

Abstract
The aging process is characterized by gradual changes to an organism's macromolecules, which negatively impacts biological processes. The complex macromolecular structure of chromatin regulates all nuclear processes requiring access to the DNA sequence. As such, maintenance of chromatin structure is an integral component to deter premature aging. In this review, we describe current research that links aging to chromatin structure. Histone modifications influence chromatin compaction and gene expression and undergo many changes during aging. Histone protein levels also decline during aging, dramatically affecting chromatin structure. Excitingly, lifespan can be extended by manipulations that reverse the age-dependent changes to chromatin structure, indicating the pivotal role chromatin structure plays during aging.
Keywords: aging, chromatin, histone modifications, epigenetics
Introduction
The basic repeating unit of chromatin, the nucleosome, consists of just under 150bp of DNA wrapped around an octamer of histone proteins including two molecules of each core histone - H2A, H2B, H3, and H4 [1]. The addition of linker histones and non-histone proteins enables the folding of arrays of nucleosomes into 30nm fibers and higher order chromatin structures. Packaging of our DNA into chromatin regulates all the genomic processes that occur within the cell. The reason for this is that the degree of chromatin compaction determines accessibility to the underlying DNA sequences. Tightly structured chromatin, or heterochromatin, minimizes genomic instability and misregulated gene expression, whereas more open chromatin, known as euchromatin, facilitates increased gene expression and genomic instability.
Aging is the prime risk factor for many human diseases such as cancer, heart disease, and diabetes. As such, elucidating the alterations to macromolecules that promote aging will be critical to develop treatments to delay or minimize age-related diseases and potentially extend lifespan. Aging is accompanied by changes in the transcriptional profile of cells and increased genomic instability [2,3]. The reasons for these age-related changes remain unclear but given that chromatin structure regulates genomic integrity and gene expression, potential changes to the chromatin structure during aging are likely to play an important role. Indeed, it has been previously suggested that changes to chromatin structure may partially explain the age-related changes to biological functions in cells and the increased incidence of disease states with age [4,5].
Chromatin structure is modified by the cell in a variety of ways to facilitate or limit access to the DNA. The most profound alteration to chromatin structure consists of the removal of histones from DNA and the opposite process of re-deposition of histones onto the DNA to re-establish chromatin. Another way to alter chromatin structure is via the addition or removal of post-translational modifications (PTMs) on specific amino acid residues on the histones. Histone PTMs exist in complex patterns including phosphorylation, acetylation, methylation and ubiquitination. PTMs on histones and DNA methylation of cytosines (collectively referred to as epigenetic marks) influence the ability of proteins to bind to the chromatin [6] which subsequently regulates the degree of compaction of the chromatin structure and the activities of the genome. For example, normal patterns of epigenetic marks are required for growth, development and the prevention of human disease. Growing evidence indicates that the patterns of epigenetic marks are altered during aging and human disease states including cancer [7]. In contrast to the genetic changes to the DNA sequence, epigenetic changes are reversible and therefore represent a promising therapeutic target for the treatment of human disease and hold equally promising potential for delaying the aging process.
Long-term maintenance of chromatin structure is required to promote normal biological functions during the aging process. Individuals with Hutchinson-Gilford progeria syndrome (HGPS), exhibit early aging characteristics such as hair-loss, decreased joint mobility, and heart disease that represent at least a subset of age-related problems [8]. Noteworthy, HGPS is accompanied by disrupted chromatin structure and nuclear organization. Moreover, the accelerated changes to the chromatin structure that occur in HGPS partially mirror the changes that occur normally in aged humans, suggesting a causal link between altered chromatin structure and aging.
In this review we will discuss the changes to chromatin structure that occur with age in multiple organisms and discuss recent publications that further demonstrate how chromatin structure impacts aging. Aging is accompanied by gross changes in DNA methylation, but this will not be discussed further here as this subject has been covered comprehensively in recent reviews [9,10]. Rather, we will summarize the changes to histone post-translational modifications, and changes in the abundance of histone proteins, histone variants and non-histone chromatin proteins during aging. These and future studies will enable the field to answer many of the remaining questions, such as what are all the age-related changes to chromatin? Which of the age related changes to chromatin cause aging? Which of the age related changes to chromatin function to delay premature aging? Which age related chromatin changes are simply a consequence rather than a cause of aging? We will also summarize very recent reports showing manipulations to chromatin structure can reverse age related changes and delay the aging process, demonstrating that the changes to the chromatin structure during aging are indeed a cause of aging.
Chromatin Modulation and Aging in Yeast
The budding yeast Saccharomyces cerevisiae has become a leading model organism for the study of aging as a consequence of the ease of its genetic manipulation, short cell division cycles, accuracy of lifespan determination and conservation of aging mechanisms across eukaryotes. One measurement of aging in budding yeast is the replicative lifespan (RLS), which is the number of times mother cells can divide to form daughter cells, modeling the finite mitotic divisions that occurs in metazoan cells. One of the first hints that chromatin structure changes during aging came from the realization that replicative aging in yeast is accompanied by the loss of transcriptional silencing which is thought to reflect a decrease in the degree of chromatin compaction [11].
Budding yeast maintain silencing in three main regions of the genome: the telomere proximal regions, the mating-type loci, and the ribosomal DNA (rDNA). Silencing of mating-type loci prevents sterility from occurring, which happens when gene products for both mating types are simultaneously expressed, preventing the cell from responding to mating pheromone. Aged yeast experience increased sterility due to the loss of silencing of the mating-type loci and subsequent expression of both mating-types [11]. Regions that contain repetitive DNA sequences such as the rDNA and telomere proximal DNA are silenced, which helps to prevent recombination, genomic instability and the formation of extra ribosomal chromosomal circles (ERCs). Silencing of reporter genes inserted near telomeres is lost upon replicative aging [12]. This occurs concomitantly with the movement of silencing proteins, including the NAD-dependent histone deacetylase (HDAC) Silencing information regulator 2 (Sir2), away from telomeric regions to the rDNA [13]. Failure to silence the rDNA results in higher levels of recombination and the formation of ERCs. Noteworthy, ERCs accumulate with age and can limit the RLS of yeast [14].
Sir2 mediated silencing of chromatin influences the RLS. Deletion of SIR2 reduces the RLS, while introducing an extra genomic copy of SIR2 extends the RLS, indicating that Sir2 is a limiting factor during aging [15]. Although the exact nature of the changes to the chromatin structure during aging were not clear, the increased genomic instability, the accumulation of ERCs, the loss of silencing with age and the ability of the Sir2 HDAC to influence yeast replicative lifespan were highly suggestive of a more relaxed chromatin structure during aging, at least at the silent regions of the genome.
Sir2 and Telomeric Histone Acetylation
Recently, Sir2's function as a histone deacetylase has been shown to directly affect aging in budding yeast. Sir2 is the primary HDAC for acetylated lysine 16 of histone 4 (H4 K16ac) [16]. Old yeast cells were found to have decreased amounts of Sir2 protein compared to young cells and this is the likely reason for increased global levels of H4 K16ac in old cells [17] (Table 1; Fig. 1). H4 K16ac is a unique epigenetic mark in that it inhibits the formation of the 30nm chromatin fiber and impedes the ability of chromatin to form cross-fiber interactions [18]. As such, increased levels of H4 K16ac in old cells presumably lead to a more open chromatin structure. Examination of H4 K16ac levels on the chromatin of old cells demonstrated increased levels of H4 K16ac at the X core and X elements within the telomeric regions examined [17]. The increased level of H4 K16ac with age also correlated with decreased silencing of reporter genes inserted near these telomere proximal DNA elements. Prevention of H4 K16ac by deletion of the gene encoding the relevant histone acetyl transferase (HAT), Something About Silencing 2 (SAS2), extended the RLS of yeast [17]. Conversely, mutation of H4 K16 to glutamine, which mimics permanent acetylation, decreased the RLS, directly showing that H4 K16ac modulates lifespan in budding yeast. As such, the ability of Sir2 to reduce H4 K16Ac levels partially explains the mechanism by which Sir2 promotes lifespan in budding yeast, though how lifespan is shortened as a consequence of loss of telomere-proximal silencing due to increased H4 K16ac remains unclear. As H4 K16ac inhibits higher order chromatin formation [18], the shortened lifespan of yeast with excess H4 K16Ac (achieved by deletion of the SIR2 gene or the K16Q mutation to mimic acetylation) could be the result of a potentially more open global chromatin structure. Conversely, the extended lifespan of cells with reduced levels of H4 K16ac (achieved by overexpression of the Sir2 HDAC or deletion of the gene encoding the Sas2 HAT) could be due to a potentially more closed global chromatin structure. Indeed, analysis of histone H4 occupancy by chromatin immunoprecipitation (ChIP) analysis on DNA in mutants with altered H4 K16ac levels is consistent with these predictions [17].
Table 1. Comparison of Histone Variants and Modifications that Change with Age.
Compilation of known changes to the abundance of histone modifications and histone variants that change from young to old organisms (in vivo) or cells (in vitro).
| Modification | Change with age | Organism | Citation |
|---|---|---|---|
|
| |||
| Global histone acetylation | Decrease | Human (in vitro) | [49] |
|
| |||
| Bulk H4 ac | Decrease | Rat (in vivo) | [65] |
|
| |||
| H3 K9me | Increase | Human (in vitro) | [52] |
|
| |||
| H3 K9me2 | Decrease | Human (in vitro) | [52] |
|
| |||
| H3 K9me3 | Decrease | Human (in vivo) | [44] |
| (in vitro) | [52] | ||
| Increase | Mouse (in vivo) | [66] | |
| Increase | Fly (in vivo) | Wood, 2010 | |
|
| |||
| H3 K9ac | Decrease | Rat (in vivo) | [67] |
| Increase | Human (in vitro) | [52] | |
|
| |||
| H3 S10ph | Increase, | Rat (in vivo) | [67] |
| Decrease | Human (in vitro) | [52] | |
|
| |||
| H3 K14ac | Increase | Mouse (in vivo) | [68] |
|
| |||
| H3 K27me3 | Decrease | Human (in vitro), Mouse (in vitro) | [69] |
|
| |||
| H4 K8ac | Increase | Mouse (in vivo) | [68] |
|
| |||
| H4 K12ac | Increase | Mouse (in vivo) | [68] |
|
| |||
| H3 K56ac | Decrease | Yeast (in vivo), | [17] |
| Human (in vitro) | [52] | ||
|
| |||
| H4 K16ac | Decrease | Yeast (in vivo), | [17] |
| Human (in vitro) | [52] | ||
|
| |||
| H4 K20me | Increase | Human (in vitro) | [52] |
|
| |||
| H4 K20me2 | Increase | Human (in vitro) | [52] |
|
| |||
| H4 K20me3 | Increase | Rat (in vivo) | [50] |
| Decrease | Human (in vitro) | [52] | |
|
| |||
| H3.1 | Decrease | Human (in vitro) | [58] |
| Decrease | Rat (in vivo) | [70] | |
|
| |||
| H3.2 | Decrease | Rat (in vivo) | [70] |
|
| |||
| H3.3 | Increase | Human (in vitro) | [58] |
| Increase | Rat (in vivo) | [70] | |
|
| |||
| H2A.1 | Decrease | Human (in vitro) | [58] |
| Decrease | Rat (in vivo) | [70] | |
|
| |||
| H2A.2 | Increase | Human (in vitro) | [58] |
| Increase | Rat (in vivo) | [70] | |
Figure 1. Alterations to Chromatin Structure During Aging in Eukaryotes.
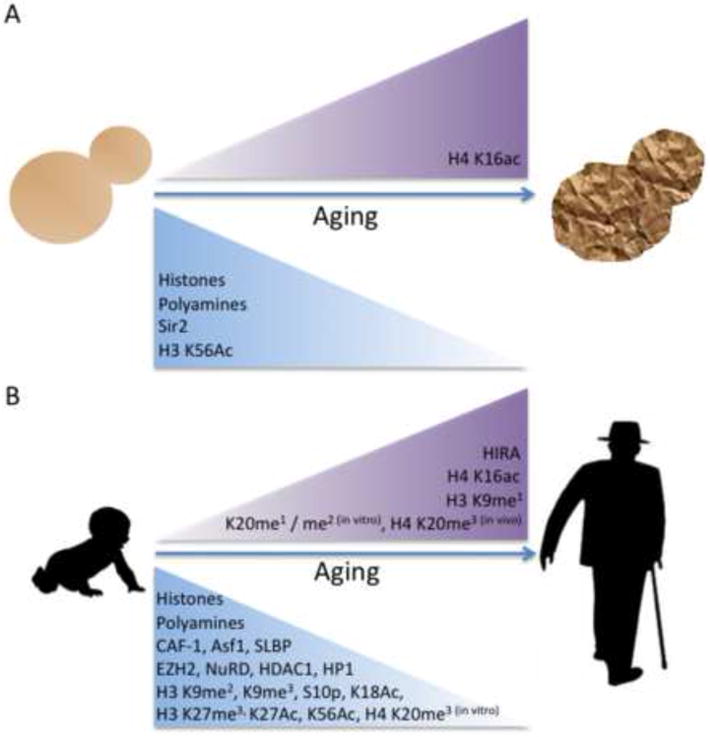
A) Budding yeast undergo changes to histone levels and histone modifications with increased replicative age. Additionally, changes to chromatin modifying proteins and organic molecules that impact chromatin structure occur with increased age. B) Mammalian cells experience changes in histone modifications during successive mitotic divisions in vitro. Levels of histones, histone modifications, and chromatin modifying proteins are altered during the aging process.
Aging and Loss of Histones
Global age-related transcriptional changes have been documented in budding yeast. These changes in gene expression during aging correlate with the shifts in the metabolic profile, elevated stress responses and induction of DNA repair genes [19,20]. Some of these transcriptional changes are likely responses to the stresses of aging itself. However, given that loss of silencing is a characteristic of aging in budding yeast, the changes in transcription during aging could also stem from a general reduction in the stringency of transcriptional regulation due to chromatin alterations.
Another example of altered transcription during aging occurs at the genes encoding the core histones. Histone transcript levels increase during aging in wild type yeast [20]. By contrast, short-lived yeast deficient in telomerase activity or dna2-1 mutants have decreased histone transcript levels in old cells [20]. Similarly, short-lived yeast mutants lacking the ability to acetylate H3 lysine 56 (H3 K56ac) also have reduced levels of histone transcripts. For example, mutants of the histone chaperone Anti-silencing function 1 (Asf1) or the HAT, Repressor of Ty 1 transposition 109 (Rtt109), which are essential for acetylation of H3 K56, or strains with an H3 K56A substitution that prevents acetylation of H3 K56, all exhibit a similar decrease of histone transcripts levels and dramatically reduced RLS [17,21,22]. Normally, H3 K56ac is enriched at the promoters of histone genes and is required for normal transcription of the cell cycle regulated histone genes [21]. Studies at model yeast promoters have revealed the likely mechanism whereby H3 K56ac promotes transcription of the histones genes - H3 K56ac promotes the disassembly of histone proteins from promoters to facilitate access by the transcriptional machinery [23]. Furthermore, while aged wild type yeast have elevated histone transcript levels, cells lacking the ability to acetylate H3 K56 fail to increase histone transcripts with age [22]. Counterintuitive to the role of H3 K56ac in promoting histone transcription and the increase in histone transcription that occurs during aging in wild type cells, bulk levels of acetylated H3 K56 (H3 K56ac) actually decreased with age [17] (Fig. 1, Table 1). This could suggest that the bulk levels of H3 K56ac in the cell are not reflecting the occupancy of H3 K56ac at the promoters of the histone genes, or that other mechanisms are acting to promote histone transcription in old cells despite the reduced level of H3 K56ac. Notwithstanding, given the role of H3 K56ac in promoting rapid and efficient gene expression, the reduction in bulk H3 K56ac levels in old cells could exacerbate and potentially contribute to other age-related transcriptional changes.
In contrast to the increase in histone transcript levels during aging, investigation of total histone protein levels in aged cells found H3, H4 and H2A protein levels to be greatly reduced relative to young cells [17,22] (Fig. 1, Fig. 2). Perhaps the increase in histone transcripts that normally occurs during aging reflects a feed back mechanism to attempt to replace the histone proteins that are lost during aging. Additionally, the occupancy of histones on DNA declines in aged yeast relative to young populations as demonstrated by ChIP and chromatin fractionation experiments [17,22]. The loss of histone occupancy during aging occurred from many different genomic locations and included the silent regions near telomeres, the rDNA, and the mating-type loci but also included the promoter of a repressed gene (PHO5) (JF and JT unpublished data) and open reading frame of an active gene ACT1. Although this loss of histones during aging has not yet been demonstrated at every region of the genome, all regions sampled to date show a profound loss of histones during aging. Additionally, it is unlikely that the loss of histones that has been observed during aging merely reflects the accumulation of a dead cell population in the culture that for some reason completely lack any histones, because yeast cells exist in a metabolically active senescent state for an extremely long time after ceasing replicative divisions [24]. The transcriptional consequences of loss of histone proteins from the genome are likely to be profound. For example, experimental depletion of H4 alters transcription of many genes, particularly those near telomeric regions [25].
Figure 2. Histones Levels Decline During Aging.
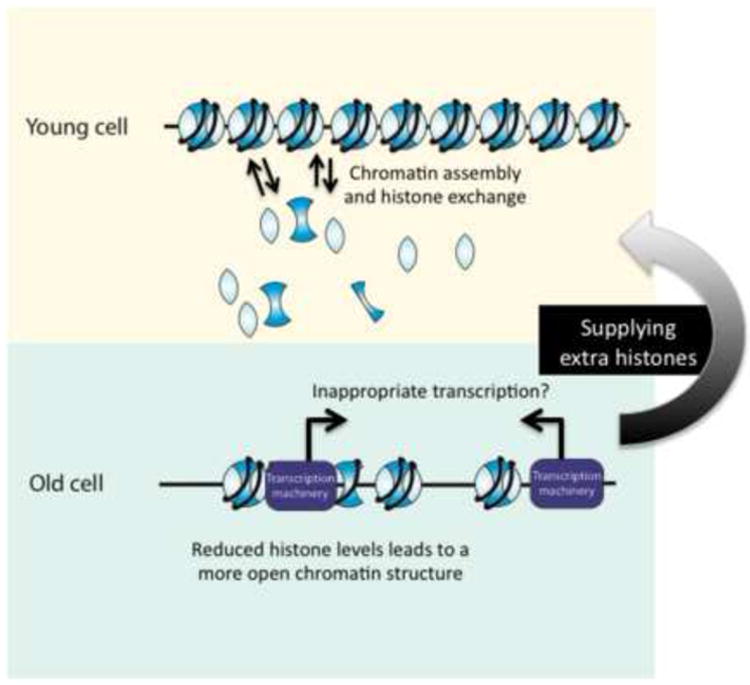
Young budding yeast cells have high levels of histones that facilitates proper chromatin assembly and histone exchange. With increasing age, histone levels decline, which may cause chromatin to be more open and have reducing histone exchange. This potentially leads to inappropriate access to the DNA, disrupting transcriptional regulation. Increasing the histone supply may allow the formation of tighter chromatin structure, thereby restoring transcriptional regulation and causing lifespan to be extended.
The loss of histone proteins during aging raises the question of whether loss of histones during aging is a cause of aging. Clearly there is a correlation between histone protein levels and lifespan in some mutants. For example the loss of histone protein occupancy on the DNA occurs after fewer generations in the asf1Δ and rtt109Δ mutants than in aged wild type yeast [22], providing a correlation between shortened lifespan and having a lower histone occupancy on the genome. Indeed, increasing the expression of H2A and H2B in short-lived asf1Δ mutants partially restored the lifespan [22], indicating that the low histone level is partially responsible for the decreased lifespan of the asf1Δ mutant. Furthermore, disruption of the histone information regulator complex (Hir), which is a repressor of histone transcription, causes elevated histone transcript levels and results in higher levels of H3 protein in aged hir1Δ mutants [22]. Indeed, the RLS of hir1Δ mutants is increased relative to wild type yeast [22]. The ultimate demonstration of a causal link between histone levels and aging was provided by the almost 50% increase in RLS that was achieved by inducing elevated levels of histones H3/H4 driven by the divergent pGAL1/10 promoters (Fig. 2) [22]. Noteworthy, this degree of lifespan extension is greater than that achieved by any known single manipulation in yeast, including calorie restriction.
The profound loss of histone proteins in aged yeast is expected to lead to a more open chromatin structure. A more open chromatin structure in turn would enable inappropriate access of the transcription machinery and DNA damaging agents to the DNA [26,27] (Fig. 2). Noteworthy, given that daughter yeast are rejuvenated, i.e. their age clock is set back to 0, the accumulation of mutations in the mother's genome is unlikely to be the major driving force that determines the replicative lifespan of yeast. The increased access of the transcription machinery to the DNA that likely results from histone loss with age may lead to elevated transcription of some genes (this could also explain the age-dependent increase in histone transcription), transcription of genes that should normally be repressed and the use of cryptic initiation sites in old cells. Clearly, these ideas now need to be thoroughly tested.
Throughout the cell cycle, histone proteins constantly leave the chromatin and are replaced, or “exchanged”, with free histone proteins. One could imagine that the lower concentration of histones in old cells is likely to lead to a smaller pool of free histones, which would limit the extent of histone exchange (Fig. 2). If histone exchange is indeed reduced in old cells, this would limit the cells capacity to refresh the histone modification pattern on the genome, and may contribute to the alterations in patterns of epigenetic marks with age (Table 1, Fig. 1). The consequence of reduced histone exchange would again be altered gene expression during aging.
Given that overexpressing histones can extend lifespan [22], it is tempting to speculate that the additional histones can restore the occupancy of histones on the DNA in old cells in order to reinstate the stringency of transcriptional regulation (Fig. 2). Extra histones may also restore the rates of histone exchange in old cells, in order to prevent the age-dependent accumulation of detrimental histone modifications such as H4 K16ac on the genome. In this case, overexpression of histones would be expected to rejuvenate the chromatin structure and genomic functions of old cells back to that of young cells. Regardless of the mechanism, it is clear that increasing histone expression and reducing levels of H4 K16ac improves lifespan, indicating that the maintenance of chromatin structure is central to the aging process in budding yeast.
Spermidine Treatment Reduces N-terminal H3 Acetylation to Extend Lifespan
Further evidence indicating a link between histone modifications, chromatin maintenance, and aging is found through the effect of the polyamine spermidine on lifespan. Polyamines are small organic molecules derived from amino acids and are synthesized by all organisms. Polyamines promote cell proliferation and inhibit apoptosis and have been implicated in cancer because polyamine levels are increased in cancer [28]. Aged mammalian cells and old yeast have decreased levels of polyamine synthesis and subsequently decreased levels of polyamines (Fig. 1, Fig. 3). Yeast subjected to spermidine supplementation have increased chronological lifespan (CLS) and RLS [29] (Fig. 3). Treatment with spermidine resulted in hypoacetylation of H3 K9, K14, and K18 during chronological aging by an apparent inhibition of HAT activity. Inactivation of HATs that acetylate H3 K9, K14, and K18 also extended RLS and reduced the response to spermidine treatment, indicating that lowering the levels of H3 N-terminal acetylation partially explains the mechanism whereby spermidine treatment extends lifespan [29]. The ability of reduced levels of H3 N-terminal acetylation and reduced levels of H4 K16Ac to extend lifespan is in contrast to the extended lifespan that results from increasing levels of H4 K5Ac and H4 K12Ac by deletion of the gene encoding the HDAC Rpd3 [30]. It is clear that not all lysine acetylations within the N-termini of histones are equivalent for lifespan extension, and this is probably explained by differences in the genes whose transcription are influenced by different histone acetylation marks.
Figure 3. Spermidine Treatment Extends Lifespan via Chromatin Alterations.
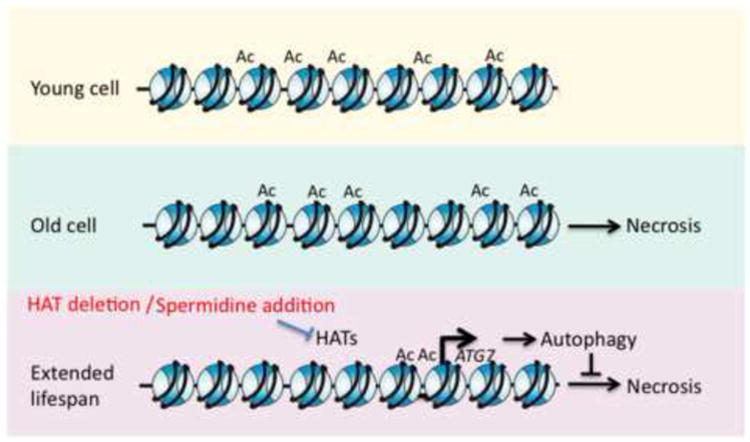
During aging, yeast experience a decline in polyamine levels that disrupts cellular functions and leads to necrotic death. The addition of spermidine causes a global decrease in H3 N-terminal acetylation and increases the expression of autophagic genes. Autophagic responses reduce the accumulation of damaged molecules and delay the onset of necrosis, extending lifespan.
In order to better understand the mechanism of lifespan extension, microarray analyses were performed in an attempt to identify genes whose expression is altered by spermidine treatment [29]. It was noted that several genes required for autophagy, such as ATG7, were upregulated (Fig. 3). Autophagy is the major lysosomal degradation pathway that recycles damaged and potentially harmful cellular material [31]. Autophagy counteracts cell death and extends lifespan in various models of aging [32,33]. Indeed, yeast treated with spermidine exhibited elevated signs of autophagy and a reduction of necrosis, which is implicated in regulating lifespan via the Tor pathway [29,34] (Fig. 3). Importantly, lifespan extension by spermidine treatment was not specific to yeast but also extended lifespan in Drosophila melanogaster, Caenorhabditis elegans, mice, and human cultured cells [29]. Furthermore, autophagy was required for lifespan extension by spermidine treatment in yeast, flies, and worms while spermidine treatment also induced autophagy in human cultured cells, indicating the conserved link between spermidine treatment, lifespan extension, and autophagy [29]. It is unclear how spermidine treatment leads to the elevated expression of the autophagy genes, because their promoters actually become enriched in H3 K18ac marks upon spermidine treatment (Fig. 3) [29]. Clearly the situation is complicated, but these studies present intriguing causal links between chromatin structure, aging and lifespan extension..
Chromatin Modulation during Mammalian Aging
Aging research in yeast has indicated that changes to the nuclear architecture and chromatin structure occur during aging and that lifespan can be positively or negatively affected by multiple types of chromatin manipulations. These lessons may also be relevant to mammalian aging. Micrococcal nuclease (MNase) digestion ladders become less discrete during aging in mammals [35,36]. MNase cleaves DNA between nucleosomes and blurring of the digestion ladder pattern indicates that the spacing of the nucleosomes may become more irregular with age. Furthermore, visual analysis of the 30nm fibers in aging fibroblasts indicates a progressive loss of density, suggesting loosening chromatin structure with age [35,36]. However, the analysis of changes to chromatin structure during aging in mammals should be revisited using improved modern day technologies, given that early studies reported reduced, increased or no change in MNase digestion during aging of metazoan cells [37]. Part of the reason for the contradictory results may depend on the fraction of senescent cells in the population and the genomic regions being studied, as some regions of the genome in senescent cells clearly becomes heterochromatinized, as indicated by the formation of SAHF (senescence associated heterochromatin foci) [38]. Presumably other regions of the genome become less compact during senescence because senescence is also accompanied by global loss of DNA methylation and loss of perinuclear heterochromatin [39,40].
Similar to the loss of silencing that occurs in old yeast cells, the ability to maintain heterochromatin also decreases in old mammals, shown by less efficient X-inactivation in old mice [41]. Genetic diseases, termed progeria, exist in mammals that partially mimic the aging process at an accelerated rate. These diseases are caused by mutations of genes involved in DNA repair, telomere maintenance, and nuclear architecture [42]. Representing a model for the aging process, lessons learned from progeria syndromes can be insightful for understanding how aging occurs.
Progeria, Aging, and Nuclear Architecture Changes
Hutchinson-Gilford progeria syndrome (HGPS) greatly disrupts the nuclear architecture. HGPS can be caused by a mutation in the gene encoding Lamin A, that leads to defective splicing and results in a C-terminally truncated protein termed progerin [43]. Lamin A is a component of nuclear laminae and comprises part of the nuclear envelope. Progerin acts as a dominant gain-of-function protein, accumulating in the nuclear envelope and causing a number of nuclear defects including a decline in heterochromatin, including loss of heterochromatin protein 1 (HP1), decreased levels of trimethylated H3 K9 (me3) and H3 K27 me3 (markers of heterochromatin), lower levels of the H3 K27 methyltransferase EZH2, increased transcription from pericentric repeats, shortened telomere length, elevated levels of DNA damage foci, and a general disruption of nuclear organization [40,43].
Progerin expression is even detectable at low levels in many cell types from healthy young and old individuals [44]. Cells from aged humans exhibit elevated levels of DNA damage foci and altered localization of Lamin A that was similar to observations of HGPS cells. Cells from aged humans also experience a decline in the levels of HP1 protein and loss of H3 K9me3 [44], indicating that several components of HGPS and normal aging are shared, albeit occurring at an earlier age in HGPS. Expression of progerin is sufficient to induce these nuclear changes in normal cells, which in turn recapitulates several of the phenotypes observed in HGPS [45]. Intriguingly, by correcting the splicing defect of Lamin A mRNA, accomplished by blocking the cryptic splice site, the HGPS-like phenotypes of cells were reversed to resemble their WT counterparts [46]. The abnormal nuclear shape of the cells reverted to a normal morphology and markers of heterochromatin, HP1 protein and H3 K9me3, were restored to near wild type levels in the majority of cells. Strikingly, cells from old healthy individuals also experienced a reversal in cellular phenotype upon reducing the level of progerin by removing the cryptic splice site [44]. The capacity to revert the cellular function of progerin-containing cells and old donor cells by reducing the level of progerin strongly indicates that maintenance of the nuclear architecture contributes to delaying the aging process. In light of the phenotypic reversal of old cells, which have had time to accumulate deleterious mutations to DNA, the phenotypic changes observed upon reducing progerin levels point to reversible changes to macromolecules rather than permanent mutations to DNA as being the major cause of aging in mammals.
Reduction of NURD and HDAC1 Protein Levels Cause HGPS Phenotypes
Further characterization of HGPS cells has elucidated additional changes to chromatin modifying proteins that contribute to the accelerated aging phenotype observed in HPGS. Yeast-two hybrid screens with the region lacking in progerin identified interactions with the histone binding proteins RBBP4 and RBBP7 [47]. RBBP4 and RBBP7 function in the NUcleosome Remodeling Deacetylase (NURD) complex, which is involved in chromatin modulation. HGPS cells exhibited decreased RBBP4/7 protein levels, along with lower levels of the NURD subunits HDAC1 and MTA3 [48]. Limiting NURD function via knockdown of various subunits in HeLa or fibroblast cells caused a loss of HP1 and H3 K9me3 along with elevated levels of DNA damage foci. These phenotypes are characteristic of HGPS, highlighting the importance of NURD function and chromatin modification in HGPS. Analysis of healthy aged cells also demonstrates a decline in levels of the NURD subunits RBBP4/7 and HDAC1 [47]. Additional research will be required to determine exactly how decreased NURD activity causes aging phenotypes. However, it is possible that reduced HDAC1 levels could lead to elevated histone acetylation, which would lead to altered transcription.
Histone Modifications During Mammalian Aging and Senescence
Changes in the abundance of histone post translational modifications (PTMs) during aging has been investigated (Table 1). Evidence for changes in histone PTMs was gained from a wide range of organisms and tissue types including mitotically dividing cells and post-mitotic cells. These results have sometimes been contradictory given the variety of organs studied and variation between individuals. Solid studies examining histone acetylation levels found a bulk reduction of H4 acetylation in rat cerebral cortex neurons and cultured human cells [49]. Levels of H4 K20 trimethylation, a marker of constitutive heterochromatin, have been shown to increase in kidney and liver tissue with age [50]. This mimics the increase in H4 K20 trimethylation levels that is observed in HGPS [40]. An interesting characterization of monozygotic twins revealed that histone acetylation of H3 and H4 is similar for twin pairs at a young age but can change in abundance between the twins with increased age [51]. These studies clearly indicated that the abundance of histone modifications changes during aging in mammals, meriting further study.
Improvements in mass spectrometry and modification-specific antibodies enabled a recent comprehensive characterization of histone modifications of early passage and late passage human fibroblast cells (Table 1, Fig. 1) [52]. Despite the fact that senescent cells do increase in abundance with increasing organismal age as observed in baboon fibroblast cells [53], the relevance of the study of entry into cell senescence in tissue culture to the process of aging in metazoans is hotly debated and has been reviewed elsewhere [54]. As such, a distinction is made in Table 1 as to whether the studies of histone modifications in mammals were performed in tissue culture models (in vitro) or in organisms (in vivo). The decreased levels of H3 K56ac and the increased level of H4 K16ac observed in old yeast cells was also apparent in aged cycling human fibroblasts [52]. Furthermore, aged cycling human fibroblasts experience many changes to the levels and cell cycle patterns of specific acetylated and methylated histone residues (Table 1, Fig. 1) [52]. As seen in aged mammalian tissue, levels of trimethylated H3 K9 decreased during aging in vitro [52]. Interestingly, there was a complete loss of H4 K20 me3 in aged cycling fibroblasts, in contrast to the increase observed in mammalian tissues and in HGPS. In general, many of the modifications that changed abundance during in vitro aging are involved in heterochromatin formation or DNA damage responses. Indeed, the large number of histone modifications whose abundance changes during aging complicates predictions of how, or if, each histone PTM change contributes to alteration of nuclear processes during aging. It will also be critical to determine whether each of these histone PTM changes occur as a consequence of aging or play a causal role in aging.
The abundance of histone modifications during aging is directly affected by changes in the levels of chromatin modifying proteins during age. In addition to the altered abundance of particular histone modifications during aging, chromatin structure is likely experiencing changes in the establishment and maintenance of chromatin due to the reduced levels of the histone chaperones Asf1A/B and CAF-1 during aging [52] (Figure 4). Given that Asf1A/B and CAF-1 participate in chromatin assembly and / or chromatin disassembly during DNA replication, DNA repair, and transcription [55], the declining histone chaperone levels during aging could cause cells to less efficiently alter chromatin structure to facilitate nuclear processes.
Figure 4. Persistent DNA Damage Signals From Shortened Telomeres Disrupt Chromatin Structure.
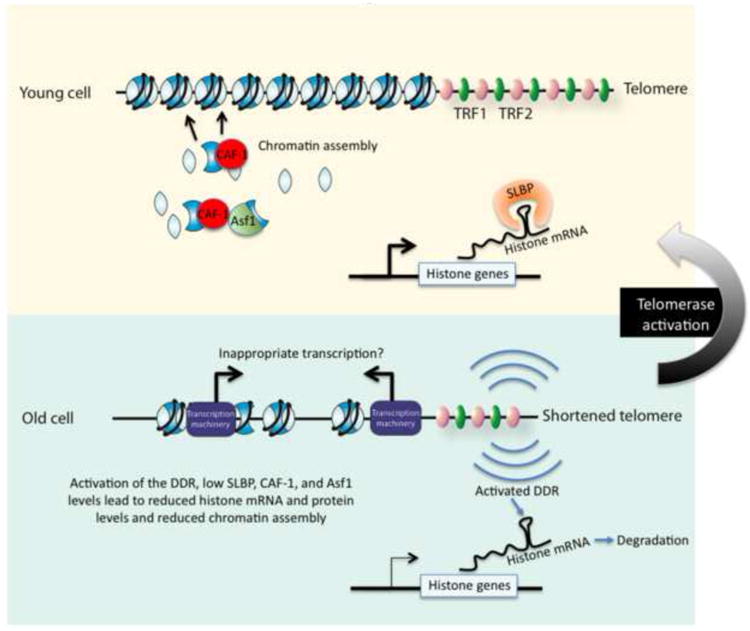
Young fibroblast cells have long telomeres and high levels of histones and chromatin modifying proteins. Successive mitotic divisions leads to shortened telomeres that cause persistent DNA damage signals. Asf1 and CAF-1 levels are reduced which disrupts chromatin structure. Reduced SLBP levels reduce the stability of the histone mRNAs during aging. Chromatin structure is likely to be more open in some regions of the genome, resulting in inappropriate access to the DNA and misregulated transcription. Reactivating telomerase extends the telomeres and halts the DNA damage signal, causing protein levels and chromatin structure to be restored.
Altered Histone Variant Abundance and Reduced Histone Protein Levels during Metazoan Aging
Histone variants have differences in amino acid residues that affect a diverse range of nuclear functions including DNA repair, transcription, and recombination [56]. The relative abundance of histone variants has been examined in various metazoans during aging [57-59]. The most notable change with likely functional consequences is the loss of the canonical histone H3.1 and H3.2 and the increase in the replication-independent variant H3.3 during aging. H3.3 exists in labile, more easily removable nucleosomes [60], and its enrichment in old cells could itself cause transcriptional changes during aging. Interestingly, aging in baboons is accompanied by an increase in the levels of HIRA [61], the histone chaperone involved in specifically incorporating H3.3 into chromatin.
Reinforcing the relevance of the loss of histone proteins from the genome observed during yeast aging, the similar result has been observed in aging human fibroblasts. Replicatively aged cells have markedly decreased levels (∼50%) of H3 and H4 relative to young cells (Figure 4) [52]. Why would histone levels decrease during mammalian aging? SLBP, a protein required for specific stabilization of the histone mRNA, decreases in abundance in cells aged in vitro, which could contribute to the decreased levels of histone proteins in high population doubling fibroblasts [52]. It is not yet clear whether the reduced levels of SLBP1, histone chaperones and histone modifying enzymes in old cells reflects a defect in transcription and / or translation. Histone transcription occurs primarily during S-phase to coincide with DNA replication. This occurs as a consequence of cyclinE-Cdk2 mediated phosphorylation of the NPAT transactivator protein of histone gene expression.
During successive replicative cycles of the human fibroblast cells, telomeres undergo gradual shortening, which causes DNA damage signals to be persistently activated. Activation of the DNA damage checkpoint blocks phosphorylation of NPAT, inhibiting histone transcription [63]. Activation of the DNA damage checkpoint also results in degradation of histone mRNAs [64]. With a dramatically altered chromatin structure as a consequence of reduced histone levels and altered patterns of histone modifications, aged cells would presumably experience many changes to all genomic processes that are likely to be detrimental for the cell. Strikingly, the reactivation of telomerase in old fibroblasts not only increased the length of telomeres but also restored the abundance of Asf1, SLBP and histones and restored the pattern of histone modifications to that seen in young fibroblasts [52]. This very exciting result indicates that the telomere length mitotic ”age clock” is controlling the deterioration of the global chromatin structure with age, and that chromatin is likely to be a key mediator of the aging process.
Conclusions
Growing evidence strongly implicates chromatin structure in the aging process. Aging is accompanied by changes in the chromatin structure, which is likely to impact all nuclear processes. As such, maintenance of chromatin structure is integral for maintaining proper cellular function during the aging process. Recent studies in budding yeast and human fibroblast cells show that chromatin is profoundly altered during aging due to the decreased level of histone proteins and changes to abundance of histone modifications. These alterations are likely to result in a more open chromatin structure during aging, presumably allowing inappropriate transcription. The identification of the specific transcripts that are misregulated as a consequence of the altered chromatin structure during aging that are causative for aging will provide a better understanding of the fundamental molecular basis of the aging process. Restoration of the aging chromatin structure back to a more youthful state by elevating histone expression, reducing H4 K16 acetylation, reducing H3 N-terminal acetylation, inactivation of the HDAC Rpd3, or blocking the signaling of persistent DNA damage from shortened telomeres, is sufficient to extend lifespan or revert the aged phenotype to a more youthful state. We expect that future studies will focus on understanding the molecular consequences of these manipulations that tighten the loose chromatin structure of old cells, in order to extend lifespan. Only in this way will the field be able to develop specific therapeutics to extend lifespan without the side effects that are likely to occur as a consequence of tightening the chromatin structure of the entire genome. It is certainly an exciting time for research at the intersection of aging and chromatin structure.
Abbreviations
- PTMs
Post-translational modifications
- rDNA
Ribosomal DNA
- H4 K16ac
Histone 4 lysine 16 acetylation
- Sir
Silencing information regulator
- HDAC
Histone deacetylase
- HAT
Histone acetyltransferase
- SAS
Something about silencing
- H3 K56ac
Histone 3 lysine 56 acetylation
- Asf1
Anti-silencing function 1
- Rtt109
Repressor of Ty 1 transposition
- WT
Wild type
- ChIP
Chromatin immunoprecipitation
- Hir
Histone information regulator
- RLS
replicative lifespan
- ERCs
Extrachromosomal rDNA circles
- HGPS
Hutchinson-Gilford progeria syndrome
- NURD
NUcleosome Remodeling Deacetylase
- CLS
Chronological lifespan
- SAHFs
Senescent-associated heterochromatin foci
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Busuttil R, Bahar R, Vijg J. Genome dynamics and transcriptional deregulation in aging. Neuroscience. 2007;145:1341–7. doi: 10.1016/j.neuroscience.2006.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maslov AY, Vijg J. Genome instability, cancer and aging. Biochim Biophys Acta. 2009;1790:963–9. doi: 10.1016/j.bbagen.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villeponteau B. The heterochromatin loss model of aging. Exp Gerontol. 1997;32:383–94. doi: 10.1016/s0531-5565(96)00155-6. [DOI] [PubMed] [Google Scholar]
- 5.Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol. 2007;8:692–702. doi: 10.1038/nrm2238. [DOI] [PubMed] [Google Scholar]
- 6.Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:R546–51. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Fraga MF, Agrelo R, Esteller M. Cross-talk between aging and cancer: the epigenetic language. Ann N Y Acad Sci. 2007;1100:60–74. doi: 10.1196/annals.1395.005. [DOI] [PubMed] [Google Scholar]
- 8.Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–24. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 9.Gravina S, Vijg J. Epigenetic factors in aging and longevity. Pflugers Arch. 2009;459:247–58. doi: 10.1007/s00424-009-0730-7. [DOI] [PubMed] [Google Scholar]
- 10.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–8. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Smeal T, Claus J, Kennedy B, Cole F, Guarente L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell. 1996;84:633–42. doi: 10.1016/s0092-8674(00)81038-7. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Villeponteau B, Jazwinski SM. Effect of replicative age on transcriptional silencing near telomeres in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1996;219:370–6. doi: 10.1006/bbrc.1996.0240. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy BK, et al. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell. 1997;89:381–91. doi: 10.1016/s0092-8674(00)80219-6. [DOI] [PubMed] [Google Scholar]
- 14.Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–42. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 15.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–80. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 17.Dang W, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–7. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–7. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 19.Yiu G, et al. Pathways change in expression during replicative aging in Saccharomyces cerevisiae. J Gerontol A Biol Sci Med Sci. 2008;63:21–34. doi: 10.1093/gerona/63.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesur I, Campbell JL. The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol Biol Cell. 2004;15:1297–312. doi: 10.1091/mbc.E03-10-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–85. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 22.Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–35. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams SK, Truong D, Tyler JK. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc Natl Acad Sci U S A. 2008;105:9000–5. doi: 10.1073/pnas.0800057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortimer RK, Johnston JR. Life span of individual yeast cells. Nature. 1959;183:1751–2. doi: 10.1038/1831751a0. [DOI] [PubMed] [Google Scholar]
- 25.Wyrick JJ, Holstege FC, Jennings EG, Causton HC, Shore D, Grunstein M, Lander ES, Young RA. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402:418–21. doi: 10.1038/46567. [DOI] [PubMed] [Google Scholar]
- 26.Ljungman M, Hanawalt PC. Efficient protection against oxidative DNA damage in chromatin. Mol Carcinog. 1992;5:264–9. doi: 10.1002/mc.2940050406. [DOI] [PubMed] [Google Scholar]
- 27.Enright HU, Miller WJ, Hebbel RP. Nucleosomal histone protein protects DNA from iron-mediated damage. Nucleic Acids Res. 1992;20:3341–6. doi: 10.1093/nar/20.13.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerner EW, Meyskens FL., Jr Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4:781–92. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 29.Eisenberg T, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–14. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 30.Kim S, Benguria A, Lai CY, Jazwinski SM. Modulation of lifespan by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:3125–36. doi: 10.1091/mbc.10.10.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–6. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–91. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 34.Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–84. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishimi Y, Kojima M, Takeuchi F, Miyamoto T, Yamada M, Hanaoka F. Changes in chromatin structure during aging of human skin fibroblasts. Exp Cell Res. 1987;169:458–67. doi: 10.1016/0014-4827(87)90206-0. [DOI] [PubMed] [Google Scholar]
- 36.Macieira-Coelho A, Puvion-Dutilleul F. Evaluation of the reorganization in the high-order structure of DNA occurring during cell senescence. Mutat Res. 1989;219:165–70. doi: 10.1016/0921-8734(89)90011-8. [DOI] [PubMed] [Google Scholar]
- 37.Medvedev ZA. Age changes of chromatin. A review. Mech Ageing Dev. 1984;28:139–54. doi: 10.1016/0047-6374(84)90014-9. [DOI] [PubMed] [Google Scholar]
- 38.Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 39.Shin DM, Kucia M, Ratajczak MZ. Nuclear and Chromatin Reorganization during Cell Senescence and Aging - A Mini-Review. Gerontology. doi: 10.1159/000281882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shumaker DK, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cattanach BM. Position effect variegation in the mouse. Genet Res. 1974;23:291–306. doi: 10.1017/s0016672300014932. [DOI] [PubMed] [Google Scholar]
- 42.Schumacher B, Garinis GA, Hoeijmakers JH. Age to survive: DNA damage and aging. Trends Genet. 2008;24:77–85. doi: 10.1016/j.tig.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 43.Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 11:567–78. doi: 10.1038/nrm2944. [DOI] [PubMed] [Google Scholar]
- 44.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–63. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–9. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–5. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pegoraro G, Misteli T. The central role of chromatin maintenance in aging. Aging (Albany NY) 2009;1:1017–22. doi: 10.18632/aging.100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009;11:1261–7. doi: 10.1038/ncb1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan JM, Cristofalo VJ. Histone acetylation during aging of human cells in culture. Biochem Biophys Res Commun. 1972;48:735–42. doi: 10.1016/0006-291x(72)90668-7. [DOI] [PubMed] [Google Scholar]
- 50.Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem. 2002;277:39195–201. doi: 10.1074/jbc.M205166200. [DOI] [PubMed] [Google Scholar]
- 51.Fraga MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Sullivan R, Kubicek S, Schreiber S, Karlsleder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. 2010 doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 54.Campisi J, Sedivy J. How does proliferative homeostasis change with age? What causes it and how does it contribute to aging? J Gerontol A Biol Sci Med Sci. 2009;64:164–6. doi: 10.1093/gerona/gln073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eitoku M, Sato L, Senda T, Horikoshi M. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell Mol Life Sci. 2008;65:414–44. doi: 10.1007/s00018-007-7305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–75. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 57.Pina B, Suau P. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev Biol. 1987;123:51–8. doi: 10.1016/0012-1606(87)90426-x. [DOI] [PubMed] [Google Scholar]
- 58.Rogakou EP, Sekeri-Pataryas KE. Histone variants of H2A and H3 families are regulated during in vitro aging in the same manner as during differentiation. Exp Gerontol. 1999;34:741–54. doi: 10.1016/s0531-5565(99)00046-7. [DOI] [PubMed] [Google Scholar]
- 59.Urban MK, Zweidler A. Changes in nucleosomal core histone variants during chicken development and maturation. Dev Biol. 1983;95:421–8. doi: 10.1016/0012-1606(83)90043-x. [DOI] [PubMed] [Google Scholar]
- 60.Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet. 2009;41:941–5. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007;128:36–44. doi: 10.1016/j.mad.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berkowitz EM, Sanborn AC, Vaughan DW. Chromatin structure in neuronal and neuroglial cell nuclei as a function of age. J Neurochem. 1983;41:516–23. doi: 10.1111/j.1471-4159.1983.tb04769.x. [DOI] [PubMed] [Google Scholar]
- 63.DeRan M, Pulvino M, Greene E, Su C, Zhao J. Transcriptional activation of histone genes requires NPAT-dependent recruitment of TRRAP-Tip60 complex to histone promoters during the G1/S phase transition. Mol Cell Biol. 2008;28:435–47. doi: 10.1128/MCB.00607-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaygun H, Marzluff WF. Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat Struct Mol Biol. 2005;12:794–800. doi: 10.1038/nsmb972. [DOI] [PubMed] [Google Scholar]
- 65.Pina B, Martinez P, Suau P. Differential acetylation of core histones in rat cerebral cortex neurons during development and aging. Eur J Biochem. 1988;174:311–5. doi: 10.1111/j.1432-1033.1988.tb14099.x. [DOI] [PubMed] [Google Scholar]
- 66.Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 67.Kawakami K, Nakamura A, Ishigami A, Goto S, Takahashi R. Age-related difference of site-specific histone modifications in rat liver. Biogerontology. 2009;10:415–21. doi: 10.1007/s10522-008-9176-0. [DOI] [PubMed] [Google Scholar]
- 68.Huang JC, et al. Changes in histone acetylation during postovulatory aging of mouse oocyte. Biol Reprod. 2007;77:666–70. doi: 10.1095/biolreprod.107.062703. [DOI] [PubMed] [Google Scholar]
- 69.Bracken AP, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–30. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pina B, Suau P. Core histone variants and ubiquitinated histones 2A and 2B of rat cerebral cortex neurons. Biochem Biophys Res Commun. 1985;133:505–10. doi: 10.1016/0006-291x(85)90935-0. [DOI] [PubMed] [Google Scholar]