

Abstract
Paracetamol (acetaminophen) is a widely used analgesic drug. It interacts with various enzyme families including cytochrome P450 (CYP), cyclooxygenase (COX), and nitric oxide synthase (NOS), and this interplay may produce reactive oxygen species (ROS). We investigated the effects of paracetamol on prostacyclin, thromboxane, nitric oxide (NO), and oxidative stress in four male subjects who received a single 3 g oral dose of paracetamol. Thromboxane and prostacyclin synthesis was assessed by measuring their major urinary metabolites 2,3-dinor-thromboxane B2 and 2,3-dinor-6-ketoprostaglandin F1α, respectively. Endothelial NO synthesis was assessed by measuring nitrite in plasma. Urinary 15(S)-8-iso-prostaglanding F2α was measured to assess oxidative stress. Plasma oleic acid oxide (cis-EpOA) was measured as a marker of cytochrome P450 activity. Upon paracetamol administration, prostacyclin synthesis was strongly inhibited, while NO synthesis increased and thromboxane synthesis remained almost unchanged. Paracetamol may shift the COX-dependent vasodilatation/vasoconstriction balance at the cost of vasodilatation. This effect may be antagonized by increasing endothelial NO synthesis. High-dosed paracetamol did not increase oxidative stress. At pharmacologically relevant concentrations, paracetamol did not affect NO synthesis/bioavailability by recombinant human endothelial NOS or inducible NOS in rat hepatocytes. We conclude that paracetamol does not increase oxidative stress in humans.
1. Introduction
Nitric oxide (NO), prostaglandin (PG) I2, that is, prostacyclin (PGI2), and thromboxane A2 (TxA2) are important short-lived signaling molecules involved in many physiological and pathological processes. Thus, PGI2 and NO are potent vasodilators and inhibitors of platelet aggregation. Contrarily, TxA2 is a strong vasoconstrictor and inductor of platelet aggregation. NO is synthesized from L-arginine (Arg) by constitutive and inducible NO synthase (NOS) isoforms. Prostaglandin H synthase (PGHS) isoforms, generally termed cyclooxygenase (COX), convert arachidonic acid (AA) to the collectively named prostanoids. The L-arginine/NO pathway is generally accepted to interact with the COX pathway and to modulate its activity [1–4]. For instance, the inducible NOS (iNOS) isoform has been shown to bind to the inducible COX isoform (COX-2) and to S-nitrosylate and activate COX-2 [2]. The role of NO in prostaglandin biology has been recently updated by Kim [4]. Potential mechanisms of direct NOS-COX cross-talk may include (1) binding of NO to the iron atom of the heme group of COX, (2) reaction of the nitrosyl cation (NO+) with sulfhydryl (SH) groups of cysteine (Cys) moieties of COX to form S-nitroso-COX, and (3) reaction of peroxynitrite (ONOO−), that is, the reaction product of NO radical (∙NO) and superoxide radical anion (O2 ∙−) produced either by NOS itself or by other enzymes including COX and CYP [3], with SH groups of Cys residues or with tyrosine (Tyr) residues of COX being involved in the catalytic process [2]. S-Nitrosylation of COX-Cys moieties by higher oxides of NO, notably dinitrogen trioxide (N2O3), and by ONOO− and S-transnitrosylation of COX-Cys moieties by low-molecular-mass S-nitrosothiols have been shown to both enhance and inhibit COX activity. Nitration of Tyr residues located in the catalytic domain of COX is assumed to inhibit COX activity [2, 4–6]. On the other hand, ONOO− has been reported to enhance COX activity presumably by increasing the peroxide concentration that is required for the peroxidase activity of COX [7].
Paracetamol (acetaminophen, APAP) is one of the most frequently applied drugs worldwide and is considered generally a safe analgesic and antipyretic drug in therapeutic dosage, which lacks however appreciable anti-inflammatory and antiplatelet activity [10]. The mechanism of the analgesic and antipyretic effects of paracetamol is not fully established, yet inhibition of PGHS activity by paracetamol in different cell and tissue types is generally assumed to be the main mode of paracetamol's analgesic and antipyretic action. PGHS possesses both peroxidase and cyclooxygenase activity. Paracetamol is believed to inhibit the peroxidase catalytic site of PGHS, unlike the majority of nonsteroidal anti-inflammatory drugs (NSAIDs) and the PGHS2 inhibitors. In vitro, paracetamol is a much stronger inhibitor of prostanoid synthesis in endothelial cells than in platelets. In particular, paracetamol is a weak inhibitor of TxA2 synthesis in platelets. It is also remarkable that the inhibitory potency of paracetamol is inversely correlated with the PGHS concentration (for a review, see [10]). These particular characteristics distinguish paracetamol from NSAIDs including acetylsalicylic acid (ASA).
In vitro, PGHS activity can be assessed by measuring the production rate of various primary prostanoids, such as PGE2, PGI2, and TxA2. Because of the remarkable chemical instability of PGI2 and TxA2, their stable hydrolysis products, that is, 6-keto-PGF1α and TxB2, respectively, are measured instead of PGI2 and TxA2 [11]. In vivo, measurement of PGE2, 6-keto-PGF1α, and TxB2 in plasma is associated with artefactual prostanoid synthesis during blood sampling and may lead to incorrect conclusions with regard to PGHS activity [12]. This especially applies to TxA2 which is produced in high amounts in activated platelets [13]. Measurement of PGE2 in the urine reflects renal PGE2 synthesis. By far more reliable is the measurement of major urinary metabolites of prostanoids, such as 2,3-dinor-TxB2 for TxA2, 2,3-dinor-6-keto-PGF1α for PGI2, and the major urinary metabolite of PGE2 (PGE-MUM) for systemic PGE2 production [11]. This can be best accomplished by means of analytical technologies which have high inherent sensitivity and selectivity such as gas chromatography-mass spectrometry (GC-MS) and more so gas chromatography-tandem mass spectrometry (GC-MS/MS) (for a review, see [11]).
Recently, Sudano and colleagues [9] reported that paracetamol (1 g TID for 2 weeks on top of standard cardiovascular therapy) increased ambulatory mean systolic and diastolic blood pressure by about 3 and 2 mmHg, respectively, without changing endothelium and platelet function in patients with coronary artery disease (CAD). Sudano et al. [9] concluded that, particularly in patients at increased cardiovascular risk, use of paracetamol should be evaluated as rigorously as traditional NSAIDs and selective COX2 inhibitors. In that study, plasma and urine PGE2 as well as plasma TxB2 did not change upon paracetamol administration [9]. However, as mentioned above, measurement of PGE2 and TxB2 in plasma is prone to artefactual prostanoid synthesis [12, 13], whereas measurement of PGE2 in the urine does not provide information about PGHS-catalyzed synthesis of the two antagonists TxA2 and PGI2 [11].
In humans, oral administration of paracetamol (500 mg) has been reported not to result in decreased excretion rate of 2,3-dinor-TxB2, unlike aspirin (500 mg) or indomethacin (50 mg), as measured by GC-MS [14]. Also, in contrast to aspirin (3 g for 2 days), oral administration of paracetamol (3 g for 2 days) has been reported not to reduce urinary excretion of PGE2 but to weakly reduce PGE-MUM excretion indicating inhibition of systemic PGE2 synthesis [15]. On the other hand, a single oral dose of 500 mg paracetamol has been shown to reduce urinary excretion rate of 2,3-dinor-6-keto-PGF1α for 6–8 h by maximally 60% (i.e., inhibition of PGI2 synthesis), without reducing urinary excretion rate of 2,3-dinor-TxB2 (i.e., no inhibition of TxA2 synthesis) [16]. The results of these in vivo studies in human subjects suggest that orally administered paracetamol, at a single dose of 500 mg or at a cumulative dose of 3000 mg per day, does not inhibit remarkably TxA2 synthesis, but it may temporarily inhibit PGI2 synthesis.
The ramifications between NOS and COX pathways have been frequently investigated in the past (reviewed in [4]), but results are inconsistent. For instance, in murine macrophages, paracetamol, at pharmacologically relevant plasma concentrations (60–120 μM), has been reported not to affect iNOS activity [17]. At suprapharmacological concentrations (2, 5, and 10 mM), paracetamol has been reported to inhibit iNOS gene expression and iNOS activity in RAW 264.7 cell line macrophages [18]. By contrast, paracetamol (up to 10 mM) has been reported not to affect neuronal NOS (nNOS) and iNOS activity in rat cerebellum and HUVECs [19]. Others have reported that paracetamol (100 μM) did not affect nNOS activity in cerebellum but inhibited NOS activity in murine spinal cord slices as measured by the radiolabelled L-citrulline assay [20]. The effect of paracetamol on in vivo in humans is elusive.
Because paracetamol, when applied at pharmacological doses, inhibits the synthesis of the vasodilatatory and antiaggregatory PGI2 much stronger and sustainably than the synthesis of the vasoconstrictory and thrombogenic TxA2 in humans, we wondered whether the paracetamol-induced shifting of the balance between vasodilatatory/antiaggregatory and vasoconstrictory/thrombogenic COX-related homeostasis may induce processes that lead to enhanced synthesis of the vasodilatatory/antiaggregatory NO, thus counteracting blood pressure fall and platelet activation. Preliminary investigations of our group showed that paracetamol, administered in therapeutic doses to healthy humans (up to 10 mg/kg), did not change whole body NO synthesis (data not shown), suggesting that a potential effect of paracetamol on NOS activity is likely to require much higher, suprapharmacological doses of this drug. In consideration of the toxicological potency of high paracetamol doses, we investigated the effects of a single oral 3 g dose in four healthy volunteers. To our knowledge, the effects of such a high single oral dose of paracetamol on PGI2, TxA2, and NO synthesis in humans have not been investigated so far. Because of the high dose used in the human study, paracetamol may induce oxidative stress and decrease NO bioavailability [21]. We therefore measured the oxidative stress biomarker 15(S)-8-iso-PGF2α [22] in plasma and urine. Nitrite in plasma was measured as a biomarker of NO synthesis and bioavailability (reviewed in [23]). In addition, we performed in vitro studies on recombinant endothelial NOS (eNOS) and inducible NOS (iNOS) in rat hepatocytes to test potential effects of paracetamol on NO synthesis and bioavailability.
2. Materials and Methods
2.1. Subjects and Study Performance
Four healthy nonsmoking male adults (aged 39, 40, 44, and 64 years) participated in the study and gave their informed consent to the study. The volunteers received orally six 500 mg paracetamol tablets (Ratiopharm) at once. Dosage was each 29 mg/kg for volunteer A and volunteer B, 37 mg/kg for volunteer C, and 52 mg/kg for volunteer D. Volunteers were not fasting but they did not eat in the first three hours following paracetamol administration. Before and after paracetamol administration, venous blood and urine were collected in 30 and 60 min intervals over an observation period of 6 h for analysis of biochemical parameters as described below. Venous blood (8 mL) was drawn by using 9 mL EDTA vacutainers (Sarstedt, Germany) and centrifuged immediately (800 ×g, 4°C, 5 min). Plasma was decanted, portioned in 0.1 and 1.0 mL aliquots as required for each biochemical parameter, and stored frozen at −80°C until analysis. Urine from spontaneous micturition was collected in 45 mL polypropylene tubes, aliquoted in 0.1 and 1.0 mL portions according to the requirement of the individual biochemical parameters, and stored at −20°C until analysis.
2.2. Analysis of Biochemical Parameters in the Human Study
All samples of this study were analyzed within 10 days after collection. In GC-MS and GC-MS/MS methods, stable-isotope labelled analogs were used as internal standards as reported in the respective references cited below. We found that paracetamol added to pooled human plasma at concentrations of 10, 25, 50, 75, and 100 mg/L did not interfere with the analysis of the biochemical parameters measured in the study plasma samples (data not shown). Data from this study are reported as mean ± standard error of the mean (SEM).
2.2.1. Measurement of Paracetamol
Plasma paracetamol concentration was determined by reverse phase HPLC (250 × 4 mm i.d., 5 μm particle size) with isocratic elution (mobile phase: 45 mM ammonium sulphate-acetonitrile, 10 : 1, v/v; flow rate: 1 mL/min) with UV absorbance detection at 236 nm.
2.2.2. Measurement of Prostanoids and Creatinine
PGI2 and TxA2 synthesis was assessed by GC-MS/MS by measuring in 1 mL urine aliquots the respective major urinary metabolites [11], that is, 2,3-dinor-6-keto-PGF1α and 2,3-dinor-TxB2, exactly as described elsewhere [24]. PGE2 and free nonconjugated 15(S)-8-iso-PGF2α in urine (1 mL) and free 15(S)-8-iso-PGF2α in plasma (1 mL) were measured by GC-MS/MS after extraction by immunoaffinity column chromatography as described previously [25]. Urinary excretion rate of the eicosanoids was corrected for creatinine excretion [11] and is expressed in nmol prostanoid/mol creatinine. Urine creatinine was measured in 10 μL urine aliquots by GC-MS as reported elsewhere [26].
2.2.3. Analysis of the L-Arginine/NO Pathway
Nitrite and nitrate were measured simultaneously in 100 μL aliquots of plasma or urine by GC-MS as described elsewhere [27]. Urinary excretion rate of nitrite and nitrate was corrected for creatinine excretion as well. Arginine and the endogenous NOS activity inhibitor asymmetric dimethylarginine (ADMA) were measured by GC-MS and GC-MS/MS, respectively, in 100 μL aliquots of ultrafiltrate obtained from plasma by centrifugation according to previously reported procedures [8].
2.2.4. Additional Analyses
Total homocysteine (hCys) in plasma (0.1 mL) was measured by a commercially available fluorescence polarimetry immunoassay (FPIA). In vivo CYP activity [28] was assessed by measuring oleic acid oxide (cis-EpOA) in 1 mL aliquots of plasma as described elsewhere [29].
2.2.5. Quality Control
Quality control (QC) samples were analyzed alongside study samples for all biochemical parameters. Accuracy and precision in the QC samples were within generally accepted ranges; that is, bias and imprecision levels were below 20%.
2.3. Effect of Paracetamol on Recombinant Human eNOS Activity
The effect of paracetamol on NOS activity in vitro was investigated by using a commercially available (ALEXIS, Grünberg, Germany) recombinant human endothelial NOS (heNOS) and by measuring simultaneously formation of [15N]nitrite and [15N]nitrate from L-[guanidine- 15N2]arginine by means of a GC-MS assay [30]. Incubations were performed at 37°C in 50 mM potassium phosphate buffer (1000 μL, pH 7) containing heNOS (50 μg/mL), L-[guanidine-15N2]-arginine (20 μM, Cambridge Isotope Labs, Andover, MA, USA), and all NOS cofactors (all purchased from Sigma-Aldrich, Steinheim, Germany) and prosthetic groups (10 μM tetrahydrobiopterin, 800 μM NADPH, 5 μM FAD, 5 μM for FMN, 500 nM calmodulin, and 500 μM CaCl2 (Merck, Darmstadt, Germany)). Reactions were terminated by addition of 400 μL aliquots of ice cold acetone and samples were processed for GC-MS analysis of [15N]nitrite and [15N]nitrate. Unlabeled nitrite and nitrate were used as internal standards for [15N]nitrite and [15N]nitrate, respectively. Data are shown as mean ± standard deviation (SD) from two independent experiments.
2.4. Effect of Paracetamol on iNOS Activity in Cultured Rat Hepatocytes Proliferating In Vitro
The effect of paracetamol on iNOS activity was investigated in primary rat hepatocytes proliferating in vitro as described recently by measuring formation of [15N]nitrite and [15N]nitrate by GC-MS [31]. In some experiments, LiCl (10 mM) was used to enhance expression of iNOS-mRNA and cell growth [31]. Incubations were performed at 37°C in the presence of 5 mM L-[guanidine-15N2]-arginine added at the time -22 h. Reactions were terminated by addition of 400 μL aliquots of ice cold acetone, and samples were processed for GC-MS analysis of [15N]nitrite and [15N]nitrate. Data are shown as mean ± SD from three independent experiments.
2.5. Statistical Analysis
Because of considerable differences in the baseline concentrations of some of the biochemical parameters measured in the four subjects, changes and statistical significance were calculated by setting the respective baseline levels to 100%. Statistical significance (P < 0.05) was evaluated by using unpaired t-test and comparing the data obtained at various times to the baseline values or to the 0.5 h values when percentage changes were compared.
3. Results
3.1. Effect of High-Dose Paracetamol on COX, NOS, and CYP Activity and on Oxidative Stress in Humans
Mean maximum paracetamol plasma concentration (C max) was 30.2 mg/L (200 μmol/L) (Figure 1). This value is consistent with a C max value of about 20 mg/L that has been reached after oral administration of 2000 mg of paracetamol [32]. In the urine samples, we measured by reverse phase HPLC with UV absorbance detection comparable creatinine-corrected excretion rates of the paracetamol glucuronide and sulphate metabolites (data not shown).
Figure 1.
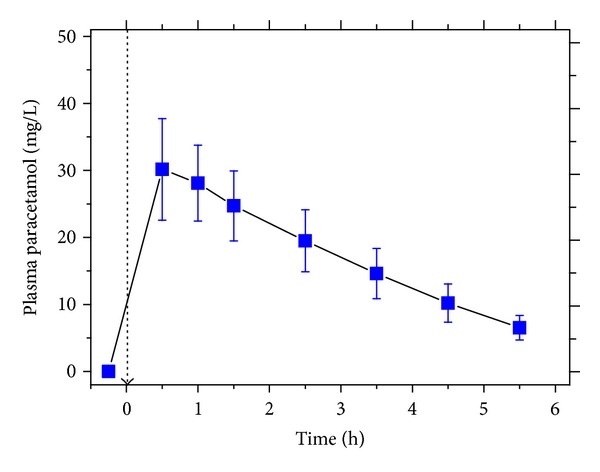
Plasma concentration of paracetamol before and after administration (time zero is indicated by the dashed arrow) of a single oral 3 g dose of paracetamol to four male subjects. Data are shown as mean ± SEM.
Upon 3 g paracetamol intake, considerable and sustained decrease in creatinine-corrected 2,3-dinor-6-keto-PGF1α excretion rate was seen, suggesting strong PGI2 inhibition by paracetamol (Figure 2(a)). Maximum and statistically significant PGI2 inhibition of about 60 to 70% was reached 1 h, 1.5 h, and 2.5 h after paracetamol administration. The extent of the decrease seen in the 2,3-dinor-6-keto-PGF1α excretion rate in the present study is comparable to that seen upon administration of a 500 mg oral paracetamol dose [14]. Paracetamol caused only a moderate, statistically insignificant decrease in 2,3-dinor-TxB2 excretion in the four volunteers (Figure 2(b)). In three out of the four volunteers, maximum TxA2 inhibition of about 70% was reached 1.5 h after administration, but the duration of TxA2 inhibition was relatively short (not shown). Figure 2(c) shows that the PGI2/TxA2 molar ratio decreased by a factor of 2 to 3 upon paracetamol administration, although statistical significance failed by a hair 1.5 h (P = 0.069) and 2.5 h (P = 0.056) after paracetamol ingestion. Paracetamol seemed to decrease very weakly the excretion of PGE2 in the urine (Figure 2(d)), suggesting that even 3 g of paracetamol taken at once is not able to inhibit renal synthesis of PGE2 in the four volunteers enrolled in the study.
Figure 2.
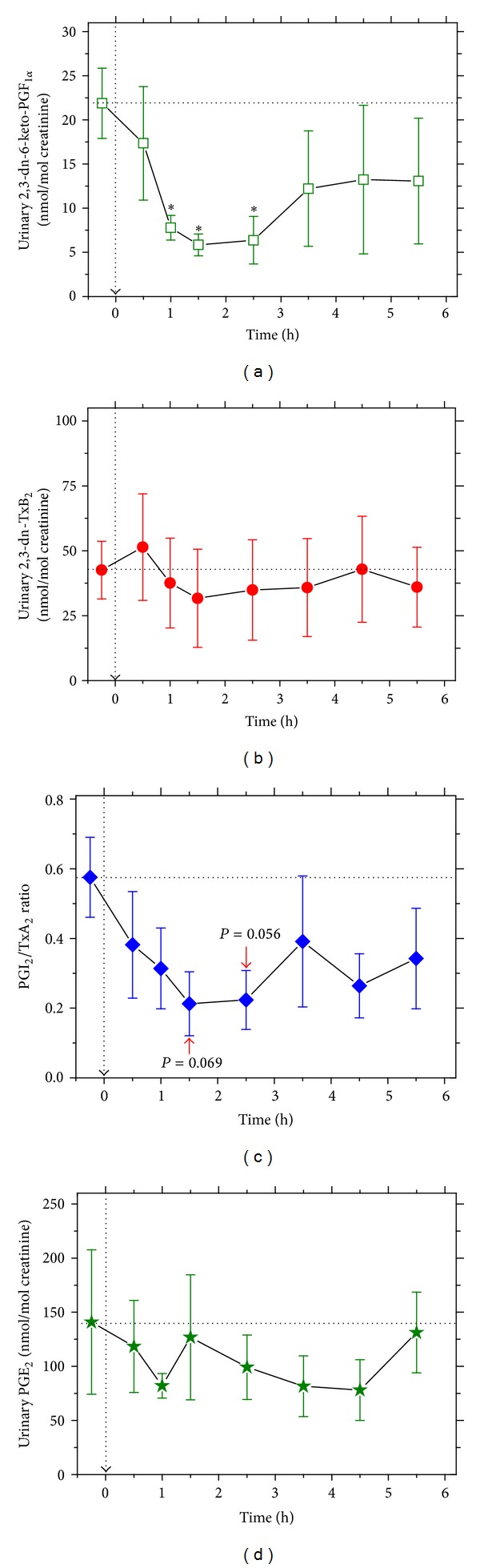
Effect of a single oral 3 g dose of paracetamol on systemic prostacyclin and thromboxane synthesis and on renal synthesis of PGE2 in four healthy volunteers (time zero and baseline values are indicated by dashed arrows). (a) Creatinine-corrected urinary excretion of 2,3-dinor-6-keto-prostaglandin F1α (2,3-dn-6k-PGF1α) as a measure of systemic PGI2 synthesis. (b) Creatinine-corrected urinary excretion of 2,3-dinor-thromboxane B2 (2,3-dn-TxB2) as a measure of systemic TxA2 synthesis. (c) PGI2/TxA2 molar ratio calculated from the 2,3-dn-6k-PGF1α and 2,3-dn-TxB2 excretion rates shown in (a) and (b), respectively. (d) Creatinine-corrected urinary excretion of PGE2 as a measure of renal PGE2 synthesis. An asterisk in (a) indicates statistical significance (P < 0.05) compared to basal values. Data are shown as mean ± SEM.
Paracetamol-induced changes in systemic prostacyclin synthesis were not accompanied by noteworthy changes in the plasma concentration of total hCys (Figure 3(a)) or in the urinary excretion rate of free 15(S)-8-iso-PGF2α (Figure 3(b)), suggesting no elevation or reduction of oxidative stress upon high-dosed paracetamol administration.
Figure 3.
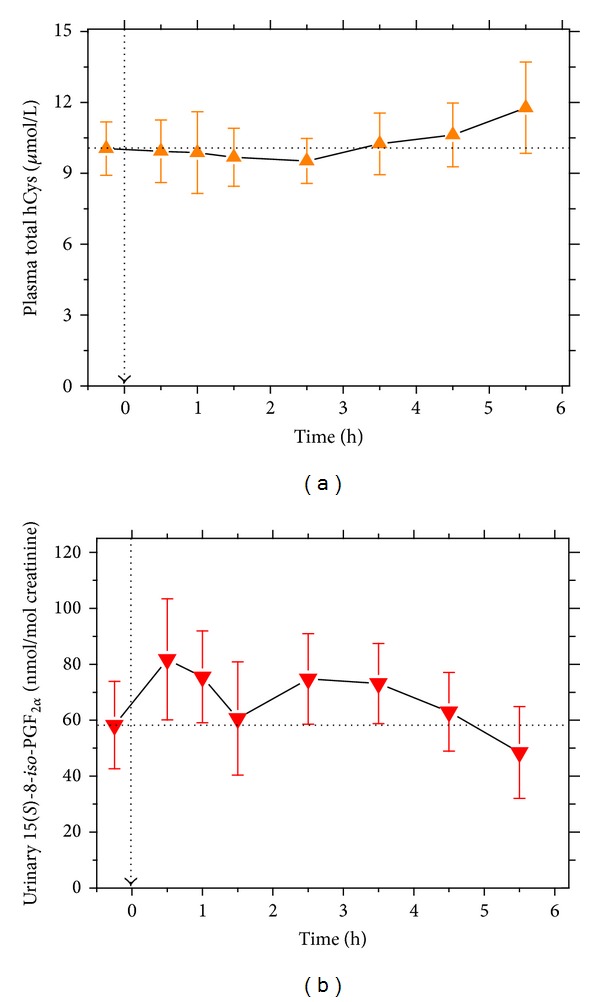
Effect of a 3 g single oral dose of paracetamol on oxidative stress in four healthy volunteers (time zero and baseline value are indicated by dashed arrows). (a) Plasma total homocysteine (hCys) concentration and (b) creatinine-corrected urinary excretion rate of 15(S)-8-iso-prostaglandin F2α (15(S)-8-iso-PGF2α) as a measure of oxidative stress. Data are shown as mean ± SEM.
Because of the considerable difference in the baseline plasma nitrite and nitrate concentrations measured in the four subjects, changes in plasma nitrite and nitrate were calculated and presented as percentage of the respective baseline levels. Figure 4(a) shows moderate increases in plasma nitrite concentration which were statistically significantly higher when the 2.5 and 3.5 h values were compared with the 0.5 h values. Changes in plasma nitrate concentrations (Figure 4(b)) and urine nitrite (Figure 4(c)) and urine nitrate (Figure 4(d)) excretion were not statistically significantly different. Finally, plasma Arg and ADMA concentrations did not change upon paracetamol ingestion (Figure 4(e)).
Figure 4.
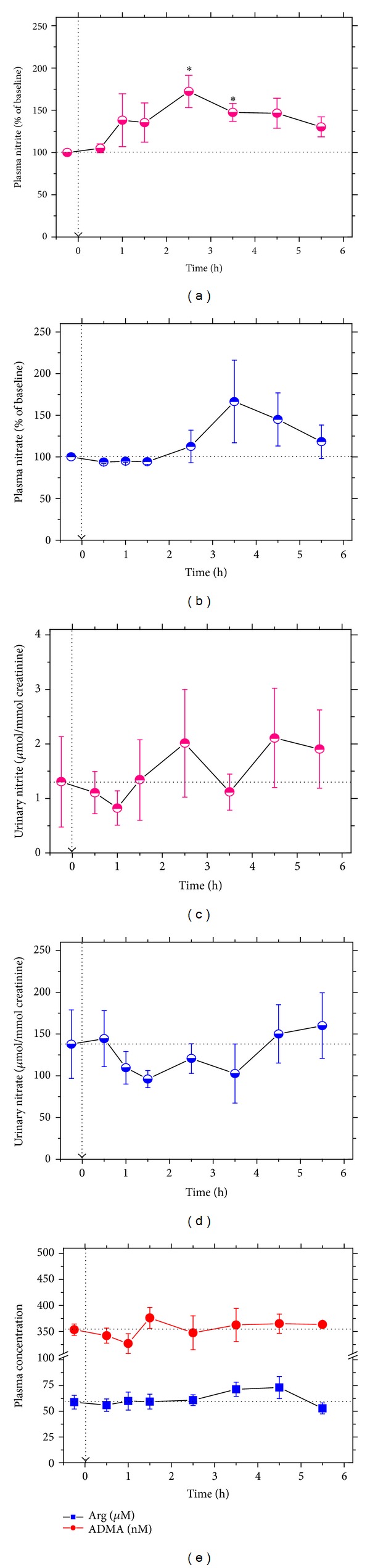
Effect of a 3 g single oral dose of paracetamol on plasma (a) and urine (c) nitrite, plasma (b) and urine (d) nitrate, and plasma arginine (Arg) and asymmetric dimethylarginine (ADMA) (e) in four healthy volunteers (time zero and baseline values are indicated by dashed lines). Data in plasma are shown as percentage changes of the baseline plasma nitrite concentrations (1.26, 3.04, 3.76, and 4.05 μM) and baseline plasma nitrate concentrations (37.3, 42.5, 26.8, and 52.9 μM), respectively. Data are shown as mean ± SEM. An asterisk indicates statistical significance (P < 0.05) compared to the 0.5 h values.
Previously, we showed that the whole body activity of CYP isoforms can be assessed by measuring the concentration of the free, that is, nonesterified, oleic acid oxide cis-EpOA in plasma [28]. Figure 5 shows an abrupt increase in mean plasma cis-EpOA concentration 2.5 h after paracetamol administration followed by an abrupt fall to baseline level 1 h later. This finding may suggest a very short-term paracetamol-induced elevation of CYP activity. However, we also found a very similar change in the plasma concentration of free 15(S)-8-iso-PGF2α (Figure 5). Previously, we observed that addition of phospholipase A2 (PLA2) to human serum increased in parallel the concentration of both free cis-EpOA and free 15(S)-8-iso-PGF2α [25]. Therefore, the temporary short-time increases in cis-EpOA and 15(S)-8-iso-PGF2α seen 2.5 h after paracetamol administration may have resulted from release of presumably hepatic PLA2 into the blood.
Figure 5.
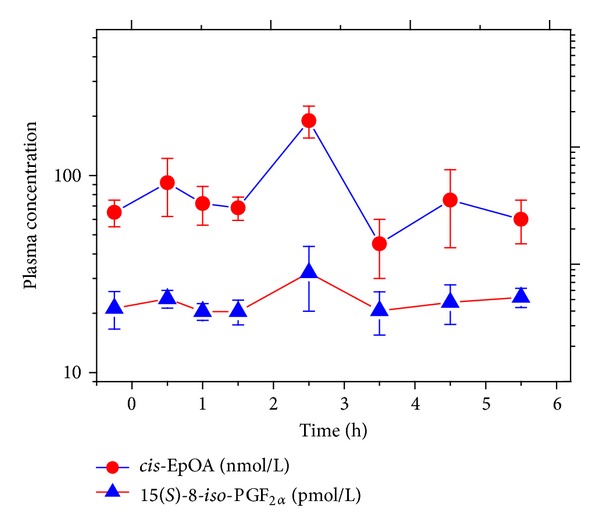
Effect of a 3 g single oral dose of paracetamol on serum cis-epoxyoctadecanoic acid (cis-EpOA) and 15(S)-8-iso-PGF2α in four healthy volunteers (time zero and baseline value are indicated by dashed lines). Data are shown as mean ± SEM. Note the logarithmic scale on the y-axis. Only the 2.5 h concentration of cis-EpOA was statistically significantly different (P < 0.05) compared to the baseline.
3.2. Effects of Paracetamol on Recombinant heNOS Activity and iNOS in Rat Hepatocytes
At the therapeutically relevant concentration of 100 μM (i.e., 15 mg/L), paracetamol had only a weak effect on the formation of [15N]nitrite and [15N]nitrate in incubation mixtures of a recombinant heNOS (Figure 6). Linear regression of analysis between the concentrations of [15N]nitrite and [15N]nitrate in the presence of paracetamol versus the concentrations of [15N]nitrite and [15N]nitrate in the absence of paracetamol revealed a slope value of 0.834 (Figure 6(b)). This finding suggests that paracetamol inhibited heNOS-catalyzed 15NO formation (i.e., [15N]nitrite and [15N]nitrate) from L-[guanidine-15N2]-arginine in average by 16.6%, notably for incubation times longer than 10 min. Similar small effects of paracetamol on iNOS were also seen in experiments with adult rat hepatocytes proliferating in vitro independent of the presence of LiCl (Figure 7). It is well established that peroxynitrite can nitrate paracetamol to 3-nitroparacetamol [33]. In the paracetamol-containing samples from both the recombinant heNOS and the rat hepatocytes iNOS, no 3-[15N]nitroparacetamol was detected by GC-MS/MS above the limit of quantitation (about 1 nM), suggesting no formation of peroxynitrite (data not shown).
Figure 6.
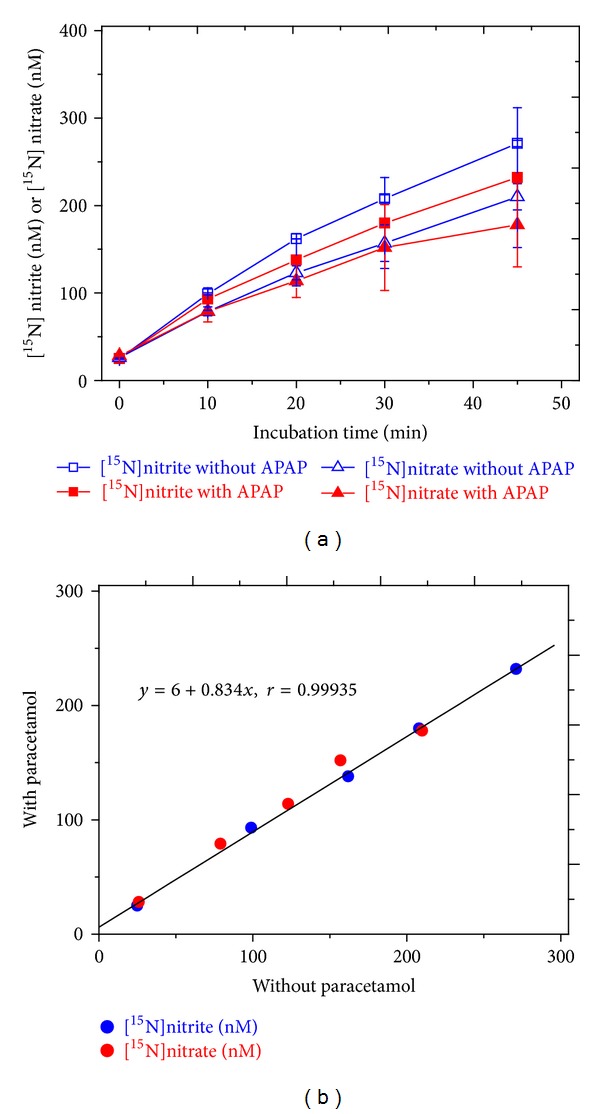
Effect of paracetamol (APAP) at 100 μM (15 mg/L) on the formation of [15N]nitrite and [15N]nitrate in an incubation mixture of recombinant heNOS upon incubation time (a) and linear regression analysis between [15N]nitrite and [15N]nitrate concentrations measured in the presence and absence of paracetamol (b). Data in (a) are shown as mean ± SD from two independent experiments; no statistical analysis was performed.
Figure 7.
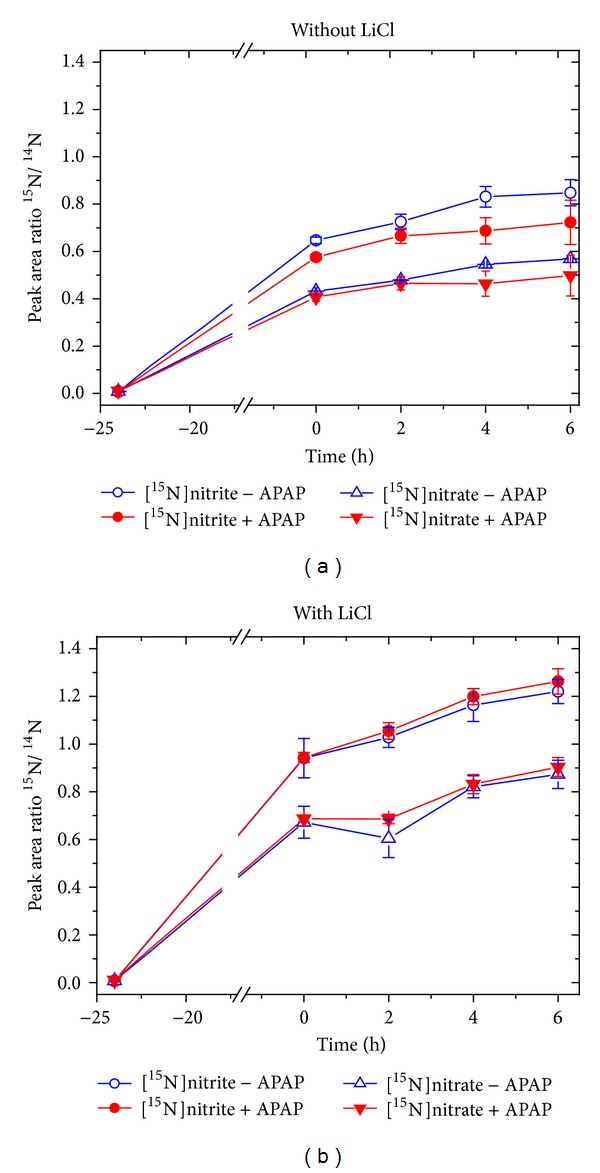
Effect of paracetamol (APAP) at 100 μM (15 mg/L) on the peak area ratio of m/z 47 for [15N]nitrite to m/z 46 for [14N]nitrite (a) and on the peak area ratio of m/z 63 for [15N]nitrate to m/z 62 for [14N]nitrate (b) upon incubation of adult rat hepatocytes with L-[guanidine-15N2]-arginine (5 mM) in the absence and in the presence of LiCl (1 mM) for the indicated times at 37°C as described elsewhere [8]. Reactions were terminated by addition of 400 μL aliquots of ice cold acetone and samples were further processed for GC-MS analysis. Data are shown as mean ± SD from three independent experiments; no statistical analysis was performed.
4. Discussion
4.1. General Remarks and Aim of the Study
Paracetamol is generally assumed to increase oxidative stress and is therefore commonly used in animal models of oxidative stress, in which paracetamol is administered in exorbitant high doses [21]. Whether paracetamol, when administered at therapeutic doses, also acts as a prooxidant is unknown. Paracetamol is known to interact with many enzymes such as CYP, COX, and NOS, which themselves are known to contribute to oxidative stress, for instance, by producing superoxide radical anions. While the inhibitory effect of paracetamol on prostacyclin synthesis in vivo in humans is well established [16], its effects on thromboxane and NO synthesis as well as on CYP activity are incompletely understood. This may be due to insufficiently high intracellular paracetamol concentrations when this drug is administered in therapeutic doses, for instance, by oral administration of a 500 mg paracetamol tablet. The aim of the present work was to investigate in healthy humans the effects of high-dosed paracetamol (i.e., 3 g) on the activity of the COX, NOS, and CYP, as well as on oxidative stress. Given the well-known hepatotoxicity of paracetamol, only four healthy subjects were enrolled in the human study. By using paracetamol concentrations that are expected to prevail for a considerable period of time after administration of a single 3 g oral dose to humans, we investigated the effects of paracetamol at suprapharmacological concentrations on the activity of two NOS isoforms in vitro, that is, on recombinant human eNOS and iNOS in rat hepatocytes.
4.2. Effects of Paracetamol on the Cyclooxygenase Pathway
Considering a mean fraction (oral bioavailability, F) value of 88% for paracetamol [32], its mean distribution volume (V D) in the volunteers of the human study described in this paper is estimated to be 88 L. This value is almost 25 times higher than the estimated volunteers' plasma volume and suggests that paracetamol may reach concentrations up to about 5000 μM in other body compartments except for red blood cells. Such high concentrations would be high enough to inhibit prostacyclin (PGI2) and thromboxane (TxA2) synthesis in endothelial cells and platelets, respectively [10].
Indeed, paracetamol, at the high single oral dose of 3 g, potently inhibited PGHS-catalyzed synthesis of PGI2, a potent vasodilator and inhibitor of platelet aggregation. By contrast, the synthesis of TxA2, a potent vasoconstrictor and platelet activator, was found not to be significantly inhibited by paracetamol in four subjects. Relative effects of drugs on PGI2 and TxA2 synthesis are commonly estimated by using the molar ratio of the prostanoids [34]. In our study, oral administration of a single 3 g oral paracetamol dose decreased the average PGI2/TxA2 molar ratio from about 0.6 before administration to values ranging between 0.4 and 0.2 after administration, thus shifting the vasodilatatory (PGI2)/vasoconstrictory (TxA2) balance at the cost of vasodilatation. A consequence of such a shift may be an increase of blood pressure. Indeed, Sudano and colleagues found that chronic administration of paracetamol to CAD patients at a lower dose (1 g TID for 2 weeks) than in the present study resulted in small blood pressure increase [9]. It is worth mentioning that in the study by Sudano et al. [9] considerably lower plasma paracetamol concentrations had been reached in comparison to those we measured in our study. In confirmation of previous studies [9, 15], we found that excretion of PGE2 did not change upon paracetamol administration, suggesting that even a high dose of 3 g paracetamol did not alter significantly renal PGE2 production in the volunteers.
4.3. Effects of Paracetamol on the L-Arginine/NO Pathway
The effects of paracetamol on NOS expression and activity have been studied by several groups. Yet, the observations are contradictory [17–20]. In our human study, paracetamol increased temporarily the concentration of nitrite in plasma. As the major fraction of circulating nitrite may originate from NO produced in the endothelium [23], our in vivo results may indicate that paracetamol increased eNOS activity and/or eNOS expression 2.5 to 3.5 h after administration. Yet, alternative ways such as paracetamol-induced reduction of nitrate to nitrite may have also increased plasma nitrite concentrations in the volunteers. Paracetamol did not change the plasma concentration of two other main parameters of the L-arginine/NO pathway, that is, L-arginine and ADMA. In vitro, paracetamol had only a very weak inhibitory effect on isolated recombinant heNOS and on iNOS in adult rat hepatocytes proliferating in vitro. LiCl that is known to induce expression and activity of iNOS in rat hepatocytes [31] increased iNOS activity but did not alter NO bioavailability. Thus, neither paracetamol nor LiCl influenced iNOS-related oxidative stress in rat hepatocytes.
4.4. Effects of Paracetamol on the Cytochrome P450 Pathway
Paracetamol is oxidized by the CYP family to NAPQI (N-acetyl-p-benzoquinone imine), the toxic intermediate of paracetamol. Unsaturated fatty acids including arachidonic acid and oleic acid are substrates for CYP enzymes [28, 35], and some of the arachidonic acid epoxides are vasoactive compounds [35]. At high concentrations (e.g., 1000 μM), paracetamol may inhibit the activity of CYP isoforms. In our human study, paracetamol increased temporarily the plasma concentration of the oleic acid oxide cis-EpOA. As cis-EpOA is a marker of CYP activity in humans [28], this finding may suggest that paracetamol increases CYP activity for a very short period of time. Another explanation for the very short-lasting increase in plasma cis-EpOA concentration could be activation of extracellular phospholipase A2 (PLA2) activity or release of hepatic PLA2 into the blood stream by paracetamol, because a considerable fraction of cis-EpOA is found esterified to human serum lipids [28]. The latter explanation is supported by the finding that the plasma concentration of free 15(S)-8-iso-PGF2α displayed a similar course including the sharp maximum like cis-EpOA in the present human study. It is worth mentioning that both 15(S)-8-iso-PGF2α and cis-EpOA are released in parallel from serum lipids upon incubation with PLA2 [28]. At present, very little is known about paracetamol effects on PLA2 activity and/or expression. In contrast to indomethacin, paracetamol (at 1000 μM) was found not to inhibit extracellular PLA2 activity as measured using radiolabelled oleic acid esterified to E. coli membranes [36]. In mice, hepatotoxicity induced by paracetamol at a dose of 400 mg/kg, that is, about 10 times higher than in our human study, was found to be associated with a time-dependent mode with increased secretion of hepatic PLA2 which was exacerbated in the absence of hepatic COX-2 [37]. Thus, the temporary increase in cis-EpOA and 15(S)-8-iso-PGF2α observed in our study may be due to paracetamol-induced short-term hepatotoxicity in the healthy subjects.
4.5. Effects of Paracetamol on Oxidative Stress
In the human study, paracetamol (3 g) did not increase oxidative stress as assessed by measuring urinary excretion of the oxidative stress biomarker 15(S)-8-iso-PGF2α [21, 22]. As discussed above, the sharp and short-lasting increase in the plasma concentration of free 15(S)-8-iso-PGF2α is likely to be due to temporary release of PLA2 from the liver and/or due to activation of extracellular PLA2. At this high dose, paracetamol did not increase plasma total hCys which is generally assumed to be associated with oxidative stress. Given the ROS-scavenging phenolic moiety of paracetamol (N-acetyl-p-aminophenol), the failure of paracetamol to enhance oxidative stress seems reasonable. The F2-isoprostane 15(S)-8-iso-PGF2α is known to be produced from AA by the catalytical action of COX [38]. In contrast to acetylsalicylic acid, indomethacin, and celecoxib [25, 39], our study indicates that paracetamol (3 g) does not inhibit COX-dependent formation of 15(S)-8-iso-PGF2α in humans.
5. Conclusion
We investigated in vitro and in vivo effects of paracetamol, an analgesic and antipyretic phenolic drug, on the L-Arg/NO, AA/COX, and CYP biochemical pathways and on oxidative stress. At the high single oral dose of 3 g, paracetamol did not alter oxidative stress in vivo. At suprapharmacological concentrations, paracetamol also did not alter oxidative stress in vitro as revealed by the unchanged nitrite-to-nitrate molar ratios measured in incubation mixtures of recombinant heNOS and in cultures of adult rat hepatocytes that express iNOS. The potent PGI2 inhibition by high-dosed paracetamol in the healthy subjects of the present study suggests that the relatively small increase in blood pressure seen in CAD patients by others [9] is likely to be due to compensatory mechanisms that involve enhanced formation of vasodilators. Potential candidates are NO and epoxyeicosatrienoic acids (EETs). In the circulation, NO can be produced from L-arginine by the catalytic action of eNOS and/or from nitrite/nitrate. EETs are produced from arachidonic acid by the catalytic action of the CYP family [34]. Our results suggest that in healthy subjects NO may compensate the loss of the vasodilatory and antiaggregatory prostacyclin caused by high-dosed paracetamol (Figure 8). Also, paracetamol does not increase oxidative stress even when given at suprapharmacological doses. We assume that in healthy humans the paracetamol-induced shift of the PGI2/TxA2 balance is counteracted by concomitant increase in circulating NO production. The underlying mechanisms remain elusive. Possible contributing mechanisms may include elevation of NO in the endothelium and conversion of nitrate to nitrite and its consecutive reduction to NO. In patients suffering from cardiovascular diseases, that is, with dysfunctional endothelium, paracetamol only partially counteracts its unfavorable vasodilatory/vasoconstrictory effect via NO.
Figure 8.
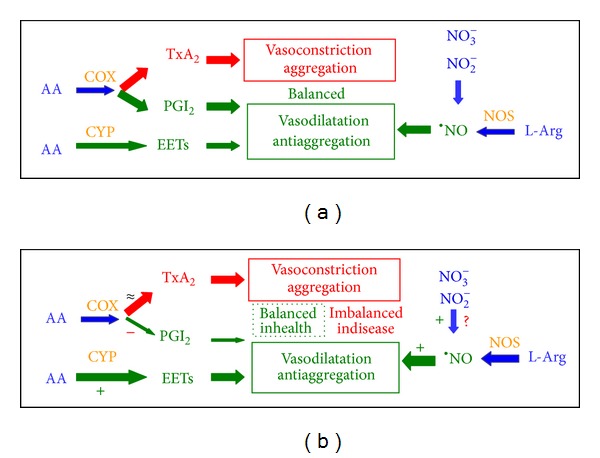
Simplified scheme showing the proposed contribution of three major enzymatic pathways to the vasoconstriction, vasodilatation, and aggregation in the vasculature without (a) and with (b) administration of paracetamol (acetaminophen) in health and disease. In platelets, arachidonic acid (AA) is converted by COX-1 to the vasoconstrictor and aggregator TxA2. In endothelial cells, AA is converted by COX-2 to PGI2, and L-Arg is oxidized by NOS to ∙NO; PGI2 and ∙NO are both vasodilatators and antiaggregators. AA is converted by CYP epoxygenases (CYP) to the vasodilatators epoxyeicosatrienoic acids (EETs). (a) In the absence of COX inhibitors including paracetamol and in health, production of TxA2, PGI2, ∙NO, and EETs guarantees a balance between vasoconstriction/aggregation and vasodilatation/antiaggregation. (b) Paracetamol and other COX inhibitors shift this balance in favour of COX-dependent vasoconstriction/aggregation. In response to this shift, ∙NO and EETs formation is increased in order to compensate the imbalance. In health, this compensation succeeds and blood pressure and platelet aggregation do not change. In endothelium dysfunction-related diseases, such as coronary artery disease (CAD), this compensation is insufficient and leads to moderate increase in blood pressure [9]. –, +, and ≈ mean inhibition, activation, and no remarkable change, respectively. The thickness of the arrows is quantitative but not true to scale measure of the contribution of the individual pathways and paracetamol.
Acknowledgments
The study was in part supported by the Deutsche Forschungsgemeinschaft (Grant TS 60/4-1). The authors thank B. Beckmann and A. Mitschke for excellent laboratory assistance and F.-M. Gutzki for performing GC-MS and GC-MS/MS analyses.
Abbreviations
- AA:
Arachidonic acid
- ADMA:
Asymmetric dimethylarginine
- APAP:
Acetaminophen (i.e., paracetamol)
- Arg:
Arginine
- ASA:
Acetylsalicylic acid
- CAD:
Coronary artery disease
- CYP:
Cytochrome P450
- COX:
Cyclooxygenase
- EETs:
Epoxyeicosatrienoic acids
- FPIA:
Fluorescence polarimetry immunoassay
- GC-MS:
Gas chromatography-mass spectrometry
- GC-MS/MS:
Gas chromatography-tandem mass spectrometry
- HUVECs:
Human umbilical vein endothelial cells
- tHyc:
Total homocysteine
- NO:
Nitric oxide
- NOS:
Nitric oxide synthase
- eNOS:
Endothelial nitric oxide synthase
- heNOS:
Human endothelial nitric oxide synthase
- iNOS:
Inducible nitric oxide synthase
- nNOS:
Neuronal nitric oxide synthase
- NSAIDs:
Nonsteroidal anti-inflammatory drugs
- PG:
Prostaglandin
- PGE2:
Prostaglandin E2
- PGE-MUM:
Prostaglandin E-major urinary metabolite
- PGHS:
Prostaglandin H synthase
- PGI2:
Prostacyclin
- PLA2:
Phospholipase A2
- QC:
Quality control
- ROS:
Reactive oxygen species
- SD:
Standard deviation
- SEM:
Standard error of the mean
- TID:
ter in di (three times a day)
- TxA2:
Thromboxane A2.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Dimitrios Tsikas conceived and designed all experiments described in the paper. Arne Trettin and Anke Böhmer performed in vitro experiments on recombinant enzymes and hepatocytes, carried out biochemical analyses, and wrote parts of the paper. Maria-Theresia Suchy performed biochemical analyses of the samples obtained in the human study. Irmelin Probst managed and supervised the experiments on hepatocytes. All authors read and approved the final version of the paper.
References
- 1.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(15):7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim SF, Huri DA, Snyder SH. Medicine: inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310(5756):1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 3.Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacological Reviews. 2005;57(2):217–252. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- 4.Kim SF. The role of nitric oxide in prostaglandin biology; Update. Nitric Oxide. 2011;25(3):255–264. doi: 10.1016/j.niox.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Förstermann U. Endothelial NO synthase as a source of NO and superoxide. European Journal of Clinical Pharmacology. 2006;62(13):5–12. [Google Scholar]
- 6.Zou MH, Martin C, Ullrich V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biological Chemistry. 1997;378(7):707–713. doi: 10.1515/bchm.1997.378.7.707. [DOI] [PubMed] [Google Scholar]
- 7.Hajjar DP, Lander HM, Pearce SFA, Upmacis RK, Pomerantz KB. Nitric oxide enhances prostaglandin-H synthase-1 activity by a heme-independent mechanism: evidence implicating nitrosothiols. Journal of the American Chemical Society. 1995;117(12):3340–3346. [Google Scholar]
- 8.Tsikas D, Schubert B, Gutzki F-M, Sandmann J, Frölich JC. Quantitative determination of circulating and urinary asymmetric dimethylarginine (ADMA) in humans by gas chromatography-tandem mass spectrometry as methyl ester tri(N-pentafluoropropionyl) derivative. Journal of Chromatography B. 2003;798(1):87–99. doi: 10.1016/j.jchromb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Sudano I, Flammer AJ, Périat D, et al. Acetaminophen increases blood pressure in patients with coronary artery disease. Circulation. 2010;122(18):1789–1796. doi: 10.1161/CIRCULATIONAHA.110.956490. [DOI] [PubMed] [Google Scholar]
- 10.Aronoff DM, Oates JA, Boutaud O. New insights into the mechanism of action of acetaminophen: its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases. Clinical Pharmacology and Therapeutics. 2006;79(1):9–19. doi: 10.1016/j.clpt.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Tsikas D. Application of gas chromatography-mass spectrometry and gas chromatography-tandem mass spectrometry to assess in vivo synthesis of prostaglandins, thromboxane, leukotrienes, isoprostanes and related compounds in humans. Journal of Chromatography B: Biomedical Applications. 1998;717(1-2):201–245. doi: 10.1016/s0378-4347(98)00210-2. [DOI] [PubMed] [Google Scholar]
- 12.Schweer H, Kammer J, Kuhl PG, Seyberth HW. Determination of peripheral plasma prostanoid concentration: an unreliable index of ’in vivo’ prostanoid activity. European Journal of Clinical Pharmacology. 1986;31(3):303–305. doi: 10.1007/BF00981128. [DOI] [PubMed] [Google Scholar]
- 13.Schweer H, Meese CO, Furst O. Tandem mass spectrometric determination of 11-dehydrothromboxane B2, an index metabolite of thromboxane B2 in plasma and urine. Analytical Biochemistry. 1987;164(1):156–163. doi: 10.1016/0003-2697(87)90380-0. [DOI] [PubMed] [Google Scholar]
- 14.Vesterqvist O, Green K. Urinary excretion of 2,3-dinor-thromboxane B2 in man under normal conditions, following drugs and during some pathological conditions. Prostaglandins. 1984;27(4):627–644. doi: 10.1016/0090-6980(84)90098-4. [DOI] [PubMed] [Google Scholar]
- 15.Bippi H, Frolich JC. Effects of acetylsalicylic acid and paracetamol alone and in combination on prostanoid synthesis in man. British Journal of Clinical Pharmacology. 1990;29(3):305–310. doi: 10.1111/j.1365-2125.1990.tb03640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green K, Drvota V, Vesterqvist O. Pronounced reduction of in vitro prostacyclin synthesis in humans by acetaminophen (paracetamol) Prostaglandins. 1989;37(3):311–315. doi: 10.1016/0090-6980(89)90001-4. [DOI] [PubMed] [Google Scholar]
- 17.Amin AR, Vyas P, Attur M, et al. The mode of action of aspirin-like drugs: effect on inducible nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(17):7926–7930. doi: 10.1073/pnas.92.17.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryu YS, Lee JH, Seok JH, et al. Acetaminophen inhibits iNOS gene expression in RAW 264.7 macrophages: differential regulation of NF-κB by acetaminophen and salicylates. Biochemical and Biophysical Research Communications. 2000;272(3):758–764. doi: 10.1006/bbrc.2000.2863. [DOI] [PubMed] [Google Scholar]
- 19.Raffa RB. Acetaminophen and metabolite: lack of effect on nitric oxide synthase (constitutive or inducible) Headache. 2002;42(3):237–238. doi: 10.1046/j.1526-4610.2002.02063.x. [DOI] [PubMed] [Google Scholar]
- 20.Godfrey L, Bailey I, Toms NJ, Clarke GD, Kitchen I, Hourani SMO. Paracetamol inhibits nitric oxide synthesis in murine spinal cord slices. European Journal of Pharmacology. 2007;562(1-2):68–71. doi: 10.1016/j.ejphar.2007.01.075. [DOI] [PubMed] [Google Scholar]
- 21.Giustarini D, Dalle-Donne I, Tsikas D, Rossi R. Oxidative stress and human diseases: origin, link, measurement, mechanisms, and biomarkers. Critical Reviews in Clinical Laboratory Sciences. 2009;46(5-6):241–281. doi: 10.3109/10408360903142326. [DOI] [PubMed] [Google Scholar]
- 22.Schwedhelm E, Benndorf RA, Böger RH, Tsikas D. Mass spectrometric analysis of F2-isoprostanes: markers and mediators in human disease. Current Pharmaceutical Analysis. 2007;3(1):39–51. [Google Scholar]
- 23.Grau M, Hendgen-Cotta UB, Brouzos P, et al. Recent methodological advances in the analysis of nitrite in the human circulation: nitrite as a biochemical parameter of the l-arginine/NO pathway. Journal of Chromatography B. 2007;851(1-2):106–123. doi: 10.1016/j.jchromb.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Tsikas D, Gutzki F-M, Böhme M, Fuchs I, Frölich JC. Solid- and liquid-phase extraction for the gas chromatographic-tandem mass spectrometric quantification of 2,3-dinor-thromboxane B2 and 2,3-dinor-6-oxo-prostaglandin F(1α) in human urine. Journal of Chromatography A. 2000;885(1-2):351–359. doi: 10.1016/s0021-9673(99)00967-x. [DOI] [PubMed] [Google Scholar]
- 25.Tsikas D, Schwedhelm E, Suchy M-T, et al. Divergence in urinary 8-iso-PGF2α (iPF2α -III, 15-F2t-IsoP) levels from gas chromatography-tandem mass spectrometry quantification after thin-layer chromatography and immunoaffinity column chromatography reveals heterogeneity of 8-iso-PGF2α . Journal of Chromatography B. 2003;794(2):237–255. doi: 10.1016/s1570-0232(03)00457-4. [DOI] [PubMed] [Google Scholar]
- 26.Tsikas D, Wolf A, Mitschke A, Gutzki F-M, Will W, Bader M. GC-MS determination of creatinine in human biological fluids as pentafluorobenzyl derivative in clinical studies and biomonitoring: inter-laboratory comparison in urine with Jaffé, HPLC and enzymatic assays. Journal of Chromatography B. 2010;878(27):2582–2592. doi: 10.1016/j.jchromb.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 27.Tsikas D. Simultaneous derivatization and quantification of the nitric oxide metabolites nitrite and nitrate in biological fluids by gas chromatography/mass spectrometry. Analytical Chemistry. 2000;72(17):4064–4072. doi: 10.1021/ac9913255. [DOI] [PubMed] [Google Scholar]
- 28.Thum T, Batkai S, Malinski PG, et al. Measurement and diagnostic use of hepatic cytochromeP450 metabolismof oleic acid in liver disease. Liver International. 2010;30(8):1181–1188. doi: 10.1111/j.1478-3231.2010.02310.x. [DOI] [PubMed] [Google Scholar]
- 29.Tsikas D, Mitschke A, Gutzki F-M, Meyer HH, Frölich JC. Gas chromatography-mass spectrometry of cis-9,10-epoxyoctadecanoic acid (cis-EODA): II. Quantitative determination of cis-EODA in human plasma. Journal of Chromatography B. 2004;804(2):403–412. doi: 10.1016/j.jchromb.2004.01.055. [DOI] [PubMed] [Google Scholar]
- 30.Tsikas D, Sandmann J, Savva A, et al. Assessment of nitric oxide synthase activity in vitro and in vivo by gas chromatography-mass spectrometry. Journal of Chromatography B. 2000;742(1):143–153. doi: 10.1016/s0378-4347(00)00142-0. [DOI] [PubMed] [Google Scholar]
- 31.Krause P, Wätzig E, Acil H, et al. Role of carbon monoxide and nitric oxide in adult rat hepatocytes proliferating in vitro: effects of CAS 1609. Nitric Oxide. 2010;23(3):220–226. doi: 10.1016/j.niox.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Rawlins MD, Henderson DB, Hijab AR. Pharmacokinetics of paracetamol (acetaminophen) after intravenous and oral administration. European Journal of Clinical Pharmacology. 1977;11(4):283–286. doi: 10.1007/BF00607678. [DOI] [PubMed] [Google Scholar]
- 33.Lakshmi VM, Hsu FF, Davis BB, Zenser TV. Nitrating reactive nitric oxygen species transform acetaminophen 3-nitroacetaminophen. Chemical Research in Toxicology. 2000;13(9):891–899. doi: 10.1021/tx000115g. [DOI] [PubMed] [Google Scholar]
- 34.Förstermann U, Kühn K, Vesterqvist O, et al. An increase in the ratio of thromboxane A2 to prostacyclin in association with increased blood pressure in patients on cyclosporine A. Prostaglandins. 1989;37(5):567–575. doi: 10.1016/0090-6980(89)90072-5. [DOI] [PubMed] [Google Scholar]
- 35.Giles TD, Sander GE, Nossaman BD, Kadowitz PJ. Impaired vasodilation in the pathogenesis of hypertension: focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. Journal of Clinical Hypertension. 2012;14(4):198–205. doi: 10.1111/j.1751-7176.2012.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lobo IB, Hoult JRS. Groups I, II and III extracellular phospholipases A2: selective inhibition of group II enzymes by indomethacin but not other NSAIDs. Agents and Actions. 1994;41(1-2):111–113. doi: 10.1007/BF01986409. [DOI] [PubMed] [Google Scholar]
- 37.Bhave VS, Donthamsetty S, Latendresse JR, Cunningham ML, Mehendale HM. Secretory phospholipase A2-mediated progression of hepatotoxicity initiated by acetaminophen is exacerbated in the absence of hepatic COX-2. Toxicology and Applied Pharmacology. 2011;251(3):173–180. doi: 10.1016/j.taap.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 38.Tsikas D, Suchy MT, Niemann J, et al. Glutathione promotes prostaglandin H synthase (cyclooxygenase)-dependent formation of malondialdehyde and 15(S)-8-iso-prostaglandin F2α . FEBS Letters. 2012;586:3723–3730. doi: 10.1016/j.febslet.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Tsikas D, Stichtenoth DO. Endothelial nitric oxide synthase, cyclooxygenase-2, and essential hypertension: is there an interaction? Hypertension. 2013;62, article e15 doi: 10.1161/HYPERTENSIONAHA.113.01947. [DOI] [PubMed] [Google Scholar]