

Iron containing monooxygenases play diverse roles in nature, ranging from the primary metabolic functions of alkane hydroxylases to the xenobiotic detoxification and secondary metabolic roles of cytochrome P450 enzymes..[1] Common to these enzymes is the ability to reductively activate molecular oxygen to generate highly electrophilic oxygen species, whose reactivity is comparable with that of ‘oxenes’ (oxygen atoms containing six-valence electrons).[2] P450 enzymes in particular possess the remarkable ability to insert oxygen atoms at virtually any position within otherwise unreactive carbon skeletons leading to the synthesis of alcohols or epoxides in diverse natural products.
Whereas enzymes are capable of inserting oxygen atoms into even unactivated C-H bonds, the sites into which nitrogen can be incorporated are more constrained. Transaminases, ammonia lyases, and amino acid dehydrogenases, for example,[3] target oxidized or otherwise chemically activated carbons for reaction. Enzymes that catalyze the concerted oxidative amination of C-H bonds are apparently absent from nature's repertoire of chemical catalysts.
Synthetic chemists, who are not limited to biologically accessible reagents and metals, have developed highly useful methods for the oxidative formation of C-N bonds.[4] These C-H amination reactions often proceed through a nitrenoid intermediate that has no parallel in natural enzymes. Although these reactions do not require preoxidized or otherwise activated carbon atoms, they do require specialized nitrene-precursors such as azides, haloamines, or iminoiodinanes. Of the many transition-metal catalysts based on Rh, Ru, Mn, Co, and Fe that catalyze intra- and intermolecular C-H amination, we were especially interested in the metal-porphyrin systems,[5] which react with iminoiodinanes in the +3 oxidation state to catalyze nitrene transfers that are isoelectronic with the well-established formal oxene transfer reaction of ferric-P450 enzymes with iodosylbenzene.[5a, 6] Trace levels (3 total turnovers; TTNs) of intramolecular C-H amination were reported more than 25 years ago for mammalian cytochrome P450 preparations reacting with iminoiodinanes.[7] We decided to revisit the possibility of finding or engineering an enzyme catalyst for this useful and challenging transformation.
Iminoiodinanes, however, are problematic for biocatalytic application, given their polymeric nature[8] and insolubility[9] in aqueous media. As an alternative to iminoiodinanes, we decided to focus on the synthetic reaction of sulfonylazides with reduced (+2 oxidation state) metal porphyrins.[10] We reasoned that such a "chemomimetic" approach to achieving direct C-H to C-N conversion could provide a biocatalytic route to amines and amides using biochemically compatible and atom-efficient azide-based nitrene precursors, with the usual enzyme advantages of selectivity and mild reaction conditions. Here we report the first highly active enzyme catalysts of C-H amination.
In initial experiments, we tested a panel of 24 purified cytochrome P450BM3 variants developed for monooxygenation reactions. Enzymes were reacted with 2,4,6-triethylbenzene-1-sulfonylazide (1) under anaerobic, reducing conditions at an enzyme loading of 0.5 mol% in aqueous media (phosphate buffer, 2.5% v/v DMSO). Most reactions gave sulfonamide 2 as the major product, though all of the tested enzymes, including wild-type (4 TTN), yielded small amounts of the C-H amination product, 3. The most active enzyme for C-H amination in the initial screen contained only a single mutation (T268A) relative to wild-type P450BM3 (28 TTN). Although mutations to the active site residue T268 are deleterious to monooxygenation activity given its role in promoting O-O bond scission, the T268A mutation was recently shown to promote P450BM3-catalyzed carbene transfers yielding cyclopropanes.[11] Thus, in spite of the significant differences between carbene and nitrene chemistry, we proceeded to test several P450BM3-based cyclopropanation catalysts, including several serine-heme ligated ‘P411’[12] enzymes (so called because the Soret peak in the ferrous CO-bound spectrum is shifted to 411 nm rather than 450 nm for cysteine ligated enzymes) in which the axial coordinating cysteine C400, absolutely required for monooxygenation activity, is mutated to serine. Given that loss of dinitrogen from azides is much more facile than O-O bond scission catalyzed by P450s, we reasoned that the ‘thiolate push’ of the axial cysteine would be unnecessary for C-H amination.[13] The most active enzyme here was the C400S mutant P411BM3-CIS (14 mutations from wild-type), which also contained the T268A mutation and supported over 140 total turnovers (73% yield of 3 by HPLC). Variant P450BM3-CIS, which lacks the C400S mutation at the axial heme ligand, was significantly less active (9 TTN), indicating that serine-heme ligation strongly enhances BM3-catalyzed C-H amination. The P450BM3-C400S single mutant (P411BM3) also exhibited markedly improved activity (49 TTN) relative to its cysteine-ligated counterpart, P450BM3 (4 TTN).
To clarify the roles of the T268A and C400S mutations in BM3-catalyzed amination, we performed further experiments at 0.1 mol% catalyst loading with the P450BM3-T268A and P411BM3 (BM3-C400S) single mutants as well as the T268A/C400S double mutant in reaction with sulfonyl azide 1 (Table 1). We found that the T268A and C400S mutations combined to yield a highly active enzyme (120 TTN for the double mutant versus 310 TTN for P411BM3-CIS, Table 1), indicating that the T268A and C400S mutations were major contributors to the high activity of P411BM3-CIS. In fact, reverting the T268A mutation in P411BM3-CIS markedly reduced activity (82 TTN).
Table 1.
Comparison of activities (TTN) and enantioselectivies of purified P450 and P411 variants with azide 1 at 0.1 mol% catalyst loading giving sulfonamide 2 and benzosultam 3. Reaction conditions described in supporting information. TTNs and enantioselectivies determined by HPLC analysis.
| In vitro catalyst | TTN[a] | % ee[b] |
|---|---|---|
| P450BM3 | 2.1 | nd |
| P450BM3-T268A | 15 | 36 |
| P411BM3 | 32 | 20 |
| P411BM3-T268A | 120 | 58 |
| P411BM3-CIS | 310 | 67 |
| P411BM3-CIS-A268T | 82 | 47 |
| P411BM3-CIS-T438S | 383 | 73 |
TTN = total turnover number
(S−R)/(S+R).
nd = not determined.
Control experiments revealed that the enzyme-catalyzed reaction was inhibited by carbon monoxide, air, and heat denaturation of the enzyme, all of which suggests that catalysis occurs at the enzyme-bound heme (Table S2). Hemin also was capable of catalyzing this reaction when reduced with dithionite (Table S3, Figure 1). That hemin could catalyze this reaction is consistent with earlier work investigating the activity of metal porphyrins in the reaction with sulfonyl azides.[14] However, whereas in vitro enzyme reactions with prochiral substrate 1 resulted in asymmetric induction (up to 73% ee, Table 1), reaction with hemin unsurprisingly yielded only racemic 3, strongly suggesting that BM3-catalyzed amination occurs within the chiral environment of the enzyme active site. Addition of sub-stoichiometric amounts of NADPH was sufficient for activity (Table S4), supporting the hypothesis that ferrous-heme is the azide-reactive state, akin to P450-catalyzed cyclopropanation.[11] Dithionite could be used in place of NADPH to drive catalysis, where its effect was comparable to that of NADPH for both cysteine- and serine-ligated enzymes P450BM3-T268A and P411BM3-T268A (Table S5), indicating that reduction to ferrous heme was not limiting.
Figure 1.

Substrate selectivity of P411 enzymes compared with free hemin. Compounds used to test the dependence of amination activity on C-H bond strength in reactions catalyzed by enzyme (0.1 mol%) or hemin (1 mol%) are shown. Reaction conditions described in supporting information.
To examine the effect of C-H bond strength on amination activity, P411BM3-CIS and P411BM3-T268A were reacted with the trimethyl and triisopropyl analogs of 1 (substrates 4 and 6, respectively). The desired benzosultam products were obtained, though the productivity was lower with both substrates (Figure 1, Table S3). Free hemin activity was inversely correlated with the substrate C-H bond strength, consistent with earlier work on cobalt porphyrin catalysts,[14] showing no measurable activity on substrate 4, minimal activity on substrate 1 (3 TTN), and the highest activity on substrate 6 (55 TTN). The differing patterns of activity observed with hemin and enzyme-catalyzed reactions suggest that the enzyme itself plays a critical role in catalyzing C-N bond formation beyond providing a chiral active site that guides the stereochemical outcome of the reaction. In particular, enzyme reactions with triethyl and trimethyl sulfonylazide substrates 1 and 4 were markedly more productive than the corresponding hemin reactions. The reduced activity of the enzyme toward triisopropyl substrate 6 suggests that the active site structure currently favors smaller substrates, though it is likely that this can be modulated by further enzyme engineering.
To determine whether this new reactivity could be exploited in vivo, we next investigated whether these enzymes expressed in intact E. coli cells could catalyze amination reactions when provided with azide substrate. Remarkably, both the P411BM3-T268A and P411BM3-CIS enzymes were highly active on 1, catalyzing hundreds of turnovers (245 TTN, 89% ee P411BM3-T268A, 680 TTN, 60% ee P411BM3-CIS) under anaerobic conditions with added glucose (Tables 2, S6). Lyophilized cells containing P411BM3-CIS could also support catalysis, with productivity similar to freshly-prepared cell suspensions (750 TTN, 61% ee). Enantioselectivity was comparable or enhanced for whole-cell catalysts relative to purified enzymes (Table 2). Including the previously characterized T438S mutation in P411BM3-CIS strongly increased enantioselectivity (430 TTN, 86% ee).[11, 15] Optimization of expression conditions increased the productivity of whole-cell C-H amination catalysts, enabling conversions to 3 of up to 66% in small-scale reactions (Table S7). Inspired by the simplicity of employing whole cells as amination catalysts, we performed a preparative scale reaction (50 mg) using anaerobic resting cells expressing the P411BM3-CIS-T438S catalyst, affording sultam 3 (77% yield by HPLC, 69% isolated yield, 87% ee). The level of stereoselectivity attained with whole-cell catalysts compared favorably with a previously reported chiral C-H amination catalyst, which gave 88% ee in reaction with substrate 1.[16]
Table 2.
Comparison of C-H amination activities (TTN) of intact E. coli cells expressing P450 and P411 variants. Reaction conditions described in supporting information.
| In vivo catalyst | [P450] or [P411] µM |
% yield[a] (3) |
TTN[b] | % ee[c] |
|---|---|---|---|---|
| pCWori-empty | 0 | 0 | 0 | nd |
| P450BM3 | 6.6 | 0.5 | 5.1 | nd |
| P450BM3-T268A | 5.8 | 7.8 | 26 | 84 |
| P411BM3 | 4.3 | 6.7 | 29 | 16 |
| P411BM3-T268A | 2.2 | 30 | 250 | 89 |
| P411BM3-CIS | 1.4 | 46 | 680 | 60 |
| P411BM3-CIS-T438S | 2.7 | 58 | 430 | 87 |
TTN = total turnover number
*(S−R)/(S+R).
nd = not determined.
The beneficial effects of the T268A and C400S mutations for C-H amination is striking in that both residues play critical roles in P450-catalyzed monooxygenation.[17] While important for protonation of iron-peroxo intermediates that occur during dioxygen activation, T268 may sterically hinder binding of bulkier azide substrates in C-H amination. Consistent with a steric role, the T268A mutation enhances the stereoselectivity of C-H amination; it also strongly impacts diastereo and enantioselectivity of styrene cyclopropanation.[11] While thiolate ligation is thought to be essential for O-O bond scission and enhancing the basicity of reactive oxygen intermediates,[13, 18] here we find that mutation of this key residue leads to high levels of in vitro amination activity (Table 1). That mutations to both T268 and C400 appear necessary for enzymatic C-H amination suggests that naturally occurring P450s, in which these two residues are highly conserved, will likely be poor catalysts of C-H amination.
Many enzyme-catalyzed reactions such as ketoreduction, monooxygenation, and transamination are increasingly useful in chemical synthesis,[19] and applications of these and other naturally-occurring reaction types will continue to develop. However, it is no longer necessary to limit biocatalysis to reactions that have natural antecedents.[11, 20] Rather, the scope of biocatalysis can be expanded by directing natural enzymes to imitate the artificial, accessing whole new chemistries by judicial choice of reaction conditions, synthetic reagents, and protein engineering.
Supplementary Material
Scheme 1.
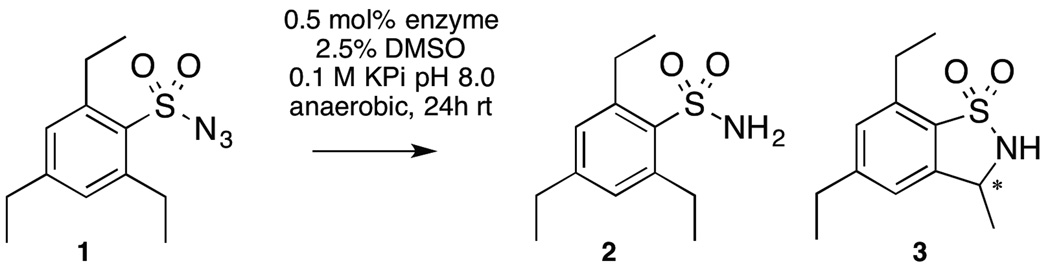
Initial reaction screening substrate and products
Acknowledgments
We thank S. Virgil and the 3CS catalysis center at Caltech for assistance with chiral HPLC and LC-MS, and D. Montgomery, Y. Liu, N. Peck, K. Rabe, R. Lauchli, and D. VanderVelde for helpful discussions. This work was supported by the Department of the Navy, Office of Naval Research (grant N00014-11-1-0205), and by the Jacobs Institute for Molecular Engineering for Medicine at Caltech. J.A.M. and Z.J.W. are supported by Ruth L. Kirschstein National Research Service Awards (F32GM101792) and (F32EB015846). C.C.F. is supported by an NSF Graduate Research Fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org
References
- 1.a) Podust LM, Sherman DH. Nat. Prod. Rep. 2012;29:1251–1266. doi: 10.1039/c2np20020a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Torres-Pazmiño DE, Winkler M, Glieder A, Fraaije MW. J. Biotechnol. 2010;146:9–24. doi: 10.1016/j.jbiotec.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 2.a) Newcomb M, Hollenberg PF, Coon MJ. Arch. Biochem. Biophys. 2003;409:72–79. doi: 10.1016/s0003-9861(02)00445-9. [DOI] [PubMed] [Google Scholar]; b) Guallar V, Ghermanm BF, Lippard SJ, Friesner RA. Curr. Opin. Chem. Biol. 2002;6:236–242. doi: 10.1016/s1367-5931(02)00310-1. [DOI] [PubMed] [Google Scholar]
- 3.a) Heberling MM, Wu B, Bartsch S, Janssen DB. Curr. Opin. Chem. Biol. 2013;17:1–11. doi: 10.1016/j.cbpa.2013.02.013. [DOI] [PubMed] [Google Scholar]; b Turner NJ. Curr. Opin. Chem. Biol. 2011;15:234–240. doi: 10.1016/j.cbpa.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Zalatan D, Du Bois J. Top. Curr. Chem. 2010;292:347–378. doi: 10.1007/128_2009_19. [DOI] [PubMed] [Google Scholar]
- 5.a) Breslow R, Gellman SH. J. Chem. Soc. Chem. Commun. 1982:1400–1401. [Google Scholar]; b Mahy JP, Bedi G, Battoni P, Masuy D. Tett. Lett. 1988;29:1927–1930. [Google Scholar]
- 6.Groves JT, Nemo TE, Myers RS. J. Am. Chem. Soc. 1979;101:1032–1033. [Google Scholar]
- 7.Svastis EW, Dawson JH, Breslow R, Gellman SH. J. Am. Chem. Soc. 1985;107:6427–6428. [Google Scholar]
- 8.Dauban P, Dodd RH. Synlett. 2003:1571–1586. [Google Scholar]
- 9.White RE, McCarthy MB. J. Am. Chem. Soc. 1984;106:4922–4926. [Google Scholar]
- 10.a) Lu H, Zhang XP. Chem. Soc. Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]; b) Caselli A, Gallo E, Ragaini F, Ricatto F, Abbiati G, Cenini S. Inorg. Chim. Acta. 2006;359:2924–2932. [Google Scholar]
- 11.Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- 12.Coelho PS, Wang ZJ, Ener ME, Baril S, Kannan A, Arnold FH, Brustad EM. Nat. Chem. Biol. 2013 doi: 10.1038/nchembio.1278. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson JH. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- 14.Ruppel JV, Kamble RM, Zhang XP. Org. Lett. 2007;9:4889–4892. doi: 10.1021/ol702265h. [DOI] [PubMed] [Google Scholar]
- 15.Huang WC, Cullis PM, Raven EL, Roberts GC. Metallomics. 2011;3:410–416. doi: 10.1039/c0mt00082e. [DOI] [PubMed] [Google Scholar]
- 16.Ichinose M, Suematsu H, Yasutomi Y, Nishioka Y, Uchida T, Katsuki T. Angew. Chem. Int. Ed. Engl. 2011;50:9884–9887. doi: 10.1002/anie.201101801. [DOI] [PubMed] [Google Scholar]
- 17.a) Meunier B, de Visser SP, Shaik S. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]; b Whitehouse CJC, Bell SG, Wong LL. Chem. Soc. Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- 18.Green MT. Curr. Opin. Chem. Biol. 2009;13:84–88. doi: 10.1016/j.cbpa.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 19.Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 20.a) Hyster TK, Knörr L, Ward TR, Rovis T. Science. 2012;338:500–503. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Köhler V, Wilson YM, Dürrenberger M, Ghislieri D, Churakova E, Quinto T, Knörr L, Häussinger D, Hollman F, Turner NJ, Ward TR. Nat. Chem. 2013;5:93–99. doi: 10.1038/nchem.1498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.