pellets were resuspended and washed in 5 mL of ice-cold 0.6 M β-mercaptoethanol and centrifuged again. Total RNA was immediately phenol extracted from the cell pellets, ethanol precipitated, and resuspended in RNase-free water as previously described (Horsey et al. 2004). “No DMS” controls were treated the same as the experimental samples except for the addition of DMS. “Stop” controls were treated the same as the experimental samples except that DMS was not added until after the addition of 5 mL of 0.6 M β-mercaptoethanol and water-saturated isoamyl alcohol. “Stop” controls serve as a measure of how effectively the DMS reactions were quenched.
Primer extension and PAGE
DMS modifications were assayed by primer extension with radiolabeled oligonucleotides as described in Liebeg and Waldsich (2009) with the following changes. For each reaction, 2.5 μL (500 ng) of whole-cell RNA was incubated with 1 μL (0.2 μM) of 32P-labeled primer (5′-AAATGACGCTCAAACAGGCATGC-3′), and 1 μL of 4.5X hybridization buffer (225 mM Hepes at pH 7.0; 450 mM KCl) at 95°C for 5 min. Reactions were then cooled to the annealing temperature of 54°C for 20 min. Fifteen microliters of prewarmed (54°C) extension mix containing 4 μL of 5X hybridization buffer (Roche Applied Science) (250 mM Tris-HCl; 150 mM KCl; 40 mM MgCl2 at pH of ∼8.5), 2.0 μL 2.5 mM dNTP mixture, 1.0 μL 0.1 mM DTT, 0.5 μL (20 units) RNasin (Promega), 0.5 μL (10 units) Transcriptor Reverse Trascriptase (Roche Applied Science), and 7.5 μL of nuclease free water was added to each reaction. For sequencing reactions, 2 μL of the appropriate ddNTP (10 mM) (Roche Applied Science) was also added. Reactions were incubated at 54°C for 1 h. RNA was degraded by the addition of 3 μL 1 M NaOH to each reaction and incubation at 55°C for 1 h. Reactions were neutralized by the addition of 3 μL 1.0 M HCl. cDNAs were precipitated with 1 μL of glycogen (10 mg/mL), 1 μL 0.5 M EDTA (pH 8.0), 2.8 μL 3.0 M NaOAc (pH 5.0), and 84 μL of ethanol. Following ethanol precipitation, dried cDNA pellets were resuspended in 6 μL of 1X loading dye (45% formamide; 0.01 M EDTA at pH 8.0), resolved on 6% polyacrylamide/7 M urea gels, and visualized by autoradiography.
Library preparation
Total RNA (3 μg) was randomly fragmented using RNA Fragmentation Reagents (Ambion) for 3 min at 70°C in a 20-μL reaction. The fragmented RNA was purified using a RNA Clean & Concentrator-5 column (Zymo Research) using the manufacturer's >17-nt-long protocol, and resuspended in 15.5 μL of nuclease free water. The RNA fragments were phosphatase-treated to remove 2′-3′ cyclic phosphate groups by incubating with 15 units of T4 Polynucleotide Kinase (PNK) (New England Biolabs) with 20 units of SUPERase-In (Life Technologies) for 1 h at 37°C in a 20-μL reaction. RNA fragments were then 5′ phosphorylated by adding 12.5 μL of nuclease free water, 2 μL 10X T4 PNK buffer, 15 units T4 PNK, and ATP to a concentration of 1 mM, and incubating at 37°C for 1 h. The RNA fragments were purified using a RNA Clean & Concentrator-5 column and resuspended in 11.75 μL of nuclease free water.
The end-prepared RNA fragments were ligated to 500 ng of universal miRNA cloning linker (5′-rAppCTGTAGGCACCATCAAT–NH2-3′) (New England Biolabs). RNA fragments and linker were heated at 80°C for 2 min and then placed on ice to disrupt secondary structures, and incubated overnight at 16°C in a 20-μL reaction containing 12.5% PEG 8000, 10% DMSO, 20 units SUPERase-In, 200 units T4 RNA Ligase 2 truncated (New England Biolabs), and 1X T4 RNA ligase reaction buffer (NEB). The RNA was purified using a RNA Clean & Concentrator-5 column (Zymo Research) using the manufacturer's >200 nt-long protocol, and resuspended in 10 μL of nuclease free water. The RNA was then ligated at its 5′ end to the 5′ linker (5′ End Acceptor Oligonucleotide 17.93R, 5′-ATCGTaggcaccugaaa-3′) (Lau et al. 2001) where uppercase is DNA and lowercase is RNA. To reduce secondary structure, the 5′ linker was incubated at 70°C for 2 min and placed immediately on ice. The RNA and 5′ linker were incubated at 22°C overnight in a 30-μL reaction containing 10% DMSO, 1X Quick Ligase Buffer (NEB), 20 units SUPERase-In, and 15 units T4 RNA ligase 1 (NEB). The RNA was purified using a RNA Clean & Concentrator-5 column (Zymo Research, >200-nt protocol) and resuspended in 26 μL of nuclease free water.
RNA fragments with ligated 5′ linkers were selectively enriched using a biotinylated oligonucleotide (5′-TTTCAGGTGCCTACGAT-3′—Biotin-TEG) complementary to the 5′ linker. The RNA and 0.67 μM oligonucleotide were denatured in a 30-μL reaction at 90°C for 90 sec in 1.3X SSC. The RNA and oligonucleotide were then annealed by cooling from 37°C to room temperature at 3°C per minute in a thermocycler, and bound to streptavidin beads (Dynabeads MyOne C1, Life Technologies) according to the manufacturer's instructions. To elute bound RNA from the beads, the sample was resuspended in 12 μL of TE, heated to 80°C for 2 min, immediately placed on a magnet, and all of the supernatant was removed.
The RNA was reverse transcribed using Superscript II Reverse Transcriptase at 50°C according to the manufacturer's instructions. The reverse transcription primer [5′-(Phos)AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC-(SpC18)-CACTCA-(SpC18)-TTCAGACGTGTGCTCTTCCGATCTATTGATGGTGCCTACAG-3′] as described in Brar et al. (2012) is complementary to the Universal miRNA cloning linker (NEB) and also contains sequence complementary to Illumina forward and reverse PCR primers. After reverse transcription, the RNA template was destroyed by adding NaOH to a concentration of 100 mM and incubating the reaction at 98°C for 20 min. The cDNA was ethanol precipitated and resuspended in 10 μL TE. Fragments in the size range of from 150 to 330 bp were extracted from an 8% denaturing urea gel and resuspended in 15 μL of TE.
The size-selected cDNA fragments were circularized by incubating with 100 units CircLigase ssDNA Ligase (Epicentre) at 60°C for 1.5 h according to the manufacturer's instructions. Under these conditions, circularization proceeds to near-completion (Supplemental Fig. S2). Because the reverse transcription terminates at DMS-modified bases, cDNA that contains the 5′ linker would not have contained a DMS-modified base. In order to enrich for cDNA that contain information on DMS stops, the cDNA was first annealed a biotinylated oligonucleotide complementary to the 5′ linker cDNA sequence (Biotin–TEG–5′-ATCGTAGGCACCTGAAA-3′) and then sequences containing the 5′ linker sequence were removed using streptavidin beads (Dynabeads MyOne C1, Life Technologies) according to the manufacturer's instructions. The remaining cDNA was ethanol precipitated and resuspended in 20 μL TE.
The cDNA was amplified by PCR using forward and reverse primers containing sequences specific to the Illumina sequencing platform. The forward primer (5′-AATGATACGGCGACCACCGAGATCTACAC-3′) was universal, while reverse primers [5′-CAAGCAGAAGACGGCATACGAGAT-(6 nt barcode)-TGTACTGGAGTTCAGACGTGTGCTCTTCCG-3′] contained a 6-nt barcode specific to each sample. In a 20-μL reaction, 1 μL of cDNA template, 0.5 μM each forward and reverse primer, 0.5 mM dNTPs, 0.4 units Phusion DNA polymerase (Finnzymes), and 1X Phusion HF buffer, were cycled for 12 rounds using an annealing temperature of 64°C. The libraries were purified and eluted in 10 μL TE using a MinElute PCR purification column (Qiagen) according to the manufacturer's instructions. The libraries were assessed and quantified using a Agilent 2200 TapeStation System. Libraries were pooled and sequenced on and Illumina HiSeq 2000 at the University of Southern California Epigenome Center.
Data analysis
Sequence data were analyzed using the Mod-seeker pipeline (Mod-seeker-map.py). Reads were first processed to remove 5′ and 3′ adapter sequences using cutadapt (Martin 2011). Reads matching the 5′ adapter (adapter reads) result from reverse-transcription termination at random fragment ends and were removed from further analyses. Reads lacking the 5′ adapter (mod-reads) were aligned to the current yeast genome sequence (SacCer3) using bowtie (version 0.12.8) (Langmead et al. 2009). Reverse transcriptase can sometimes add untemplated nucleotides to cDNA 3′ ends. Mod-read alignments often had mismatches at their 5′ ends corresponding to these untemplated nucleotides. Mod-seeker corrected these mismatches by trimming them from the alignment files. Aligned mod-reads were mapped to annotated genes using the intersectBed program from bedtools. Gene-mapped reads from treatment and control samples were then reported in files containing the numbers of read 5′ ends mapping to each nucleotide of each gene.
The resulting data were further analyzed to identify sites significantly enriched in treatment sample reverse-transcriptase stops using a custom python program, Mod-seeker-stats.py. Cochran-Mantel-Haenszel tests were used to estimate the odds ratios of enrichment. Resulting P-values were corrected using the Benjamini-Hochberg method (Benjamini and Hochberg 1995). To avoid complications from small differences in fragmentation efficiency, the last 45 nucleotides of each gene were not analyzed for statistical significance. Mod-seeker-stats.py also returns odds-ratio estimates of the fold enrichment for every significant site, given by
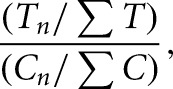 |
where Tn and Cn represent the number of RT stops at a particular nucleotide, and ∑T and ∑C represent the sum of all stops in treatment (T) and control (C) samples, respectively. Fold-enrichment values were mapped to rRNA secondary structure models (Cannone et al. 2002) by modification of previously described programs (Leshin et al. 2011).
DATA DEPOSITION
High-throughput sequencing data have been submitted to NCBI Short Read Archive under accession number SRP029192. Genome-browser track files (bedGraph) are available upon request. The mod-seeker pipeline is included as supplemental material, and is also available at the McManus lab website (https://www.bio.cmu.edu/labs/mcmanus/).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to members of the McManus and Woolford labs for helpful comments and discussions. We also thank the De la Cruz lab at the Universidad de Sevilla for providing the rpl26Δ strain. This work was supported by a grant from the National Institutes of Health (RO1 GM28301-32) and funds from the David Scaife Family Charitable Foundation to J.L.W., and start-up funding provided by the Department of Biological Sciences at Carnegie Mellon University and a Charles E. Kaufman Foundation New Investigator award to C.J.M.
REFERENCES
- Babiano R, Gamalinda M, Woolford JL Jr., de la Cruz J 2012. Saccharomyces cerevisiae ribosomal protein L26 is not essential for ribosome assembly and function. Mol Cell Biol 32: 3228–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B (Methodological) 57: 289–300 [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M 2011. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334: 1524–1529 [DOI] [PubMed] [Google Scholar]
- Brar GA, Rouskin S, McGeachy AM, Ingolia NT, Weissman JS 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7: 1534–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel C, Romby P 2000. Probing RNA structure and RNA-ligand complexes with chemical probes. Methods Enzymol 318: 3–21 [DOI] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, et al. 2002. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JA, Westhof E 2009. The dynamic landscapes of RNA architecture. Cell 136: 604–609 [DOI] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, Weeks KM 2009. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci 106: 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM 2013. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505: 696–700 [DOI] [PubMed] [Google Scholar]
- Horsey EW, Jakovljevic J, Miles TD, Harnpicharnchai P, Woolford JL Jr. 2004. Role of the yeast Rrp1 protein in the dynamics of pre-ribosome maturation. RNA 10: 813–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E 2010. Genome-wide measurement of RNA secondary structure in yeast. Nature 467: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862 [DOI] [PubMed] [Google Scholar]
- Lawley PD, Brookes P 1963. Further studies on the alkylation of nucleic acids and their constituent nucleotides. Biochem J 89: 127–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leland PA, Raines RT 2001. Cancer chemotherapy—ribonucleases to the rescue. Chem Biol 8: 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshin JA, Heselpoth R, Belew AT, Dinman J 2011. High throughput structural analysis of yeast ribosomes using hSHAPE. RNA Biol 8: 478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeg A, Waldsich C 2009. Probing RNA structure within living cells. Methods Enzymol 468: 219–238 [DOI] [PubMed] [Google Scholar]
- Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, Schroth GP, Pachter L, Doudna JA, Arkin AP 2011. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Proc Natl Acad Sci 108: 11063–11068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10–12 [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM 2005. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 127: 4223–4231 [DOI] [PubMed] [Google Scholar]
- Moazed D, Noller HF 1986. Transfer RNA shields specific nucleotides in 16S ribosomal RNA from attack by chemical probes. Cell 47: 985–994 [DOI] [PubMed] [Google Scholar]
- Peattie DA, Gilbert W 1980. Chemical probes for higher-order structure in RNA. Proc Natl Acad Sci 77: 4679–4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS 2013. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505: 701–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY 2012. RNA SHAPE analysis in living cells. Nat Chem Biol 9: 18–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Moazed D, Noller HF 1988. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Methods Enzymol 164: 481–489 [DOI] [PubMed] [Google Scholar]
- Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, Mathews DH, Lowe TM, Salama SR, Haussler D 2010. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods 7: 995–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460: 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks KM 2010. Advances in RNA structure analysis by chemical probing. Curr Opin Struct Biol 20: 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SE, Hughes JMX, Igel AH, Ares M 2000. Use of dimethylsulfate to probe RNA structure in vivo. Methods Enzymol 318: 479–493 [DOI] [PubMed] [Google Scholar]
- Westhof E, Romby P 2010. The RNA structurome: high-throughput probing. Nat Methods 7: 965–967 [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]