

Abstract
Multivalent nanoparticle platforms are attractive for biomedical applications because of their improved target specificity, sensitivity and solubility. However, their controlled assembly remains a considerable challenge. An efficient hydrazone ligation chemistry was applied to the assembly of Cowpea mosaic virus (CPMV) nanoparticles with individually tunable levels of a VEGFR-1 ligand and a fluorescent PEGylated peptide. The nanoparticles recognized VEGFR-1 on endothelial cell lines and VEGFR1-expressing tumor xenografts in mice, validating targeted CPMV as a nanoparticle platform in vivo.
Non-invasive visualization of complex cellular systems is of growing importance in many areas of research.1 Given current technologies, intravital imaging of clinically relevant solid tumors remains a challenge due to the specificity, sensitivity, and bioavailability of available agents in vivo. Molecular nanoscaffolds can be used to address these challenges by enabling the multivalent display of targeting ligands, imaging moieties, and other functional groups. The plant virus Cowpea mosaic virus (CPMV) (Fig. 1) is a viral nanoparticle (VNP) which has proven to be a robust macromolecular scaffold for numerous applications.2, 3 CPMV is ∼30 nm in diameter with a capsid composed of 60 identical copies of the large (L, 41 kDa) and small (S, 24 kDa) capsid proteins.4 The protein coat can be chemically modified at amino,5 carboxyl,6, 7 or thiol groups2 on its exterior surface, enabling a range of chemical and biological modifiers to be attached to the particles.8
Figure 1. CPMV: Strategy for labeling the virus capsid.
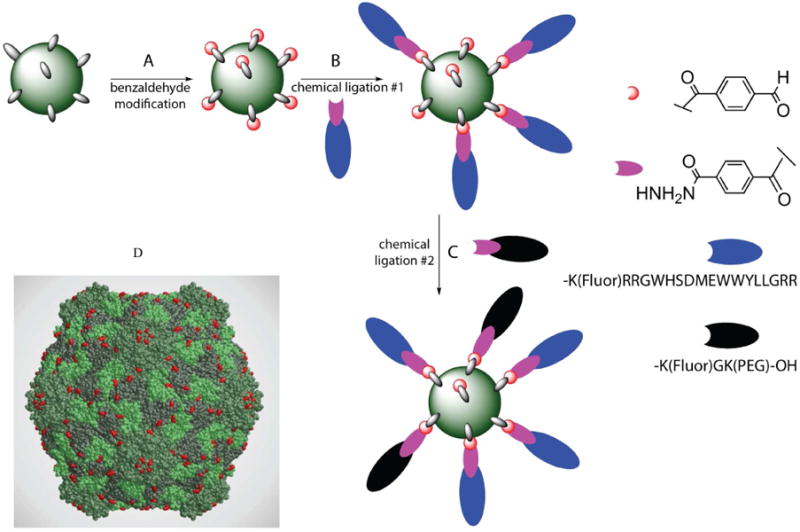
A. Surface exposed lysines on CPMV are covalently modified with a benzaldehyde group for ligation (red). B. The hydrazide group (purple) on the ligand F56f (blue) reacts specifically with some of the benzaldehydes on CPMV. C. A second ligation with hydrazide-PEG500f (black) labels remaining benzaldehyde sites on CPMV, yielding a doubly-modified nanoparticle. D. Three-dimensional structure of CPMV particle showing large subunits (light and dark green), and small subunits (medium green). Lysines available for conjugation are shown in red (image created in RasMol and DeepView using coordinates generated in VIPER 24 from 1NY7.pdb).
The general feasibility of using VNPs for applications such as biomedical imaging and targeted drug-delivery has been demonstrated for VNPs such as Cowpea chlorotic mottle virus (CCMV), Potato virus X, Hibiscus chlorotic ringspot virus (HCRSV), and bacteriophages M13, Qβ, and MS2,9-15 For example, we recently showed that a CPMV-fluorophore conjugate enables global imaging of the entire vasculature in live embryos.3 Dye-labeled CPMV particles have also been utilized to image sites of inflammation in the central nervous system.12 Toward targeted and therapeutic approaches, dual-modified targeted VNPs bearing chemotherapeutics or photosensitizers have been developed with the goal of cytotoxic activity.11, 15 13 Covalent modification of VNPs with tumor ligands such as folic acid allows targeting to cancer cells in vitro.10 Neverthless, targeted delivery of VNPs to specific molecular receptors in vivo has not yet been demonstrated.
Selective targeting and imaging of cells expressing vascular endothelial growth factor receptors (VEGFRs) has potential applications in angiogenesis research, as well as in understanding, detecting, and treating cancer.16 Because of its upregulation in tumor vasculature, a number of non-invasive imaging strategies have targeted VEGFR-2 (Flk1 or KDR).17, 18 The homologous VEGFR-1 (Flt1) is expressed in breast cancers,19 gastric cancers,20 and schwannomas.21 Importantly, VEGFR-1 has been identified as a tumor-specific vascular endothelial cell surface protein by subtractive proteomic mapping.22 A VEGFR-1 specific peptide, F56 was discovered through phage display techniques directed against the soluble receptor.23 This high-affinity peptide has been shown to suppress tumor growth and metastasis in implanted human breast cancer cells (BICR-H1) in an immune-compromised mouse model.23 These properties suggest that VEGFR-1 directed ligands may be useful for directing VNPs to tumors and tumor vasculature.
While it has been possible to conjugate small-molecule ligands to VNPs in a controlled manner, it has been more challenging to control the sequential attachment of more complex macromolecular ligands. In order to generate CPMV nanoparticles with 1) a fluorescent dye for imaging, 2) a PEG polymer for improved plasma circulation time and 3) the peptide ligand, F56, we developed a stepwise assembly strategy based on an efficient hydrazone ligation chemistry (Fig. 1 and S1; experimental details are given in the Supporting Information). The chemoselective reaction of an arylhydrazide or hydrazine with a benzyaldehyde moiety is rapid in weakly acidic buffer and forms a bisarylhydrazone product that absorbs strongly at ∼350 nm. To facilitate the ligation, CPMV was functionalized with a highly reactive, water-soluble 4-formylbenzoyl-sulfoNHS ester to convert exposed lysine residues to benzaldehyde groups (Fig. 1). The number of reactive benzaldehyde groups per virion was directly determined by reaction with excess 2-hydrazinopyridine and measuring the absorbance at 350 nm (ε=18,000 M-1cm-1), directly showing that an average of 280 of the 300 available sites on the CPMV particle were modified (Table 1). For subsequent conjugations, the level of excess substituent added to each reaction is expressed as “fold excess” over the number of attached benzaldehyde groups.
Table 1.
Attachment of substituents on CPMV. The number of substitutions per virus was measured by UV absorption of fluorescein at 495 nm with the exception of SFB, where the absorption was measured at 350 nm. For simplicity in the table PEG corresponds to PEG500f. For subsequent conjugations, the level of excess substituent added to each reaction is expressed as “fold excess” over the number of attached benzaldehydes.
| Reagents | Control C | SFB | PEG P1 |
PEG P2 |
PEG P3 |
PEG P4 |
F56f F1 |
F56f PEG FP1 |
F56f PEG FP2 |
F56f PEG FP3 |
|---|---|---|---|---|---|---|---|---|---|---|
| fold excess PEG/aldehyde | 10 | 20 | 50 | 100 | 20 | 50 | 100 | |||
| fold excess F56/aldehyde | 100 | 100 | 100 | 100 | ||||||
| fold excess fluorescein/aldehyde | 2 | |||||||||
| Substitutions per CPMV | 75 | 280 | 20 | 42 | 125 | 197 | 133 | 149 | 163 | 188 |
Unmodified CPMV particles are rapidly internalized by a wide variety of cell types in vitro and in vivo25-27. This uptake is governed primarily by binding to a cell-surface form of the intermediate filament vimentin 25, 28, and can be inhibited by PEGylation,3, 10, 29 a modification that also increases plasma circulation time and should enhance the accumulation of the nanoparticles in target tissues.30 For this purpose, PEG500f, a non-targeted peptide with a fluorescein, a monodisperse PEG polymer and a terminal hydrazido group, was synthesized (Fig. S2). PEG500f alone was incubated with benzaldehyde modified CPMV yielding 20 (P1), 42 (P2), 125 (P3) and 197 (P4) molecules of PEG500f per VNP particle, respectively (Fig. 2a, Table 1). Further analysis of CPMV-PEG500f conjugates by SDS-PAGE indicated a progressive loading of PEG500f onto the lysines of the large subunit (3) and the small subunit (2) (Fig. 2b and 2c). Using 100 fold-excess of PEG500f per benzaldehyde, an average of two lysines on L and one on S are linked to the fluorophore, in agreement with ∼200 fluorophores per viral particle (Fig. 2b).
Figure 2. CPMV conjugation with F56 peptide-fluorescein and PEG-fluorescein.
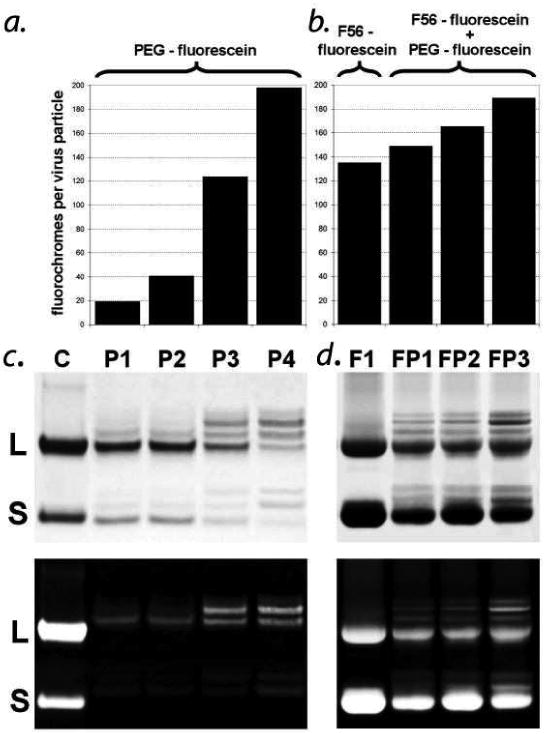
a. Relative fluorescence of PEG-conjugated CPMVs; P1, 10-fold excess; P2, 20-fold excess; P3, 50-fold excess; P4, 100-fold excess of PEG500f over benzaldehyde groups. b. Relative fluorescence of F56f and F56f/PEG500f mixed conjugated CPMVs; F1, 100-fold excess F56f; FP1, 100-fold excess F56f / 20fold excess PEG500f; FP2, 100- fold excess F56f / 50-fold excess PEG500f; FP3, 100-fold excess F56f / 100-fold excess PEG500f over benzaldehyde groups. c. SDS-PAGE of CPMV conjugates stained with Coomassie showing large (L) and small (S) subunits. d. SDS-PAGE of CPMV conjugates by UV transillumination. Lane labels in (b) correspond to those in (a) and (c).
The VEGFR-1-specific F56 peptide is extremely insoluble and had previously been studied only as a fusion protein with dihydrofolate reductase (DHFR).23 Four arginines (to improve solubility), a fluorescein and an N-terminal hydrazide were appended to form, hydrazido-K(fluorescein)RRGWHSDMEWWYLLGRR (F56f). This peptide was ligated to benzaldehyde-functionalized CPMV at pH 5.5 through a stable hydrazone linkage. A maximal loading of 133 peptides per particle (F1), as measured by fluorescein absorbance, was obtained (Fig. 2a and Table 1). The CPMV nanoparticles remained intact in the labeling procedure as monitored by gradient density ultracentrifugation and size exclusion chromatography. The high levels of labeling with the poorly soluble F56f peptide under ambient conditions demonstrates the utility of this hydrazone ligation approach for the functionalization of macromolecules and is comparable to the highest reported loadings of complex targeting molecules onto CPMV.31
Since only half of the benzaldehyde moieties on CPMV were modified by the F56f peptide ligand, we reasoned that sequential conjugation of F56f, followed by the more reactive PEG500f, would be possible (Fig. 1). Aliquots of the reaction mixture containing F56f-loaded CPMV (130/particle) were supplemented with a 20, 50 or 100 fold excess of PEG500f (over benzaldehyde groups) to generate doubly labeled FP particles. This procedure resulted in further loading of the CPMV-F56f conjugate with 16 (FP1), 30 (FP2) or 55 (FP3) PEG500f molecules as determined by fluorescein absorbance (Fig. 2a, Table 1). Analysis of these conjugates by SDS-PAGE indicated a progressive increase in loading of the CPMV surface with F56f peptide and PEG500f. Using this two-step approach, two peptides were loaded to achieve a total substitution level of over 180/virion. Despite the small Stokes shift of fluorescein, the densely labeled VNPs were highly fluorescent.32 Importantly, the modular nature of the synthetic procedures described here are compatible with other imaging moieties such as near infra-red fluorescent dyes.
To test the specificity of targeting, Ea.hy926 human endothelial cells that express high levels of VEGFR-133 and fibroblasts lacking VEGFR-1 expression 34 were treated with CPMV derivatives (Experimental details are given in the Supporting Information). Consistent with previous findings 35, cell attachment assays using fluorescent microscopy show that low levels of PEG500f (P1 and P2) did not affect the binding affinity of the particles to endothelial cells (Fig. 3a). In contrast, particles with high loading, P3 and P4, showed a 55% and 84% reduction in non-specific binding. The absence of signal in fluorescence imaging experiments with P4 confirmed minimal binding or internalization into endothelial cells (Figs. 3b, 4b).
Figure 3. Binding of CPMV conjugates to endothelial cellsin vitro.
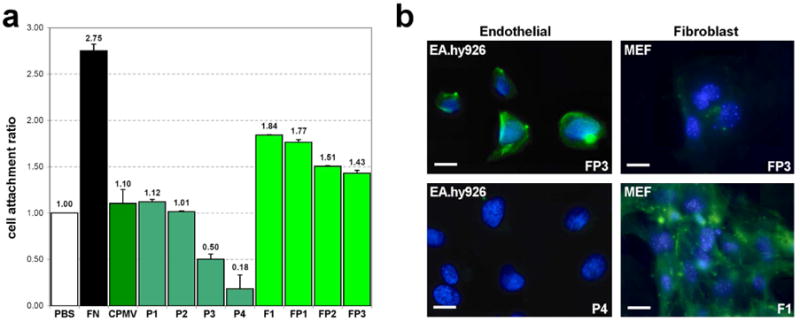
a. Cell adhesion assay with human endothelial cells (AE.hy926). Results are normalized to PBS control (y axis, Binding Efficiency: relative binding, normalized to PBS). Assay was performed in duplicate. Positive control: fibronectin (FN); negative control: native CPMV (CPMV); see Table 1 for an explanation of CPMV conjugates. b. Fluorescence visualization of CPMV conjugates FP3 (loaded with PEG500f and F56f), P4 (loaded with PEG500f only) and F1 (loaded with F55f only) bound to endothelial cells (EA.hy926) and fibroblasts (MEF). CPMV particles are shown in green. Nuclei were stained with DAPI (in blue).
Figure 4. Targeting CPMV to tumors in vivo.
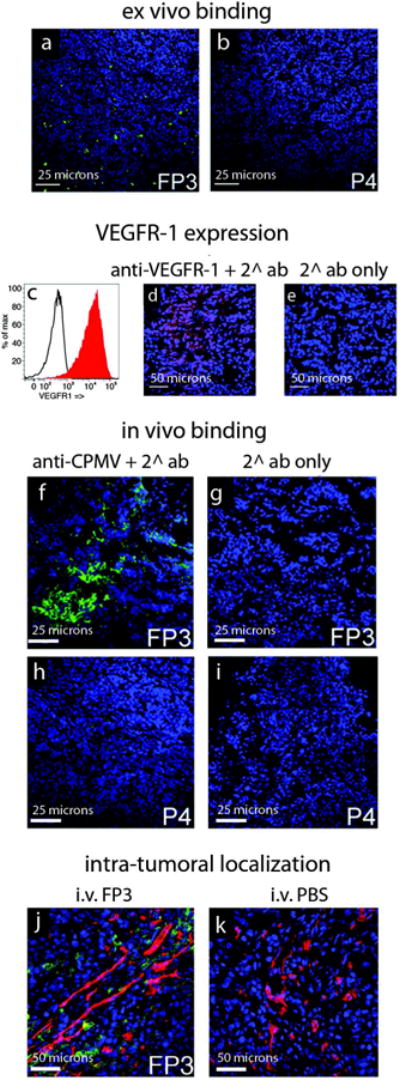
a,b. Confocal images showing that FP3 (a), but not P4 nanoparticles (b), bind to tumor tissue. CPMV was stained with fluorescently-labeled secondary antibodies. c. Expression of VEGFR-1 on HT-29 cells in vitro studied by flow cytometry. Red histogram = anti-VEGFR-1 and secondary antibody. Black histogram = secondary antibody only. d,e. HT-29 tumors stained ex vivo with anti-VEGFR-1 antibodies (d) or secondary antibody only (e). f-i. FP3 or P4 particles localize within HT-29 tumor xenografts following intravenous administration in nude/WEHI mice. CPMV was stained using specific antibodies. j,k. Intra-tumoral localization of particles (green) and endothelial cells (red) following intravenous administration of FP3 (j) or PBS (k). CPMV was stained using specific antibodies, and endothelial cells were stained using anti-CD31 antibody. Blue = DAPI stain to indicate cell nuclei.
As shown in Fig. 3a, F56f-ligand conjugated CPMV F1, bound to human endothelial cells 67% more efficiently than unlabeled virus. This binding was significantly reduced in the presence of free F56f peptide (Fig. S3). Fluorescence microscopy revealed that non-PEGylated F1 associated significantly with both endothelial cells and fibroblasts (Fig. 3b, bottom right). These results confirm that sufficient PEG500f loading is required to achieve specific targeting of VEGFR-1.
To further evaluate the specificity of PEGylated, VEGFR-1 targeted particles, the dual-labeled nanoparticle FP3 was visualized by confocal fluorescence microscopy. We found that the FP3 nanoparticles did not bind to fibroblasts (Fig. 3b top right) but bound VEGFR-1 expressing EA.hy926 endothelial cells in a specific manner (Fig. 3b top left, Fig. 4a). These results strongly suggest that VEGFR-1 peptide F56f can effectively target CPMV nanoparticles to endothelial cells, and that the addition of PEG significantly reduces non-specific binding.
We next evaluated the tumor specificity of targeted FP3 and untargeted P4 nanoparticles, by examining their ability to bind selectively to HT-29 (human colon carcinoma) tumor sections ex vivo. Using confocal microscopy imaging, significant binding to the tumor sections was observed with FP3 but not with P4 (Fig. 4a,b). Nanoparticles containing F56f-ligand but lacking PEG500f (F1) also bound the HT-29 tumor cells, but non-specific binding was also observed, as expected (data not shown).
To determine whether VEGFR-1-targeted CPMV nanoparticles could localize to tumors in vivo, nude/WEHI mice bearing HT-29 tumors, which express high levels of VEGFR-1 (Fig 4c-e), were inoculated intravenously with either FP3 or P4, and the particles were allowed to circulate for two hours before the tumors were excised. CPMV was detected by immunofluorescence staining and confocal microscopy (see Supporting Information for experimental details). No uptake was detected in tumors isolated from mice administered with P4 (Fig. 4h,i). In contrast, FP3 accumulated throughout the tumor tissue (Fig 4j,k). These results suggest that multifunctional nanoparticles can target tumors in vivo but do not necessarily accumulate on the endothelial cells lining the tumor vessels (Fig 4j). The somewhat unexpected extravasation of the targeted nanoparticles may be explained by the inherent leakiness of the tumor vasculature known as the enhanced permeability and retention effect (EPR) 36, as well as the expression of VEGFR-1 on both the endothelial and tumor cells.
In summary, we have developed a hydrazone ligation chemical strategy to generate a multivalent CPMV nanoparticle that specifically targets VEGFR-1. We developed targeted VNPs comprised of 133 copies of VEGFR-1 ligand, 55 copies of a PEGylated peptide, and a total of 188 fluorescent dyes. Importantly, the hydrazone ligation strategy enables the sequential introduction of different peptides in a ratio that is controlled by the reaction conditions, and the modular nature of the chemistry can be tailored for specific applications. For example, in order to use these nanoparticles for in vivo imaging it will be necessary to use an alternate imaging moiety with improved tissue penetration characteristics. The hydrazone ligation strategy enables design of these uniquely multifunctional CPMV nanoparticles, which may be further developed into powerful tools for the detection and the monitoring of treatment of cancer and other diseases.
Supplementary Material
Acknowledgments
This study was supported by NIH grants P01-HD09402 to P.E.D.; R01 HL 068648 to H.S.; R01 CA112075 to M.M., K99 EB009105 to N.F.S.; as well as CIHR MOP-84535 to J.D.L., American Heart Association Postdoctoral Fellowship (to N.F.S.), and AmfAR fellowship to F.B.
Footnotes
Supporting Information Available. Experimental details and supporting Figures are given. The information is available free of charge at http://pubs.acs.org.
References
- 1.Ntziachristos V, Ripoll J, Wang LV, Weissleder R. Nat Biotechnol. 2005;23(3):313–20. doi: 10.1038/nbt1074. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Lin T, Johnson JE, Finn MG. Chem Biol. 2002;9(7):813–9. doi: 10.1016/s1074-5521(02)00166-7. [DOI] [PubMed] [Google Scholar]
- 3.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Nat Med. 2006;12(3):354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin T, Chen Z, Usha R, Stauffacher CV, Dai JB, Schmidt T, Johnson JE. Virology. 1999;265(1):20–34. doi: 10.1006/viro.1999.0038. [DOI] [PubMed] [Google Scholar]
- 5.Chatterji A, Ochoa W, Paine M, Ratna BR, Johnson JE, Lin T. Chem Biol. 2004;11(6):855–63. doi: 10.1016/j.chembiol.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Chatterji A, Ochoa WF, Paine M, Ratna BR, Johnson JE, Lin TW. Chemistry & Biology. 2004;11(6):855–863. doi: 10.1016/j.chembiol.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Steinmetz NF, Lomonossoff GP, Evans DJ. Langmuir. 2006;22(8):3488–90. doi: 10.1021/la060078e. [DOI] [PubMed] [Google Scholar]
- 8.Steinmetz NF, Evans DJ. Org Biomol Chem. 2007;5(18):2891–902. doi: 10.1039/b708175h. [DOI] [PubMed] [Google Scholar]
- 9.Brown WL, Mastico RA, Wu M, Heal KG, Adams CJ, Murray JB, Simpson JC, Lord JM, Taylor-Robinson AW, Stockley PG. Intervirology. 2002;45(4-6):371–80. doi: 10.1159/000067930. [DOI] [PubMed] [Google Scholar]
- 10.Destito G, Yeh R, Rae CS, Finn MG, Manchester M. Chem Biol. 2007;14(10):1152–62. doi: 10.1016/j.chembiol.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren Y, Wong SM, Lim LY. Bioconjug Chem. 2007;18(3):836–43. doi: 10.1021/bc060361p. [DOI] [PubMed] [Google Scholar]
- 12.Shriver LP, Koudelka KJ, Manchester M. Journal of Neuroimmunology. 2009 doi: 10.1016/j.jneuroim.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinmetz NF, Hong V, Spoerke ED, Lu P, Breitenkamp K, Finn MG, Manchester M. J Am Chem Soc. 2009;131(47):17093–5. doi: 10.1021/ja902293w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinmetz NF, Mertens ME, Taurog RE, Johnson JE, Commandeur U, Fischer R, Manchester M. Nano Lett. 2009 doi: 10.1021/nl9035753. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suci PA, Varpness Z, Gillitzer E, Douglas T, Young M. Langmuir. 2007;23(24):12280–6. doi: 10.1021/la7021424. [DOI] [PubMed] [Google Scholar]
- 16.Kargiotis O, Rao JS, Kyritsis AP. J Neurooncol. 2006;78(3):281–93. doi: 10.1007/s11060-005-9097-6. [DOI] [PubMed] [Google Scholar]
- 17.Backer MV, Levashova Z, Patel V, Jehning BT, Claffey K, Blankenberg FG, Backer JM. Nat Med. 2007;13(4):504–9. doi: 10.1038/nm1522. [DOI] [PubMed] [Google Scholar]
- 18.Cai W, Chen K, Mohamedali KA, Cao Q, Gambhir SS, Rosenblum MG, Chen X. J Nucl Med. 2006;47(12):2048–56. [PubMed] [Google Scholar]
- 19.Lee TH, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, Avraham S. PLoS Med. 2007;4(6):e186. doi: 10.1371/journal.pmed.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamaguchi T, Bando H, Mori T, Takahashi K, Matsumoto H, Yasutome M, Weich H, Toi M. Cancer Sci. 2007;98(3):405–10. doi: 10.1111/j.1349-7006.2007.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uesaka T, Shono T, Suzuki SO, Nakamizo A, Niiro H, Mizoguchi M, Iwaki T, Sasaki T. J Neurooncol. 2007;83(3):259–66. doi: 10.1007/s11060-007-9336-0. [DOI] [PubMed] [Google Scholar]
- 22.Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA, Testa JE, Schnitzer JE. Nature. 2004;429(6992):629–35. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- 23.An P, Lei H, Zhang J, Song S, He L, Jin G, Liu X, Wu J, Meng L, Liu M, Shou C. Int J Cancer. 2004;111(2):165–73. doi: 10.1002/ijc.20214. [DOI] [PubMed] [Google Scholar]
- 24.Reddy VS, Natarajan P, Okerberg B, Li K, Damodaran KV, Morton RT, Brooks CL, 3rd, Johnson JE. J Virol. 2001;75(24):11943–7. doi: 10.1128/JVI.75.24.11943-11947.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G, Manchester M. PLoS Pathog. 2009;5(5):e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Nat Med. 2006;12(3):354–60. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez MJ, Plummer EM, Rae CS, Manchester M. PLoS One. 2009;4(11):e7981. doi: 10.1371/journal.pone.0007981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koudelka KJ, Rae CS, Gonzalez MJ, Manchester M. J Virol. 2007;81(4):1632–40. doi: 10.1128/JVI.00960-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinmetz NF, Manchester M. Biomacromolecules. 2009;10(4):784–92. doi: 10.1021/bm8012742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okuda T, Kawakami S, Akimoto N, Niidome T, Yamashita F, Hashida M. J Control Release. 2006;116(3):330–6. doi: 10.1016/j.jconrel.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 31.Sen Gupta S, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Bioconjug Chem. 2005;16(6):1572–9. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 32.Soto CM, Blum AS, Vora GJ, Lebedev N, Meador CE, Won AP, Chatterji A, Johnson JE, Ratna BR. J Am Chem Soc. 2006;128(15):5184–9. doi: 10.1021/ja058574x. [DOI] [PubMed] [Google Scholar]
- 33.Fu JX, Wang W, Bai X, Wang L, Zhu ZL, Chen ZX, Ruan CG. Ai Zheng. 2002;21(11):1217–21. [PubMed] [Google Scholar]
- 34.Morishita K, Johnson DE, Williams LT. J Biol Chem. 1995;270(46):27948–53. doi: 10.1074/jbc.270.46.27948. [DOI] [PubMed] [Google Scholar]
- 35.Steinmetz NF, Manchester M. Biomacromolecules. 2009;10(4):784–92. doi: 10.1021/bm8012742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iyer AK, Khaled G, Fang J, Maeda H. Drug Discov Today. 2006;11(17-18):812–8. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.