

Abstract
Increased osteoclastogenesis and angiogenesis occur in physiologic and pathologic conditions. However, it is unclear if or how these processes are linked. To test the hypothesis that osteoclasts stimulate angiogenesis, we modulated osteoclast formation in fetal mouse metatarsal explants or in adult mice and determined the effect on angiogenesis. Suppression of osteoclast formation with osteoprotegerin dose-dependently inhibited angiogenesis and osteoclastogenesis in metatarsal explants. Conversely, treatment with parathyroid hormone related protein (PTHrP) increased explant angiogenesis, which was completely blocked by osteoprotegerin. Further, treatment of mice with receptor activator of nuclear factor-κB ligand (RANKL) or PTHrP in vivo increased calvarial vessel density and osteoclast number. We next determined whether matrix metalloproteinase-9 (MMP-9), an angiogenic factor predominantly produced by osteoclasts in bone, was important for osteoclast-stimulated angiogenesis. The pro-angiogenic effects of PTHrP or RANKL were absent in metatarsal explants or calvaria in vivo, respectively, from Mmp9−/− mice, demonstrating the importance of MMP-9 for osteoclast-stimulated angiogenesis. Lack of MMP-9 decreased osteoclast numbers and abrogated angiogenesis in response to PTHrP or RANKL in explants and in vivo but did not decrease osteoclast differentiation in vitro. Thus, MMP-9 modulates osteoclast-stimulated angiogenesis primarily by affecting osteoclasts, most probably by previously reported migratory effects on osteoclasts. These results clearly demonstrate that osteoclasts stimulate angiogenesis in vivo through MMP-9.
Introduction
Both osteoclastogenesis and angiogenesis are enhanced in pathologic conditions, such as multiple myeloma, bone metastases, and rheumatoid arthritis, which are associated with increased local expression of inflammatory cytokines.1,2 Osteoclasts and blood vessels are closely associated with a vessel present at every bone-remodeling compartment.3 However, few studies examined if and how osteoclasts may play a role in angiogenesis. Osteoclast-conditioned media have been reported to be angiogenic in vitro, and this was attributed to secretion of osteopontin by osteoclasts.4,5 However, other investigators reported that osteoclasts were not required for angiogenesis. Further, osteopetrotic mice (op/op or Fos−/−) or clodronate treatment of wild-type (WT) mice did not inhibit developmental vessel invasion into mouse caudal vertebrae.6 Similarly, op/op mice have normal levels of vessel invasion in their epiphyses.7
Early studies in matrix metalloproteinase-9 (Mmp9)−/− mice suggested that osteoclasts stimulate angiogenesis by secretion of MMP-9, but the hypothesis was not pursued. Mmp9−/− mice display delayed endochondral ossification and vessel invasion into the primary ossification center, accompanied by lengthened growth plates. However, this phenotype resolves, and adults have normal-appearing bones that are slightly shorter than WT animals.8 Likewise, lack of MMP-9 inhibits vessel invasion and healing of long bone fractures.9 In adult bone, MMP-9 is predominantly expressed by osteoclasts and committed osteoclast precursors but can be expressed at very low levels by other cell types, such as osteoblasts and hypertrophic chondrocytes during development or neutrophils and macrophages during fracture repair.8–11 Studies reported that MMP-9 stimulates angiogenesis through activation or release of growth factors from matrix. MMP-9 was implicated as the angiogenic switch in a mouse pancreatic cancer model in which MMP-9 released matrix (heparan) associated vascular endothelial growth factor (VEGF) and increased VEGF signaling.12 MMP-9 and its release of matrix-bound VEGF are important for osteoclast invasion of the long bone growth plate and VEGF-induced osteoclast migration in vitro but is not important for solubilization of bone matrix.13,14
Therefore, we determined the effect of osteoclasts and their secretion of MMP-9 on angiogenesis. Osteoclasts were stimulated in vivo or in metatarsal explants with receptor activator of nuclear factor-κB ligand (RANKL), an important and specific osteoclast differentiation factor, or parathyroid hormone related protein (PTHrP), which stimulates osteoclastogenesis primarily through induction of RANKL in osteoblasts.15,16 Osteoprotegerin (OPG), the decoy receptor for RANKL, was used to inhibit osteoclast differentiation and activity in metatarsal explants.15,17 These results show that osteoclasts contribute to angiogenesis in bone explants and in vivo through a mechanism requiring MMP-9.
Methods
Materials and mice
Monoclonal rat anti–mouse CD31 was purchased from BD Biosciences PharMingen (#550274). Polyclonal rabbit anti–mouse MMP-9 was from Abcam (#ab38898). Vector Laboratories supplied biotinylated rabbit anti–rat mouse adsorbed, or biotinylated goat anti–rabbit secondary antibodies and streptavidin-horseradish peroxidase (HRP). The RatLaps type I collagen C-terminal telopeptide (CTX) assay was from Immunodiagnostic Systems, and the CryoJane sectioning aid and adhesive slides from Instrumedics. Human in vitro angiogenesis assays were purchased from TCS CellWorks. Recombinant human OPG-Fc, RANKL macrophage colony-stimulating factor (M-CSF), and osteopontin were from R&D Systems. Recombinant mouse RANKL-GST was generously provided by Drs S. Teitelbaum and F. P. Ross (Washington University, St Louis, MO). hPTHrP(1-34) was purchased from American Peptide. RNEasy RNA isolation kit was from QIAGEN. Human leukocyte acid phosphatase kit was from Sigma-Aldrich. RT2 cDNA synthesis kit and Human Angiogenesis Q-PCR array were from SA Biosciences. The bicinchoninic acid protein assay was from Pierce Chemical. ImmunoHistoMount aqueous mounting media was from Santa Cruz Biotechnology. Mmp9−/− mice were generated as described and backcrossed to C57BL/6 mice for 10 generations.8 Timed pregnant C57BL/6 mice were from The Jackson Laboratory. C57BL/6 mice were from Harlan.
Human osteoclast culture and angiogenic factor array analysis
Bone marrow aspirates were collected in heparinized syringes from normal donors after obtaining informed consent. These studies were approved by the University of Pittsburgh Institutional Review Board. Bone marrow mononuclear cells were separated by density sedimentation on Ficoll-hypaque. A total of 10 million cells were incubated overnight in 10% fetal calf serum α-minimal essential medium (α-MEM) in 10-cm culture dishes. Nonadherent cells were removed by gentle washing. For osteoclast or macrophage culture, nonadherent cells were diluted to 2 × 106/mL in α-MEM with 20% horse serum, 10 ng/mL rh M-CSF with or without 50 ng/mL rh RANKL, and seeded at 0.4 mL per well of 48-well plates. Cultures were maintained for 17 days as previously described.18 Expression of angiogenic factors in osteoclast (RANKL + M-CSF treated) versus control cultures cultured with M-CSF only were determined using the SA Biosciences Q-PCR Human Angiogenesis array.
Mouse marrow cultures
Nonadherent bone marrow cells from age-matched male WT or Mmp9−/− C57BL/6 mice were cultured in 96-well plates at 106/mL, 0.2 mL per well in α-MEM with 10% fetal calf serum, 10 ng/mL M-CSF, and 50 ng/mL RANKL for 6 days as previously described.19 Cultures were fixed and stained for tartrate-resistant acid phosphatase (TRAP) and quantified by counting TRAP+ multinucleated cells in 5 replicate wells.
Fetal mouse metatarsal angiogenesis assay
The assay was conducted as described with minor modifications.20 Briefly, embryos were harvested from CB6 F1 × CD-1 (outbred), C57BL/6 WT, or Mmp9−/− pregnant female mice at 17.5 days postcoitum. The middle 3 metatarsals were dissected from each hind leg and cultured in 24-well plates containing 10% fetal calf serum α-MEM with the indicated treatments for 15 days. At least 10 bones were used per group. Medium volume was maintained at 150 μL for the first 3 days and 250 μL subsequently. Medium was replaced every 3 days, and spent media was stored at −80°C for measurement of CTX (RatLaps). Media from freeze-thawed bones was used as a blank for CTX measurements. Explants were then stained for CD31 as follows: Bones were fixed for 15 minutes with zinc macrodex formalin, washed twice with phosphate-buffered saline (PBS), and blocked overnight at 4°C in PBS containing 2% rabbit serum, 0.1% Triton X-100, 0.05% Tween-20, 1% bovine serum albumin, 0.1% gelatin, and 0.05% sodium azide. All PBS buffers were pH 7.2. The primary antibody was applied at a dilution of 1:50 in PBS plus 1% bovine serum albumin and 0.1% gelatin. Secondary antibody and streptavidin-HRP (Vector Laboratories) were applied in PBS at 1:100 or 1:250 dilution, respectively, and explants were stained with 3-amino-9-ethylcarbazole (AEC) HRP substrate. Images were acquired with an Olympus Multimode dissecting microscope and were quantified either with the Metamorph angiogenesis tube formation application for tube area, length, and branches, or with ImageJ for total CD31+ area and corrected for the area of bones stained with control IgG in place of primary antibody.
For comparing the angiogenic and resorptive response of Mmp9−/− versus WT metatarsals treated with PTHrP, 7 litters of C57BL/6 and 6 litters of C57BL/6 Mmp9−/− were treated with 100nM PTHrP or solvent separately. The mean fold change from control in CD31+ or area or CTX concentration with PTHrP treatment was calculated for each litter. The effect of PTHrP on each genotype was compared by calculating mean fold change from control per litter. This analysis was necessitated by the large variability in the basal level of angiogenesis and resorption between litters.
TRAP activity was extracted from metatarsal explants that were fixed and stained for CD31 and stored dry. Bones were rehydrated and homogenized with a ground glass homogenizer in 120 μL of NP-40 lysis buffer (1% NP-40, 150mM NaCl, 50mM Tris, pH 8), then rotated at 4°C overnight, and cleared by centrifugation. To assay TRAP activity, 35 μL of homogenate was added to 200 μL of TRAP substrate (50mM sodium acetate, pH 5, 25mM sodium tartrate, 0.4mM MnCl2, 0.4% N,N,-dimethylformamide, 0.2 mg/mL fast red violet, 0.5 mg/mL naphthol AS-MX phosphate), then incubated 3 hours at 37°C, and the absorbance read at 540 nm. Results were corrected for total protein, which was assayed by adding 25 μL of homogenate to 250 μL of bicinchoninic acid assay reagent, incubating 2 hours at 37°C and reading the absorbance at 562 nm.
Histology and immunohistochemistry
Bones were fixed overnight in 2% paraformaldehyde, decalcified for 4 days in 10% ethylenediaminetetraacetic acid, pH 7.4, soaked in 3 changes of 30% sucrose in PBS overnight, and snap-frozen in Optimum Cutting Temperature embedding medium by immersion in liquid nitrogen-cooled isopentane; 7-μm sections were cut on a cryostat equipped with a CryoJane tape-transfer system. Slides were stored frozen until use and then thawed and washed 3 times in PBS. For immunohistochemistry, slides were peroxidase quenched in 0.3% H2O2 in methanol, washed 3 times for 5 minutes in PBST (0.05% Tween-20), and then blocked for 30 minutes in 5% serum from the same species from which the secondary antibody was derived. Primary antibodies were applied overnight at 4°C in PBS; anti-CD31 (1:100), anti–MMP-9 (1:4000), followed by washes in PBST, and then biotinylated secondary antibodies (anti–rat 1:100, anti–rabbit 1:2500 dilutions), streptavidin-HRP (1:250 dilution), and diaminobenzidine (DAB) peroxidase substrate were added. TRAP histochemistry was performed as described previously.21 Slides were mounted in aqueous media.
In vivo studies
Six (2 male and 4 female) WT or Mmp9 −/− C57BL/6 7-week-old mice per group were injected with m-RANKL-GST (1 mg/kg in 50-65 μL of PBS) subcutaneously over the calvaria daily for 5 days under light isoflurane anesthesia. Similarly, for studies involving PTHrP, six 5-week-old male C57BL/6 mice were treated with 2 μg of hPTHrP(1-34) in 100 μL of 1% bovine serum albumin-PBS, pH 5.2, systemically by subcutaneous injection 4 times a day for 5 days.22,23 Control mice were injected with vehicle. After 5 days, blood was collected by retro-orbital puncture and mice were killed. Calvaria were sectioned in a coronal orientation anterior to the junction of the sagittal and coronal sutures and stained for CD31, TRAP, or MMP-9. All quantitative histologic analyses were performed by a blinded observer. For PTHrP-treated mice, CD31+ vessels were quantified in the inner table adjacent to the midline of 10× objective images by selecting the DAB signal with a hue saturation intensity filter (Fovea Pro software; Reindeer Graphics) selected to limit to true signal from 45 to 90 degrees area running within Adobe Photoshop 7. For PTHrP-treated mice between bone tables (parasagittal spaces) or RANKL-treated mice in the outer table, CD31+ vessels were quantified by counting vessel numbers and using a grid map projected with ImagePro software to determine total area covered by vessels (including lumens). Average vessel size was calculated from total area and vessel density. Osteoclasts were quantified in PTHrP-treated bones by calculating TRAP+ area as was described for DAB earlier in this section, using Fovea Pro and selecting between 90 and 135 degrees. Osteoclasts were quantified in RANKL-treated calvaria by calculating the TRAP+ (resorptive) surface of the subperiosteal surface of the calvarial outer table by grid map counting, as well as TRAP+ area. All animal protocols were approved by the Institutional Animal Care and Use Committee of Veterans Administration Pittsburgh Healthcare System, University of Pittsburgh, or Virginia Commonwealth University.
Statistics
The unpaired Student t test or 1-way analysis of variance with the least significant difference procedure was used for analyzing 2 or multiple groups, respectively. The ratio t test (paired t test on logarithms of vehicle and treated samples) was used to analyze fold change from control data. To analyze correlation, the Pearson correlation coefficient was calculated by linear regression, and the 1-sample F test for a correlation coefficient was used to test for significance. Two-tailed analyses were performed with SPSS software. Significance was set at α = 0.05.
Results
Osteoclasts stimulate angiogenesis in fetal mouse metatarsal explants
Angiogenesis in bone is regulated by contributions from many cell types, including osteoblasts, stromal cells, and marrow elements.24 To determine the effect of osteoclast activity on angiogenesis in a more physiologic model for bone than purified cell cultures, we determined the effects of modulating osteoclast number and activity on angiogenesis in the well-characterized fetal mouse metatarsal assay. In this assay, metatarsals from embryonic day (E) 17.5 mice are cultured in vitro. At this developmental stage, the primary ossification center is formed but not yet invaded by osteoclast precursors, which are in the periosteum. Endothelial cells form tubes in a mixed cellular outgrowth during culture.20 This assay has been used to analyze the effects of osteoblast-specific gene knockouts on angiogenesis.25 As shown in Figure 1A and B, inhibition of osteoclast formation with OPG reduced angiogenesis in a dose-dependent manner, as measured by labeling endothelial cells with anti-CD31 and quantitative image analysis of angiogenic tube formation. To verify that OPG inhibited osteoclast formation and activity, we measured type I collagen CTX levels in the conditioned media or activity of TRAP extracted from the bone explants treated with OPG (Figure 1B). There was a parallel decrease in angiogenesis, CTX concentration, and TRAP activity. Further, metatarsal explant angiogenesis was significantly correlated with TRAP activity extracted from the explants as demonstrated by regression analysis of explants from all doses of the OPG dose-response curve (Figure 1C). To verify that OPG was not toxic to endothelial cells, we treated the TCS CellWorks HUVEC/fibroblast coculture angiogenesis assay, which does not contain osteoclasts, with equivalent doses of OPG and observed a minimal increase in angiogenesis rather than any inhibition (data not shown).
Figure 1.

Osteoclasts are important for angiogenesis in bone explants. (A) Osteoclast inhibition decreases angiogenesis in metatarsal explants. Metatarsal explants stained for endothelial cells (red, CD31); 17.5 days postcoitum outbred fetal mouse metatarsals were cultured with indicated treatments for 15 days before fixation.  indicates area of osteoclast resorption that is prominent in the control bones and decreases with increasing OPG. (B) Quantification of angiogenic outgrowth and osteoclast number and activity in metatarsal explants. Number of branches and other angiogenesis tube formation parameters quantified at the end of the assay period (15 days). CTX (RatLaps) assayed from metatarsal explant-conditioned media collected from days 7 to 9 of culture. TRAP activity extracted by homogenization of bones after 15 days of culture and assayed by color development using a TRAP substrate. Data are mean ± SEM. *P < .05. Images acquired as whole mounts in water with an Olympus IX71 microscope with a UPlanFLN objective, numeric aperture (NA) of 0.13, and a Spot RTKE camera with Spot Advanced software (original magnification ×4). (C) Correlation of osteoclast formation and angiogenesis. The TRAP activity extracted from bone explants and angiogenesis (branch points) was correlated for all samples treated with various concentrations of OPG-Fc and analyzed by linear regression. r indicates Pearson correlation coefficient; P, significance from 1-sample F test for linear regression. Results are from 1 representative experiment of at least 2 performed.
indicates area of osteoclast resorption that is prominent in the control bones and decreases with increasing OPG. (B) Quantification of angiogenic outgrowth and osteoclast number and activity in metatarsal explants. Number of branches and other angiogenesis tube formation parameters quantified at the end of the assay period (15 days). CTX (RatLaps) assayed from metatarsal explant-conditioned media collected from days 7 to 9 of culture. TRAP activity extracted by homogenization of bones after 15 days of culture and assayed by color development using a TRAP substrate. Data are mean ± SEM. *P < .05. Images acquired as whole mounts in water with an Olympus IX71 microscope with a UPlanFLN objective, numeric aperture (NA) of 0.13, and a Spot RTKE camera with Spot Advanced software (original magnification ×4). (C) Correlation of osteoclast formation and angiogenesis. The TRAP activity extracted from bone explants and angiogenesis (branch points) was correlated for all samples treated with various concentrations of OPG-Fc and analyzed by linear regression. r indicates Pearson correlation coefficient; P, significance from 1-sample F test for linear regression. Results are from 1 representative experiment of at least 2 performed.
We next investigated whether angiogenesis was increased by osteoclast stimulation. As shown in Figure 2A, stimulation of osteoclast formation with PTHrP, which increases osteoclastogenesis primarily through increased RANKL expression on osteoblasts, increased the area of CD31+ endothelial cells in metatarsal explant cultures. Because PTHrP can have direct effects on osteoblast differentiation or survival, we also treated the explants with OPG to determine whether the angiogenic effect of PTHrP required osteoclasts. PTHrP failed to stimulate angiogenesis in the presence of OPG. Osteoclast stimulation and inhibition did not simply have opposite effects on explant angiogenesis but also had differing effects on the morphology of the endothelial cell outgrowth. As shown in Figure 2B, PTHrP increased CD31+ area 1.5-fold because of increased density of endothelial cells adjacent to the bone. However, parameters of endothelial tube formation, such as number of branch points, which were inhibited by OPG, were not increased by PTHrP treatment (Figure 2B second panel). The reasons for these contrasting effects on endothelial morphology are under investigation but are consistent with a mechanism of increased proteinase-mediated release of short forms of VEGF, resulting in disorganized vessels.26
Figure 2.

Osteoclast stimulation increases angiogenesis in bone explants. (A) PTHrP stimulates angiogenic outgrowth from metatarsal explants by a mechanism requiring osteoclasts. Whole-mount fetal metatarsal explants from outbred mice cultured for 15 days with 3 μg/mL rh OPG-Fc or 100nM PTHrP as indicated. Images acquired as whole mounts in water with an Olympus IX71 microscope with a Plan N 2x objective, NA of 0.06, and a Spot RTKE camera with Spot Advanced software (original magnification × 2). (B) Quantification of angiogenic response to PTHrP and OPG. Angiogenesis was quantified by measurements of total CD31+ area (i) or angiogenic tube formation (branches; ii). Data are mean ± SEM. *P < .05. Similar results were seen in 2 experiments. Results are from 1 representative experiment of 2 performed.
MMP-9 is important for osteoclast stimulation of angiogenesis in metatarsal explants
We then determined the relative expression levels for genes involved in angiogenesis in human bone marrow cultures treated with RANKL and M-CSF to induce osteoclast formation and compared them with cultures treated with M-CSF alone. MMP-9 was the most highly expressed proangiogenic factor at the mRNA level by human marrow cultures treated with RANKL and M-CSF and was expressed approximately 100-fold higher than all the other angiogenic factors examined (Table 1). MMP-9 expression was increased more than 7-fold compared with control bone marrow cultures treated only with M-CSF. Because MMP-9 can be pro-angiogenic, is secreted by osteoclasts, and its null allele delays blood vessel invasion of the growth plate, we next determined whether osteoclasts stimulated angiogenesis in part by secretion of MMP-9.
Table 1.
Angiogenic factors that were up-regulated in human bone marrow osteoclast cultures
| Angiogenic factor | RANKL + M-CSF culture expression relative to housekeeping genes* | Fold increase vs M-CSF–treated cultures† |
|---|---|---|
| MMP-9 | 47 | 7.1 |
| ANPEP (APN, CD13) | 0.48 | 4.1 |
| β3-integrin | 0.069 | 28.2 |
| MMP-2 | 0.069 | 7.1 |
| Neuropilin-2 | 0.060 | 5.7 |
| Sphingosine kinase 1 | 0.020 | 11 |
| CXCL5 (ENA-78) | 0.012 | 8.1 |
| Notch-4 | 0.0061 | 5.0 |
| COL18A1 (endostatin) | 0.0026 | 8.1 |
| Angiopoietin-2 | 0.00020 | 4.1 |
RANKL indicates receptor activator of nuclear factor-κB ligand; M-CSF, macrophage colony-stimulating factor; and MMP, matrix metalloproteinase.
Expression levels of genes involved in angiogenesis were determined with the SA Biosciences Human Angiogenesis Q-PCR array in bone marrow cultures treated with RANKL and M-CSF or M-CSF alone. Expression levels were normalized to the average Ct value of five housekeeping genes (B2M, HPRT1, RPL13A, GAPD, and ACTB) as recommended by the manufacturer. Expression levels in cultures treated with RANKL and M-CSF are reported in the middle column.
The relative, normalized expression levels of genes involved in angiogenesis were then compared between cultures treated with RANKL and M-CSF to cultures treated with M-CSF alone by dividing the expression levels in the RANKL + M-CSF culture by the expression levels in the M-CSF only culture. Results are reported in the right column as fold increase compared with cultures treated with M-CSF alone. Only genes whose expression was increased at least 4-fold are reported.
Metatarsals from 7 WT and 6 Mmp9−/− C57BL/6 litters of mouse embryos were treated with 100nM PTHrP(1-34) or vehicle, and the ability of PTHrP to stimulate angiogenesis was compared by measuring CD31+ area. Because of the large variability in the level of basal angiogenesis and resorption among litters, angiogenesis and osteoclast activity results were analyzed in terms of fold change from control for each litter. PTHrP increased angiogenesis in WT but not in Mmp9−/− metatarsal explants, and the angiogenic effects differed between the genotypes (Figure 3A-B). To verify that PTHrP stimulated osteoclasts in this assay, levels of the bone resorption marker, type I collagen CTX, were measured in the conditioned media collected from days 1 to 3 and 4 to 6 of culture from WT and Mmp9−/− explants. PTHrP significantly stimulated bone resorption in WT explants at both time points but did not significantly stimulate resorption at 3 or 6 days in MMP-9−/− explants (Figure 3C). Other investigators similarly reported that, in mice lacking Mmp9, osteoclast bone resorption was decreased at the growth plate because of a defect in osteoclast migration.13
Figure 3.

MMP-9 is important for stimulation of angiogenesis by osteoclasts in bone explants. (A) PTHrP-induced metatarsal angiogenesis is blunted in Mmp9−/− explants. Sample (original magnification ×2) images of WT or Mmp9−/− C57BL/6 metatarsal explants treated with vehicle or 100nM PTHrP as indicated and stained for CD31. Images acquired as in Figure 2. (B) Angiogenic response to PTHrP is significantly less in Mmp9−/− than in WT metatarsal explants. Metatarsals from 7 WT and 6 Mmp9−/− litters were treated with 100nM PTHrP or vehicle. The mean fold change from control in CD31+ area per litter for all litters ± SEM is reported. *P < .05, difference in treatment response between genotypes. PTHrP significantly stimulated angiogenesis in WT but not in Mmp9−/− metatarsals, as determined by the ratio t test comparing vehicle and PTHrP-treated means for each litter. (C) PTHrP increases bone resorption in WT but not in Mmp9−/− metatarsal explants. CTX assayed from conditioned media collected from days 1 to 3 or 4 to 6 of culture of all 7 WT and 6 knockout litters and reported as mean fold change from control. *P < .05, treatment response compared with vehicle. NS indicates not significant.
Increased osteoclast formation with PTHrP stimulates angiogenesis in vivo
We next determined whether osteoclasts increased angiogenesis in mice treated with PTHrP. Therefore, mice were treated with PTHrP every 6 hours subcutaneously for 5 days and then analyzed for osteoclasts and vessels histologically. This protocol dramatically induces osteoclast formation in calvaria, and the frequent dosing schedule minimizes possible osteoblastic (anabolic) effects of PTHrP.22 Osteoclasts and blood vessels, labeled for the endothelial cell marker CD31, were dramatically increased between the bone tables in calvaria after PTHrP treatment (Figure 4A). The dramatic increase in total CD31+ vessel area per tissue area was primarily the result of increased vessel number (Figure 4B). Average vessel size was not significantly increased (data not shown). Cells of the reticuloendothelial system, such as those found in sinusoids of the bone marrow or liver, do not express CD31.27 Thus, these data reflect changes in endothelial cells of capillaries and arterioles rather than the venous sinusoids. PTHrP also induced an anabolic response within the calvarial inner table and associated tissue, resembling the response seen in hyperparathyroidism, termed “osteitis fibrosa cystica.” PTHrP increased the thickness of the inner table 4-fold (Figure 4C). CD31+ vessels in the inner table increased in proportion with tissue area in PTHrP-treated bones. Thus, CD31+ vessels in the inner table were not increased when normalized for tissue area (data not shown). PTHrP treatment increased osteoclastogenesis, as measured by TRAP+ area within the whole section (Figure 4D). We also analyzed angiogenesis in long bones. The pattern of vessels in the metaphysis adjacent to the distal femoral growth plate was changed, but there were no significant differences in vessel area (data not shown).
Figure 4.
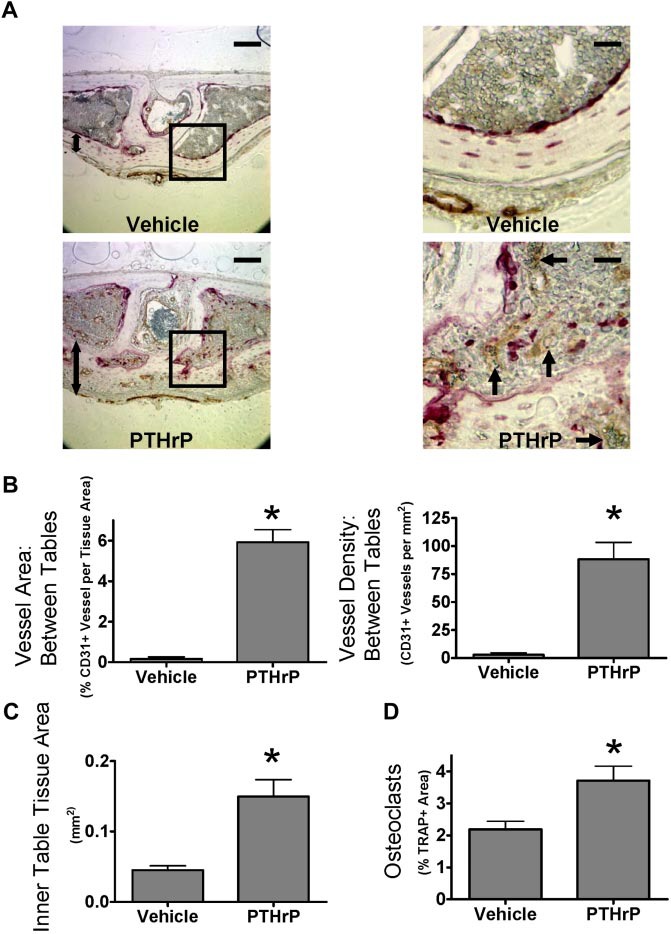
PTHrP increases bone angiogenesis in vivo. (A) Stimulation of osteoclast activity by PTHrP increases angiogenesis in mouse calvaria in vivo. Coronal sections of calvaria from mice treated with PTHrP(1-34) every 6 hours for 5 days, labeled for CD31 (brown) and TRAP (red), and lightly counterstained with hematoxylin.  indicates prominent CD31+ vessel; 2-headed arrow, inner table thickness. Outer surface is at the top of the image. Images of aqueous mounted slides acquired with a Nikon Eclipse TE300 microscope using a 20×/0.45 NA Plan Fluor objective and a Spot Pursuit camera with Spot Advanced 6.4 software. Scale bar represents 100 μm for full field, 25 μm for insets. Auto contrast and auto color adjustment were performed with Adobe Photoshop software. (B) PTHrP increases CD31+ vessel density between calvarial bone tables. Total vessel area and vessel density quantified by grid map counting by a blinded observer of the parasagittal areas of CD31-stained calvarial sections. (C) PTHrP increases thickness of inner table. Total tissue area of the inner table was quantified by image analysis by a blinded observer. (D) PTHrP induces osteoclast formation in calvaria. Total TRAP+ area as a percentage of tissue area was quantified by image analysis by a blinded observer. Data are mean ± SEM. *P < .05 compared with vehicle. Results are from 1 representative experiment of 2 performed.
indicates prominent CD31+ vessel; 2-headed arrow, inner table thickness. Outer surface is at the top of the image. Images of aqueous mounted slides acquired with a Nikon Eclipse TE300 microscope using a 20×/0.45 NA Plan Fluor objective and a Spot Pursuit camera with Spot Advanced 6.4 software. Scale bar represents 100 μm for full field, 25 μm for insets. Auto contrast and auto color adjustment were performed with Adobe Photoshop software. (B) PTHrP increases CD31+ vessel density between calvarial bone tables. Total vessel area and vessel density quantified by grid map counting by a blinded observer of the parasagittal areas of CD31-stained calvarial sections. (C) PTHrP increases thickness of inner table. Total tissue area of the inner table was quantified by image analysis by a blinded observer. (D) PTHrP induces osteoclast formation in calvaria. Total TRAP+ area as a percentage of tissue area was quantified by image analysis by a blinded observer. Data are mean ± SEM. *P < .05 compared with vehicle. Results are from 1 representative experiment of 2 performed.
Increased osteoclast formation with RANKL stimulates angiogenesis in vivo by a mechanism requiring MMP-9
Because PTHrP also stimulates osteoblasts, which can produce angiogenic factors,25 we next studied the effect of RANKL on angiogenesis because RANKL does not affect osteoblasts.16 We stimulated osteoclast formation in both WT and Mmp9−/− C57BL/6 mice by supra-calvarial injection of RANKL to determine whether osteoclasts increased angiogenesis by a mechanism requiring MMP-9. RANKL treatment induced dramatic changes in bone resorption and osteoclast formation in the outer table (Figure 5A). The resorbed area was replaced by noncalcified tissue resembling active periosteum and contained newly formed vessels not previously present in calcified tissue. Angiogenesis was induced by RANKL only in WT but not Mmp9−/− calvaria as measured by CD31+ vessel density or total CD31+ vessel area within the outer table of calvaria (Figure 5B). Angiogenesis in WT calvaria treated with RANKL was significantly different from RANKL-treated Mmp9−/− calvaria when angiogenesis was quantified by vessel density, and nearly significant when angiogenesis was quantified by total vessel area. As with calvaria treated with PTHrP, no significant differences in vessel size were detected (data not shown).
Figure 5.
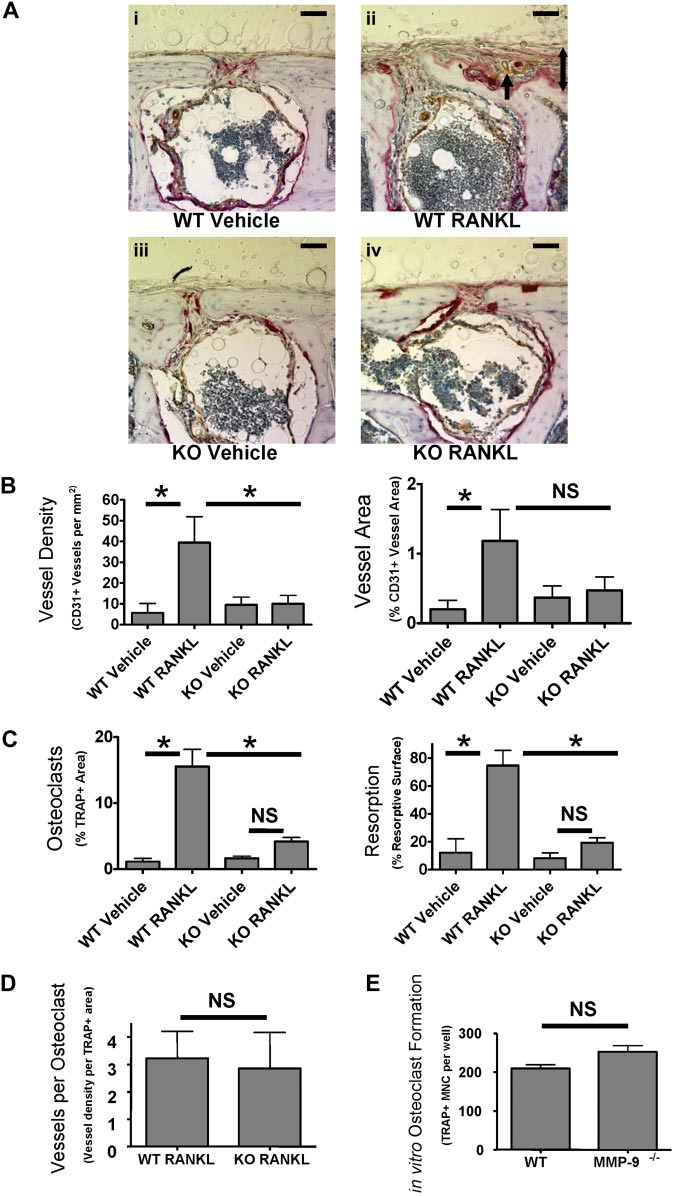
MMP-9 is important for stimulation of bone angiogenesis by osteoclasts in vivo. (A) Supra-calvarial RANKL increases angiogenesis and osteoclast formation to a greater extent in calvaria of WT than Mmp9−/− mice. Images of the outer table of calvarial sections stained for TRAP (red) and CD31 (brown) and lightly counterstained with hematoxylin. Images are WT vehicle-treated (i), WT RANKL-treated (ii), Mmp9−/− vehicle-treated (iii), or Mmp9
−/− RANKL-treated (iv). Images were acquired as in Figure 4. indicates CD31+ vessel in remodeling bone; 2-headed arrow, thickness of RANKL-induced bone remodeling. Scale bars represent 50 μm. Outer surface is at the top of the image. Auto contrast and auto color adjustment were performed with Adobe Photoshop software. (B) Quantification of RANKL-induced vessel response by calculation of CD31+ vessel density or area in the outer table of 10× objective images taken at the calvarial midline. (C) Total percentage TRAP+ area of the calvarial outer table or resorptive surface of the outer table subperiosteal surface. (D) Vessels per osteoclast were calculated by dividing vessel density by TRAP+ area for each RANKL-treated animal. (E) In vitro osteoclast formation from bone marrow from WT and Mmp9−/− C57BL/6 mice was quantified by counting TRAP+ multinucleated cells. Data are mean ± SEM. *P < .05. NS indicates not significant.
In parallel to our findings in metatarsal explants with PTHrP, osteoclast formation was stimulated by RANKL in WT but less so in Mmp9−/− animals in the calvarial outer table (Figure 5C). Resorption surface or TRAP+ area was significantly less in Mmp9−/− than WT calvaria treated with RANKL. There was no significant increase in osteoclast stimulation with RANKL among Mmp9−/− mice as measured by resorption surface or TRAP+ area. To examine the relative angiogenic capacity of Mmp9−/− osteoclasts, we calculated vessel number per osteoclast, which did not differ between genotypes (Figure 5D). Thus, Mmp9 is not of primary importance for osteoclasts to stimulate angiogenesis per se but affects osteoclast numbers at the angiogenic site. To determine whether the decrease in osteoclast numbers in Mmp9−/− mice results from decreased osteoclast differentiation or precursor number, osteoclast differentiation was determined in vitro. WT and Mmp9−/− mice formed similar numbers of osteoclasts (Figure 5E). Thus, MMP-9 most probably affects osteoclast-stimulated angiogenesis primarily through effects on osteoclast migration.
We then determined the cell type(s) expressing MMP-9 in mouse calvaria. MMP-9 expression was almost completely restricted to osteoclasts. Immunohistochemical analysis of calvaria from the studies described in Figure 5 showed that MMP-9 was almost completely colocalized with the osteoclast marker TRAP (Figure 6). Only very rare MMP-9+, TRAP− mononuclear cells were detected in the marrow and periosteum.
Figure 6.
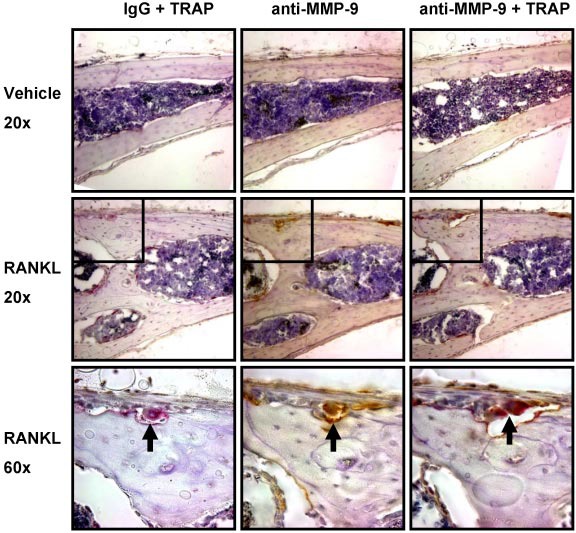
MMP-9 is predominantly expressed by osteoclasts in mouse calvaria. Serial coronal sections of vehicle or RANKL-treated mouse calvaria were stained for TRAP (red), MMP-9 (brown), or IgG as indicated. represents an osteoclast present in 3 serial sections, positive for both TRAP and MMP-9. Insets shown at original magnification ×60 are indicated by boxes. Images were acquired with a Nikon Eclipse E800 microscope fitted with a Plan Fluor 20×/0.5 NA objective or Plan Apo 60×/1.4 NA objective (oil), and an Olympus America SN CG603057-H camera with Magnifire software. Auto contrast adjustments were performed with Adobe Photoshop software (original magnifications: ×20 or ×60 as indicated). Results are from 1 representative experiment of 2 performed.
Discussion
In this report, we demonstrate that osteoclasts stimulated angiogenesis in bone explants and in vivo and that MMP-9 is required for osteoclast stimulation of angiogenesis, primarily because of its effects on osteoclast formation and/or activity. These are the first studies to show that osteoclasts stimulate angiogenesis in vivo or in explants and suggest that osteoclast formation and/or activity and angiogenesis are linked both in development and in mature remodeling bone.
We used an in vitro explant system that permitted us to study the contribution of osteoclasts to angiogenesis in a bone microenvironment. The metatarsal system allows the study of osteoclast-stimulated angiogenesis by many possible mechanisms, including direct release of angiogenic factors, proteolytic activation of matrix-bound angiogenic factors, and stimulation of angiogenic factor production in a second cell type. We found that inhibition of osteoclast formation and activity with OPG reduced angiogenesis and that stimulation of osteoclast activity with PTHrP increased angiogenesis in the explants. The proangiogenic activity of PTHrP was blocked by OPG, demonstrating that PTHrP stimulated angiogenesis primarily by its effects on osteoclasts. We then extended our studies and found that stimulation of osteoclast formation and activity with PTHrP or RANKL, the most specific stimulator of osteoclast formation and function, stimulated angiogenesis in vivo. We could not determine whether OPG blocked the proangiogenic affects of PTHrP in vivo because we were unable to obtain OPG that was active in our in vivo system.
In studies to examine the mechanism(s) responsible for osteoclast stimulation of angiogenesis, we determined that MMP-9 plays an important role in osteoclast stimulation of angiogenesis, as well as bone remodeling, with both the angiogenic and bone-resorptive effects of PTHrP being absent in Mmp9−/− metatarsal explants. Similarly, the proangiogenic, osteoclastogenic and bone-resorptive effects of RANKL were reduced in Mmp9−/− calvaria in vivo. The reduced angiogenesis seen in Mmp9−/− mice treated with RANKL most probably reflects the decreased osteoclast numbers and activity in Mmp9−/− mice at the site where angiogenesis occurs, compared with WT controls. The number of vessels per osteoclast was not different between genotypes, suggesting that osteoclasts lacking MMP-9 do not have an intrinsic angiogenic deficit once they have formed and migrated to the proper location. Similarly, PTHrP stimulated both angiogenesis and resorption in WT bone explants but did not stimulate either angiogenesis or resorption in explants lacking Mmp-9.
The observed reductions in stimulated osteoclast formation or function in Mmp9−/− mice are most probably the result of reduced osteoclast migration rather than direct effects of MMP-9 on osteoclast differentiation or matrix solubilization (Figure 7). Explants from E17 Mmp9−/− mouse metatarsals, but not more mature bones, show a lower level of basal resorption compared with WT because of delayed osteoclast invasion of the ossification center rather than a requirement for MMP-9 in biochemical matrix solubilization.13 Likewise, MMP-9 is required for osteoclast or osteoclast precursor migration in vitro.14,28,29 Mmp9−/− and WT mice did not display different levels of osteoclast formation in vitro, showing that the decreased resorption or osteoclast numbers in explants or in vivo was not the result of defective differentiation or decreased precursor numbers. MMP-9 is predominantly expressed by osteoclasts in bone and is expressed at high levels, suggesting that the MMP-9 required for osteoclast stimulation of angiogenesis is secreted by osteoclasts themselves. Because of our observations that MMP-9 is important for both bone resorption and angiogenesis under conditions of increased osteoclastogenesis, it may be possible clinically to inhibit both bone destruction and angiogenesis with an MMP-9 inhibitor.
Figure 7.

Role of MMP-9 in osteoclast-stimulated angiogenesis. In this model, osteoclasts release MMP-9, which induces osteoclast migration to increase local osteoclast numbers and activity. The osteoclasts then secrete factors that increase angiogenesis.
A potential mechanism by which osteoclast-derived MMP-9 may stimulate angiogenesis, as well as osteoclast migration, is through its capacity to release heparin-binding isoforms of VEGF-A (eg, VEGF165 and VEGF189) from the extracellular matrix. This molecular action of MMP-9 occurs in bone and is important for osteoclast recruitment into the primary ossification center.13 Further, Mmp9−/− mice have delayed vessel invasion into the primary ossification center.8 This molecular mechanism may also allow osteoclasts to stimulate angiogenesis directly by releasing matrix-bound VEGF, which acts on vessels.
Other factors in addition to MMP-9 may also be involved in osteoclast stimulation of angiogenesis. Osteopontin was reported to be required for angiogenic activity of osteoclast-conditioned media in vitro, but its effects in vivo are unknown.4 A linked osteoblastic response may also be required for osteoclast-stimulated angiogenesis because osteoblast-derived VEGF is clearly important for angiogenesis. This osteoblastic VEGF expression may contribute to osteoclast stimulation of angiogenesis when osteoclasts are treated with PTHrP. However, direct stimulation of production of VEGF from osteoblasts by PTHrP probably does not contribute to the angiogenesis seen in our studies. We found that OPG blocked PTHrP stimulation of angiogenesis in metatarsal explants and that RANKL could stimulate angiogenesis in vivo.
Our findings that MMP-9 affects both angiogenesis and osteoclasts suggests that osteoclastogenesis and angiogenesis are linked in some situations. Linkage between osteoclasts and vessels occurs most probably in situations where osteoclast invasion is required, such as fetal long bones before invasion of the growth plate or remodeling of calvaria induced by RANKL. Osteoclasts most probably invade from the periosteum in both situations. Such linkage between osteoblasts and angiogenesis occurs during development of long bones.25 In addition, linkage between osteoclasts and vessel formation could result from endothelial cell stimulation of osteoclast formation. Endothelial cells can increase osteoclast formation by several mechanisms, including increased RANKL expression on their surface, thereby stimulating osteoclast formation when cocultured with osteoclast precursors.30 Endothelial cells may also regulate the recruitment of osteoclast precursors to remodeling sites from the vascular compartment.31
It is possible that OPG, which we used to modulate osteoclast activity, may also affect endothelial cells. Several studies have reported possible direct stimulatory effects of OPG on endothelial cells, suggesting that we may have underestimated the contribution of osteoclasts to angiogenesis in our experiments using OPG.32–34 Further, OPG can stimulate angiogenesis or endothelial cell survival in cell culture or aortic ring explants.35,36 In agreement with these findings, when equivalent concentrations of OPG to those used to inhibit metatarsal angiogenesis were tested in the HUVEC/fibroblast coculture angiogenesis assay, which lacks osteoclasts, we observed a slight stimulation rather than any inhibition of angiogenesis in the HUVEC cultures (data not shown). It is unclear whether PTHrP can directly inhibit or stimulate endothelial cells. Two studies reported that PTHrP inhibited and one reported that PTHrP stimulated endothelial cells.32–34 Like PTHrP, it is unclear whether RANKL has possible direct effects on endothelial cells.36,37
Before our studies, the role of osteoclasts in angiogenesis was unclear, with no studies demonstrating that osteoclasts stimulated angiogenesis in vivo or in explants. Tanaka et al have reported that osteoclasts stimulate angiogenesis in cell culture, and attributed it to osteopontin production by osteoclasts.4 We also observed stimulation of angiogenesis with osteoclast-conditioned media in a HUVEC-fibroblast coculture angiogenesis assay (data not shown). However, in contrast to these results, we were unable to detect any proangiogenic effects of rh-osteopontin at doses up to 3-fold the reported dose. The basis for these differences is unknown. These investigators did not determine whether osteoclast-secreted osteopontin contributed to angiogenesis in vivo.
Similarly, Vu et al reported in the “Discussion” section of their paper that osteopetrotic (Fos−/− and op/op) mice have delayed blood vessel invasion into the primary ossification center and growth plate in vivo.8 However, other investigators found that osteoclasts do not play a role in angiogenesis and reported that lack of osteoclasts did not affect blood vessel invasion into the epiphyses of tibiae or mouse tail vertebrae.6,7 These results suggest that osteoclasts may be less important for angiogenesis in vertebrae and long bone epiphyses. Differences in the contribution of osteoblasts to angiogenesis at different bone anatomic sites have been reported.25 Osteoblast contributions to angiogenesis are more important in long bones than calvaria; thus, the contribution of osteoclasts to angiogenesis may differ at different anatomic sites.25
In conclusion, we have shown that osteoclasts contribute to angiogenesis in vitro and in vivo by a mechanism requiring MMP-9. MMP-9 is important for osteoclast resorption and formation, probably because of its previously reported effects on osteoclast invasion or migration rather than differentiation or matrix solubilization. Osteoclast-stimulated angiogenesis is deserving of further study to more precisely define the molecular mechanism(s) involved and to identify the physiologic and pathologic settings in which osteoclast-stimulated angiogenesis plays a role.
Acknowledgments
The authors thank University of Pittsburgh Center for Biological Imaging for use of facilities, T. Clemens and Y. Wang for advice on the metatarsal assay, R. Chambers for assistance with Mmp9−/− mice, S. Teitelbaum and F. P. Ross for mouse RANKL-GST and advice on its use, and D. Beer Stolz for developing the CD31 IHC methods.
This work was supported in part by Novartis Pharmaceuticals Corp. The materials are the result of work supported with resources and the use of facilities at the Veterans Administration Pittsburgh Healthcare System, Research and Development.
F.C.C. is an MD, PhD candidate at the University of Pittsburgh, School of Medicine. This work is in partial fulfillment of his PhD degree.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: F.C.C. designed and performed most experiments and wrote the manuscript; J.L.A. and K.D.P. helped with animal studies; R.J.C. assisted with metatarsal experiments; S.D.S. helped design and interpret MMP-9 studies and provided mice; J.J.W. helped design metatarsal experiments and provided mice; H.C.B. helped interpret and design in vivo studies; and G.D.R. designed experiments and wrote the manuscript.
Conflict-of-interest disclosure: G.D.R. is a consultant for Novartis Pharmaceuticals Corp. The remaining authors declare no competing financial interests.
Correspondence: G. David Roodman, Department of Medicine, University of Pittsburgh, Veterans Administration Pittsburgh Healthcare System, R&D 151-U, University Dr C, Pittsburgh, PA 15240; e-mail: roodmangd@upmc.edu.
References
- 1.Ribatti D, Nico B, Vacca A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene. 2006;25:4257–4266. doi: 10.1038/sj.onc.1209456. [DOI] [PubMed] [Google Scholar]
- 2.Findlay DM, Haynes DR. Mechanisms of bone loss in rheumatoid arthritis. Mod Rheumatol. 2005;15:232–240. doi: 10.1007/s10165-005-0412-z. [DOI] [PubMed] [Google Scholar]
- 3.Hauge EM, Qvesel D, Eriksen EF, Mosekilde L, Melsen F. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J Bone Miner Res. 2001;16:1575–1582. doi: 10.1359/jbmr.2001.16.9.1575. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka Y, Abe M, Hiasa M, et al. Myeloma cell-osteoclast interaction enhances angiogenesis together with bone resorption: a role for vascular endothelial cell growth factor and osteopontin. Clin Cancer Res. 2007;13:816–823. doi: 10.1158/1078-0432.CCR-06-2258. [DOI] [PubMed] [Google Scholar]
- 5.Kiesel J, Miller C, Abu-Amer Y, Aurora R. Systems level analysis of osteoclastogenesis reveals intrinsic and extrinsic regulatory interactions. Dev Dyn. 2007;236:2181–2197. doi: 10.1002/dvdy.21206. [DOI] [PubMed] [Google Scholar]
- 6.Deckers MM, Van Beek ER, Van Der Pluijm G, et al. Dissociation of angiogenesis and osteoclastogenesis during endochondral bone formation in neonatal mice. J Bone Miner Res. 2002;17:998–1007. doi: 10.1359/jbmr.2002.17.6.998. [DOI] [PubMed] [Google Scholar]
- 7.Sugiura J, Sakurai Y, Okuyama N, Yamasaki A. Vascular invasion of epiphyseal growth plate in osteopetrotic (op/op) mouse tibiae. J Hard Tissue Biol. 2006;15:96–100. [Google Scholar]
- 8.Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colnot C, Thompson Z, Miclau T, Werb Z, Helms JA. Altered fracture repair in the absence of MMP9. Development. 2003;130:4123–4133. doi: 10.1242/dev.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haeusler G, Walter I, Helmreich M, Egerbacher M. Localization of matrix metalloproteinases (MMPs), their tissue inhibitors, and vascular endothelial growth factor (VEGF) in growth plates of children and adolescents indicates a role for MMPs in human postnatal growth and skeletal maturation. Calcif Tissue Int. 2005;76:326–335. doi: 10.1007/s00223-004-0161-6. [DOI] [PubMed] [Google Scholar]
- 11.Bord S, Horner A, Hembry RM, Reynolds JJ, Compston JE. Distribution of matrix metalloproteinases and their inhibitor, TIMP-1, in developing human osteophytic bone. J Anat. 1997;191:39–48. doi: 10.1046/j.1469-7580.1997.19110039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engsig MT, Chen Q-J, Vu TH, et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 2000;151:879–890. doi: 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishibashi O, Niwa S, Kadoyama K, Inui T. MMP-9 antisense oligodeoxynucleotide exerts an inhibitory effect on osteoclastic bone resorption by suppressing cell migration. Life Sci. 2006;79:1657–1660. doi: 10.1016/j.lfs.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 15.Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao J, McCauley LK. Skeletal metastasis: established and emerging roles of parathyroid hormone related protein (PTHrP). Cancer Metastasis Rev. 2006;25:559–571. doi: 10.1007/s10555-006-9033-z. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda H, Shima N, Nakagawa N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG: a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 18.Kurihara N, Chenu C, Miller M, Civin C, Roodman GD. Identification of committed mononuclear precursors for osteoclast-like cells formed in long term human marrow cultures. Endocrinology. 1990;126:2733–2741. doi: 10.1210/endo-126-5-2733. [DOI] [PubMed] [Google Scholar]
- 19.Oba Y, Chung HY, Choi SJ, Roodman GD. Eosinophil chemotactic factor-L (ECF-L): a novel osteoclast stimulating factor. J Bone Miner Res. 2003;18:1332–1341. doi: 10.1359/jbmr.2003.18.7.1332. [DOI] [PubMed] [Google Scholar]
- 20.Deckers M, van der Pluijm G, Dooijewaard S, et al. Effect of angiogenic and antiangiogenic compounds on the outgrowth of capillary structures from fetal mouse bone explants. Lab Invest. 2001;81:5–15. doi: 10.1038/labinvest.3780207. [DOI] [PubMed] [Google Scholar]
- 21.Rao H, Lu G, Kajiya H, et al. Alpha9beta1: a novel osteoclast integrin that regulates osteoclast formation and function. J Bone Miner Res. 2006;21:1657–1665. doi: 10.1359/JBMR.060718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi SJ, Reddy SV, Devlin RD, et al. Identification of human asparaginyl endopeptidase (legumain) as an inhibitor of osteoclast formation and bone resorption. J Biol Chem. 1999;274:27747–27753. doi: 10.1074/jbc.274.39.27747. [DOI] [PubMed] [Google Scholar]
- 23.McHugh KP, Hodivala-Dilke K, Zheng M-H, et al. Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105:433–440. doi: 10.1172/JCI8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandi ML, Collin-Osdoby P. Vascular biology and the skeleton. J Bone Miner Res. 2006;21:183–192. doi: 10.1359/JBMR.050917. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wan C, Deng L, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117:1616–1626. doi: 10.1172/JCI31581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169:681–691. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura-Ishizu A, Morikawa S, Shimizu K, Ezaki T. Characterization of sinusoidal endothelial cells of the liver and bone marrow using an intravital lectin injection method. J Mol Histol. 2008;39:471–479. doi: 10.1007/s10735-008-9186-x. [DOI] [PubMed] [Google Scholar]
- 28.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J Bone Miner Res. 2003;18:1404–1418. doi: 10.1359/jbmr.2003.18.8.1404. [DOI] [PubMed] [Google Scholar]
- 29.Dong Z, Bonfil RD, Chinni S, et al. Matrix metalloproteinase activity and osteoclasts in experimental prostate cancer bone metastasis tissue. Am J Pathol. 2005;166:1173–1186. doi: 10.1016/S0002-9440(10)62337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem. 2001;276:20659–20672. doi: 10.1074/jbc.M010153200. [DOI] [PubMed] [Google Scholar]
- 31.Kindle L, Rothe L, Kriss M, Osdoby P, Collin-Osdoby P. Human microvascular endothelial cell activation by IL-1 and TNF-alpha stimulates the adhesion and transendothelial migration of circulating human CD14+ monocytes that develop with RANKL into functional osteoclasts. J Bone Miner Res. 2006;21:193–206. doi: 10.1359/JBMR.051027. [DOI] [PubMed] [Google Scholar]
- 32.Bakre MM, Zhu Y, Yin H, et al. Parathyroid hormone-related peptide is a naturally occurring, protein kinase A-dependent angiogenesis inhibitor. Nat Med. 2002;8:995–1003. doi: 10.1038/nm753. [DOI] [PubMed] [Google Scholar]
- 33.Akino K, Ohtsuru A, Kanda K, et al. Parathyroid hormone-related peptide is a potent tumor angiogenic factor. Endocrinology. 2000;141:4313–4316. doi: 10.1210/endo.141.11.7875. [DOI] [PubMed] [Google Scholar]
- 34.Diamond AG, Gonterman RM, Anderson AL, et al. Parathyroid hormone hormone-related protein and the PTH receptor regulate angiogenesis of the skin. J Invest Dermatol. 2006;126:2127–2134. doi: 10.1038/sj.jid.5700338. [DOI] [PubMed] [Google Scholar]
- 35.Pritzker LB, Scatena M, Giachelli CM. The role of osteoprotegerin and tumor necrosis factor-related apoptosis-inducing ligand in human microvascular endothelial cell survival. Mol Biol Cell. 2004;15:2834–2841. doi: 10.1091/mbc.E04-01-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGonigle JS, Giachelli CM, Scatena M. Osteoprotegerin and RANKL differentially regulate angiogenesis and endothelial cell function. Angiogenesis. 2009;12:35–46. doi: 10.1007/s10456-008-9127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim YM, Kim YM, Lee YM, et al. TNF-related activation-induced cytokine (TRANCE) induces angiogenesis through the activation of Src and phospholipase C (PLC) in human endothelial cells. J Biol Chem. 2002;277:6799–6805. doi: 10.1074/jbc.M109434200. [DOI] [PubMed] [Google Scholar]