

Abstract
A significant number of contributions to our understanding of primary immunodeficiencies in pathogenesis, diagnosis and treatment were published in the Journal in 2013. For example, deficiency of mast cell degranulation due to STAT3 deficiency was demonstrated to contribute to the difference on frequency of severe allergic reactions in AD-HIES patients, compared to atopic individuals with similar high IgE serum levels. High levels of non-glycosylated IgA were found in WAS patients and these abnormal antibodies might contribute to nephropathy in WAS. New described genes causing immunodeficiency included caspase recruitment domain 11 (CARD11), mucosa-associated lymphoid tissue 1 (MALT1) for combined immunodeficiencies, and tetratricopeptide repeat domain 7A (TTC7A) for mutations associated to multiple atresia with combined immunodeficiency. Other observations expand the spectrum of clinical presentation of specific genes. (e.g., adult onset idiopathic T-cell lymphopenia and early onset autoimmunity might be due to hypomorphic mutations of the RAG genes). Newborn screening in California established incidence of SCID at 1/66,250 live births. The use of HSCT for primary immunodeficiencies was reviewed, with recommendations to give priority to research oriented to establish best regimens to improve safety and efficacy of bone marrow transplantation. These represent only a fraction of significant research done in primary immunodeficiencies that has accelerated the quality of care of these patients. Genetic analysis of patients has demonstrated multiple phenotypic expressions of immune deficiency in patients with nearly identical genotypes, suggesting that additional genetic factors, possibly gene dosage, or environmental factors are responsible for this diversity.
Keywords: Genotype vs. phenotype expressions, immunology, immunoreconstitution, intravenous immunoglobulin, primary immunodeficiencies, newborn screening for SCID, whole genome sequencing
Reports in clinical immunology published in the Journal in 2013 continue to define all aspects of human immunodeficiencies, widening the spectrum of clinical presentation and increasing the number of genes responsible to maintain immune function.
The increasing complexity of primary immunodeficiencies (PIDs) was discussed by Maggina and Gennery,1 who argued that the current international classification2 based on the traditional compartmentalization of the immune system (T cells, B cells, innate immunity and complement) might no longer be optimal for clinical use. They described several examples of gene defects causing well known PIDs that also present with widely diverse clinical phenotypes. For example, different types of mutations (gain-of-function, null, residual function) in the Wiskott-Aldrich syndrome (WAS) protein can manifest as 3 different entities: X-linked thrombocytopenia, X-linked neutropenia or Wiskott-Aldrich syndrome. The authors propose to develop two reference catalogs: one according to primary immunological function, and a second catalog to describe specific gene mutations with genotype/phenotype correlation if available. In addition, they propose to have a treatment catalog to ensure that patients obtain the most updated and appropriate management. Notarangelo3 reviewed specific disorders of T cell characterized by incomplete loss of function that provide further insight into immune mechanisms. These included recently described gene defects: lymphocyte specific tyrosine kinase (LCK) deficiency, Ras homology family member H deficiency (RHOH), macrophage stimulating 1 (MST1) deficiency, DOCK8 deficiency, IL-2 inducible tyrosine kinase deficiency (ITK), and coronin 1A (CORO1A) deficiency.
With a different prospective based on regional and geographical differences of disease patterns in immunodeficiency disorders, Al-Herz and Al-Moussa.4 highlighted the distinct proportion of defects in recombination activating genes (RAG) 1 and 2 proteins and MHC class II antigen expression as causes of combined immunodeficiencies in the Middle East (where such defects account for over 60% of reported cases), as compared with reports from US, Australia and Asian cohorts, where IL2Rγ deficiency is the predominant cause. They discussed the difficulties for diagnosis and treatment of immunodeficiencies in areas with limited access to current technologies, and briefly explained reasons for different distribution of genetic lesions, such as the increased frequency of autosomal recessive inheritance observed in communities were the practice of consanguineous marriages is common.
Primary Immunodeficiencies: Advances in Mechanisms of Disease
Orange provided a comprehensive review5 of the current understanding of NK cell disorders, whether isolated or as part of another immunodeficiency syndrome, with diagnostic criteria and the genetic mutations that have been associated with inherited NK cell dysfunction. Compared to disorders of adaptive immunity, these are fewer in number so far. Two genes associated with low NK cell counts are gata-binding protein 2 (GATA2) and minichromosome maintenance 4 (MCM4) gene. Another gene associated with defective function of NK cells is attributed to a mutant FCGR3A gene that encodes the Fc receptor for NK cells (CD16). This defective gene has been identified in 3 unrelated families. Campbell and Hasegawa6 reviewed current concepts of NK cell biology, including the classification of NK cells as members of the group 1 innate lymphoid cells which secrete IFNγ, but not TH2 cytokines. The list of activating and inhibiting receptors for NK cell function continues to expand. This article also reviewed the crosstalk between innate and adaptive immunity through specific antibodies and cytokines resulting in the control of viral infections and malignancies.
The process of development and differentiation of human B cells was summarized by Piper and coauthors, who highlighted the contribution of case reports of genetic immunodeficiencies, which help define the function of the putative genes involved. These authors also pointed-out differences of function of homologous proteins in mice and humans. For example, deletion of B cell linker (BLNK) protein in mice results in a B cell development arrest at the pre-B cell stage, however, with BLNK deletion in humans, the arrest in in the pro-B cell stage. Other considerations were the B cell egress from bone marrow to B cell follicles, the formation of germinal centers and the homing into the marginal zones in the spleen. Also discussed in the review were B cell peripheral differentiation, somatic hypermutation and the development into memory B cells and B cells plasma cells, as well as the different mechanisms of B cell tolerance. (Figure 1) Understanding these mechanisms is important for the development of further strategies to treat autoimmunity, malignancy and immunodeficiency.
Fig 1.
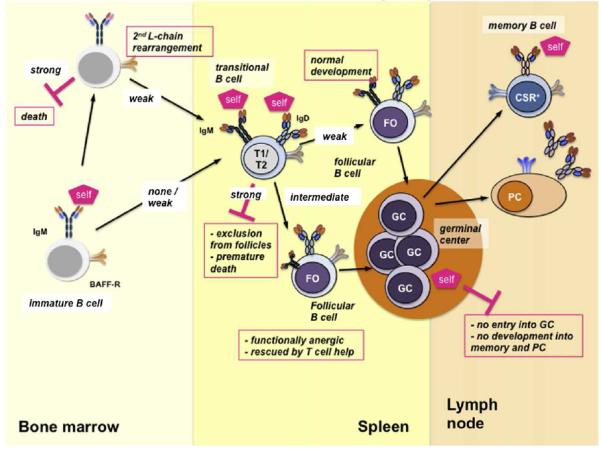
Different mechanisms of B cell tolerance.GC, germinal center. FO, Follicular cell. PC; plasma cell. (From Piper at al. J Allergy Clin Immunol 2013;131:959-969)
Mehling and collaborators8 studied the effects of fingolimod, a sphingosine-1-phosphate antagonist used in the management of multiple sclerosis. Fingolimod blocks lymphocyte egression from lymph nodes. By measuring T cell subsets in peripheral blood, they found that naïve CD4+ and CD8+ T cells were trapped in the lymph nodes within the first 3 hours, while central memory T cells were maintained at same concentration in circulation. Results from trans-well experiments with gradients of the chemokines CXCL12, CCL19 and CCL21 were also consistent with the increased migration of naïve T cells compared to memory T cells.
In the last few years, newly described subsets of CD4+ helper T cells have added complexity to the TH1/TH2 paradigm in the modulation of the immune response. These new subsets include TH17, TH9, TH22, follicular helper T cells and regulatory T cells (Tregs). Hirahara and coauthors9 discussed the characteristics of these cells, and also described the flexibility of their classical phenotypes and overlapping functions. For example, IL-10 is expressed and secreted by more than one subset of helper T cells. These changes in phenotype stability are regulated by the expression of specific transcription factors, as well as changes in the cell microenvironment.
Advances in defining the pathophysiology of well-defined syndromes focused on the Wiskott-Aldrich syndrome (WAS) and the hyper IgE syndrome (HIES). Aberrant glycosylation of immunoglobulin has been previously reported in WAS, but Shimizu et al10 studied serum levels of IgA in WAS patients over 10 years of age, and found increased concentrations of this abnormal IgA compared to control subjects. Circulating IgG-IgA complexes were also elevated. The authors postulate that these findings contribute to the autoimmune glomerulopathy seen in 40-70% of WAS patients. Of note, glomerulopathy cleared in one WAS patient who was studied before and after hematopoeitic stem cell transplant (HSCT). Using a murine model for WAS, Lang and coauthors11 demonstrated decreased viral autoimmunity as measured by interferon production in response to lymphochoriomeningitis virus (LCMV) infection. Compared to control mice, there were fewer CD8+ T cells specific to viral proteins in blood and spleen, with equal numbers of total CD8+ T cells.
The WAS protein (WASP) participates in the regulation of T cell activation and actin reorganization. Watanabe and collaborators12 demonstrated that TCR/CD3 ligation decreases intracellular WASP, an event that is blocked when a protease inhibitor is present. It was further shown that protein degradation is mediated by ubiquitination. WASP degradation limits the duration of F-actin assembly and controls T cell activation.
Dedicator of cytokinesis 8 (DOCK8) is the gene associated with autosomal recessive hyper IgE syndrome (AR-HIES). DOCK8 deficient patients have an increased incidence of viral infections, a characteristic that might be attributed to NK cell dysfunction. Mizesko and coauthors13 showed that NK cells from ten DOCK8 deficient patients and a NK cell line with reduced DOCK8 expression had severe reduction of cytotoxicity against K562 erythroleukemia cells, which could not be rescued with IL-2 stimulation. When studying the immunological synapse, NK cells from DOCK8 deficient patients did not show accumulation of F-actin, CD18 clustering or perforin polarization, suggesting an essential role of DOCK8 as a mediator of NK cell degranulation.
Autosomal dominant AD-HIES syndrome patients present with high levels of serum IgE, although with relatively mild allergy manifestations, as compared with patients with primarily allergic disorders and similar high serum IgE levels. Siegel and collaborators14 compared patients with AD-HIES, with severe atopic dermatitis and a group of healthy controls. Both patient groups presented with eczema and had similar geometric means of total serum IgE and of food specific IgE (milk, egg) levels; however, the percentage of patients reporting food induced anaphylaxis was lower in the AD-HIES group than in patients with eczema (8.5% vs 33.3%). The authors hypothesized that this observation could be explained by decreased mast cell degranulation induced by IgE. AD-HIES patients had wheals induced by histamine with sizes similar to controls, but wheal sizes were significantly decreased when induced by morphine or anti-IgE antibody. In an animal model of HIES, anaphylaxis induced by IgE was significantly attenuated, as measured by skin temperature change. Working with a human mast cell line, the authors showed that knock-down of STAT3 expression leads to reduced phosphorylation of proximal signal transduction proteins involved in degranulation pathways.
To explore the role of IL-21 in the differentiation of human CD8+ T cells, Ives and colleagues15 examined patients with mutations in STAT3, STAT1 and IL-21R. They demonstrated that the CD8+ T cell differentiation into effector cells in response to IL-21 was impaired in patients with STAT3 deficiency, but not in patients with STAT1 deficiency, confirming the role of STAT3 downstream of IL21R signaling cascade for CD8+ T cell differentiation. This impairment was corrected when IL-2 and IL-15 stimulation were added, suggesting that additional signals can replace the role of IL21. Also in this pathway, follicular T helper (CD4+) cells secrete IL-21 and CXCL13, thus participating in the generation of specific antibodies. Mazerolles and collaborators16 found that the average proportion of peripheral blood CD4+CD45RO+CXCR5+ T cells from 9 patients with AD-HIES were significantly decreased compared to healthy controls (mean 11.4 vs 3.4%).
Poliani et al.17 studied thymi from 2 infants with defects of ζ-chain associated protein of 70 kDa (ZAP70). Compared to control subjects, these patients had smaller thymic medullary areas, and reduced expression of AIRE and of markers of mature medullary thymic epithelial cells (mTECs). These observations may help explain the clinical manifestations of immune dysregulation that have been observed in some patients with ZAP70 deficiency. Perez de Diego and coworkers18 took advantage of proteome technology to identify therapeutic targets for inherited susceptibility to herpes simplex. They analyzed fibroblasts from a patient with defect in unc-93 homolog B1 (C. elegans) (UNC93B) and compared these with fibroblasts of a healthy control. The authors identified specific protein networks, and identified the upregulation of superoxide dismutase 2, as a candidate for potential intervention.
Driessen et al.19 explored mechanisms of antibody deficiency in a cohort of 15 patients with ataxia telangiectasia (AT), from which only three patients presented with hypogammaglobulinemia in infancy, and four presented with late-onset AT. T and B cell phenotyping demonstrated changes that were most significant in patients with low IgG levels: low B cell number, low percentage of naïve and transitional B cells, and increased CD21lowCD38low anergic B cells. Naive CD4+ and CD8+ T cells were also reduced. Somatic hypermutation rate was not affected, suggesting affinity maturation was conserved. However, class-switching recombination events were reduced when measured by the quantification of IGH transcripts. The authors conclude that AT patients might develop decreased immunity because of reduced B and T production secondary to deficient VDJ recombination, and reduced recombination of distal IGH constant genes (IGHG2, IGHG4, IGHA2). Because those with late onset AT did not present these findings, the degree of immune deficiency is likely dependent on residual ATM kinase activity.
The role of transactivator and calmodulin inactivator (TACI) in the pathogenesis of common variable immunodeficiency (CVID) has become unclear, with reports of relatives of CVID patients carrying the same mutations yet not presenting with hypogammaglobulinemia. Martinez-Gallo and collaborators20 showed that peripheral blood B cells isolated from members of 7 families presenting with TACI mutations and normal IgG levels were defective in vitro. TACI expression was decreased and could not be upregulated by TLR9 stimulation. In addition, a proliferation inducing ligand (APRIL) binding was impaired. The authors suggest that deleterious heterozygous mutations in TACI participate in the development of CVID, however environmental events or other genes might be necessary.
In a cohort of 193 patients with primary antibody disorders, Chew and collaborators21 investigated whether the 1858T allele of the protein tyrosine phosphatase non-receptor type 22 (PTPN22) was associated with the occurrence of autoimmunity, as this association occurs in immunocompetent population. The presence of this allele has been associated with increased risk of type I diabetes, Graves’ disease, rheumatoid arthritis and lupus. Sixteen (43%) of 37 patients with antibody disorders and autoimmunity had at least one T allele compared with 27 (1%) of 156 without autoimmune disease.
Periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis (PFAPA) syndrome is an inflammatory disease of unknown pathogenesis, with diagnosis based on clinical presentation. Kolly et al.22 studied 15 patients with PFAPA, reporting that IL-1β production by stimulated monocytes is increased at baseline and during a febrile attack.
Primary Immunodeficiencies: Reports of novel gene defects
The accelerated rate of discovering primary immune defects was illustrated by a review of 19 new gene defects responsible for immunodeficiency disorders, which were published within one year of the last international classification report. Parvaneh and coauthors23 summarized the clinical phenotypes of these gene defects: five resulted in combined immunodefiencies, four were defects of innate immunity (two presenting with herpes encephalitis), three were predominantly antibody defects, and there were two of each in the categories of well-defined syndromes with immunodeficiency, and deficiencies of immune dysregulation and autoinflammatory disorders. There was one description of a new defect in the group of congenital defects of phagocytes. Recently developed genetic technologies, such as whole-genome and whole exome sequencing, continue to uncover genes causing inherited defects. Greil et al.24 described an infant of consanguineous parents who developed P.jiroveci pneumonia at 6 months of age. Immunological investigations showed normal T and B cell counts, but T cell proliferation to mitogens was strongly impaired. Phenotyping revealed that T cells were predominantly naïve T cells. Whole genome sequencing suggested only one potential defect, identified as a premature stop codon in the gene caspase recruitment domain 11 (CARD11). Reconstitution studies using CARD11-deficient Jurkat T cell line demonstrated that the mutated CARD11 protein did not facilitate IL-2 production and cell proliferation after CD3/CD28 stimulation.
Stepensky and collaborators25 reported a 9 month-old patient with an upper respiratory infection who was found to have panhypogammaglobulinemia. Lymphocyte phenotype and T cell receptor excision circle (TREC) testing were reported to be within the normal range. Mitogen-stimulated proliferation of lymphocytes was normal. B cell phenotyping, however, suggested a block in the transitional stage of B cell development. Because of parent consanguinity, whole genome sequencing demonstrated a homozygous deletion of exon 21 in the gene CARD11. T cell proliferation induced by CD3/CD28 was non reactive. Hematopoietic stem cell transplantation from her brother resulted in complete immune reconstitution.
Jabara et al.26 studied 2 siblings from consanguinous parents who presented with recurrent pneumonias, meningitis and failure to thrive. T cell and B cells percentages were within the normal range, however T cell proliferations to mitogens and antigens were depressed. Whole genome sequencing revealed a homozygous missense mutation in MALT1 that was predicted to result in protein degradation. Reconstitution experiments were performed with cells from Malt1 knock-out mice: IL-2 secretion after PMA/ionomycin stimulation in murine T cells was detected when these cells were transduced with wild-type human MALT1 stimulation, but not when they were transduced with the mutated MALT1. The authors suggest testing for mutations in this gene in patients with combined immunodeficiencies (CID) and normal T and B cell counts.
Congenital multiple intestinal atresias (MIA) is a rare inherited condition that may associate with combined immunodeficiency (CID). Chen and collaborators27 performed whole exome sequencing in 15 subjects from 5 families with CID-MIA. Deleterious mutations in the tetratricopeptide repeat domain 7A (TTC7A) gene were found in all five families. TTC7A has been previously shown to play a role in cellular functions including cell cycle control, protein scaffolding, and intracellular signaling.
Magerous-Chatinet et al.28 reported an infant of consanguineous parents with repeated infections, hepatosplenomegaly and lymphadenopathy who had lymphocytosis, consisting mostly of double negative (i.e. CD4− CD8−) lymphocytes, suggesting autoimmune lymphoproliferative disease (ALPS). When the FAS ligand in serum was not detected, sequence analysis of tumor necrosis factor ligand superfamily, member 6 (TNFSF6 or FASLG) showed a premature stop codon in exon1 resulting in absent protein expression.
The simultaneous occurrence of two or more gene defects in a patient might modify traditional phenotypes. This case is illustrated by the report of 4 brothers of consanguineous parents who presented with severe eczema, diarrhea, prolonged failure-to-thrive, and frequent respiratory infections.29 The FOXP3 gene sequence was normal and Tregs had normal suppressive activity. Genomic-wide genotyping identified homozygous pathogenic mutations in DOCK8, and in CLEC7A, previously associated to AR-HIES and CMC, respectively. The authors postulate that the combination of gene defects was responsible for the unusual clinical presentation in this family. Hsu and collaborators30 reported two unrelated subjects with moderate clinical symptoms corresponding to AD-HIES. Molecular analysis revealed STAT3 mutations with 33% mosaicism in several tissues, including blood cells, hair follicle and stomach biopsy. The authors postulate that mosaicism might explain incomplete clinical presentation, and represents a diagnostic challenge since detection requires careful examinations of sequencing output and might be missed by automated software programs used in gene sequencing
Giancane et al.31 studied a 5 month-old girl with severe pneumonia and sepsis who was found to have low serum IgG with increased IgM levels. Opportunistic infections, female gender and hypohydrosis suggested investigation of IkBα protein, as a cause of autosomal dominant form of ectodermal dysplasia with immunodeficiency. The authors report a newly characterized missense IKBA mutation predicted to prevent IkBα from activating NFkB
Primary Immunodeficiencies: New phenotypes for known gene defects
The spectrum of immunological disorders due to recombinase activating gene 1 (RAG1) mutations continues to expand. Abraham and collaborators32 reported a patient with T cell lymphopenia presenting at 38 years of age, with a clinical syndrome including mildly elevated IgE and chronic dermatitis. RAG1 sequencing detected a heterozygous mutation previously reported to cause SCID or Omenn syndrome (OS) when present in a homozygous mutation. The authors suggested that RAG1 hypomorphic mutations might explain this patient’s T cell lymphopenia.
Henderson et al.33 reported early-onset autoimmunity as a clinical phenotype within the spectrum of RAG deficiency. In two sisters with T cell lymphopenia, preserved B cells counts and normal immunoglobulin levels. These sisters had low antibody responses to childhood vaccines and one of the sisters developed autoimmune cytopenias, polymyositis and Raynaud symptoms. Further testing demonstrated biallelic RAG1 mutations leading to reduced recombinase activity. This report adds to the list of conditions that present with autoimmunity in early infancy.
Cattaneo and coauthors34 reported 3 patients with a state of increased frequency of infections with normal numbers of T and B cells, but with different subset abnormalities, such as reduced memory B cells in two patients. Their T cell repertoires were restricted and TREC levels were undetectable. Compound heterozygous gene mutations were found in Janus-associated kinase 3 (JAK3). Of note, there was significant engraftment of maternal T cells in one of the patients over 33 months of age. Farnault et al.35 reported a 27 month-old boy with recurrent infections and with a hypomorphic mutation in JAK3, presenting with expansion of a CD56−CD16+KIR+NKG2C+ subset of NK cells. Blood NK cells were 58% of lymphocytes, and IL7R gene sequence was normal. These two cases represent unusual manifestations of JAK3 deficiency, which has traditionally been described as a T− B+NK− SCID.
Coronin-1A (CORO1A) deficiency has been associated with a SCID phenotype. Moshous et al.36 reported 3 siblings with EBV-associated B cell lymphomas presenting in infancy. Whole exome sequencing revealed a missense mutation in CORO1A, with considerably reduced protein expression. Immunological studies showed T cell lymphopenia with absent naïve T cells TCR Vβ repertoire and oligoclonality with expansion of memory T cells. The authors conclude that CORO1A defects should be suspected in infants who develop EBV-associated B cell lymphomas. The list of gene defects that might present with characteristic of Omenn syndrome is growing. Henderson and collaborators37 reported an infant who had severe desquamative eczema and was shown to have neurosensory deafness, absolute neutropenia, and lymphopenia with presence of T cells, which were found to be oligoclonal. He was the brother of a girl previously diagnosed with reticular dysgenesis, and had a homozygous mutation in adenylate kinase 2 (AK2) gene. Testing for AK2 mutations revealed the same homozygous genetic lesion.
Autosomal dominant STAT1 gain-of-function mutations are associated with chronic mucocutaneous candidiasis (CMC). Wang et al.38 reported a 6 year-old girl who had a chronic granulomas caused by Fusarium solain. Exome sequencing resulted with a previously described pathogenic STAT1 mutation. Sampaio et al.39 reported STAT1 gain-of-function mutations in 3 patients with histoplasmosis and two patients with refractory coccidioidomycosis. These articles extend the range of infectious agents affecting patients with STAT1 mutations in addition to Candida sp. Because these STAT1 mutations are also associated with autoimmunity, Uzel and collaborators40 examined a series of 73 patients with immunodeficiency, polyendocrinopathy, enteropathy, X-linked (IPEX)-like syndrome, who did not have FOXP3 mutations. They found 5 patients presenting with STAT1 gain-of-function mutations, with four of these patients also suffering of chronic candidiasis. Romberg and collaborators41 described 4 members of a family with the gain-of-function STAT1 mutation pE235A who developed, in addition to CMC, B cell lymphopenia and antibody deficiency. It is still unclear what factors determine which immunological functions are affected when STAT1 gain-of-function mutations are present.
Allenspach and coauthors42 reported a patient who presented with frequent infections and hypogammaglobulinemia in childhood and received a diagnosis of CVID. At 20 years of age, he developed pure red cell aplasia and profuse diarrhea. A colon biopsy showed massive apoptosis of crypt cells, suggesting dyskeratosis congenita. Pathogenic mutations were found in the dyskeratosis congenita (DKC1) gene, and decreased expression of dyskerin protein. This report reminds us that patients with defects in DKC1 might initially presents with hypogammaglobulinemia.
Primary Immunodeficiencies: Diagnostic issues
Two articles report patients with unusual clinical findings. Bacon et al.43 reported a patient with DNA ligase 4 deficiency who developed a B cell lymphoma that was not associated to Epstein-Barr virus (EBV). Al-Zahrani et al.44 described costochondral dysplasia in a premature infant with a diagnosis of reticular dysgenesis. Costochondral dysplasia has not previously been associated to an immune disorder other than adenosine deaminase deficiency.
Defects of the IL12/IFNγ axis might require an additional event to show susceptibility to mycobacterial infection, as shown by Tassone and collaborators.45 They reported a patient with autoimmune hepatitis who had received immunosuppression for over 10 years and was found to have intra-abdominal mycobacterial granulomas. Decreased expression of IL-12RB and IL-12-induced IFNγ led to gene sequencing studies and confirmation of IL-12RB1 deficiency.
In addition to the identification of genetic lesions, confirmation of the mode of inheritance is important for predicting the risk of recurrence. Geeden et al.46 reported an infant with recurrent infections who was found to have deficiency of adenosine deaminase (ADA). Molecular diagnosis revealed a homozygous deletion mutation in the ADA gene that produced a premature stop codon. Only the father, but not the mother, was a carrier of this mutation. A SNP array revealed uniparental disomy of chromosome 20, which carries a very low risk of recurrence should the parents consider conceiving again. Alsina et al.47 reported a patient with X-linked SCID, with a mutation pR226C in the IL2RG gene, previously known to cause SCID. His mother was a heterozygous carrier who had the mutation at different percentages according to the tissue examined. Her mosaicism was lowest at 8% in lymphocytes and highest at 20% for neutrophils and other tissues examined reflecting the selective advantage of lymphocytes expressing the wild-type IL2Rγ protein for their development. Her risk of having another boy with SCID might be decreased by this mosaicism.
By reporting the analysis of Bruton’s tyrosine kinase (BTK) gene missense mutations in 12 patients with antibody deficiencies, Abbott and collaborators48 reminded us that not every aminoacid change results in loss of function. Identification of gene mutations should be followed with assessment of stability, expression and function of the mutant protein.
CVID is characterized by late-onset B cell deficiency, with absent or reduced production of antibodies, and hypogammaglobulinemia. In addition, various T- cell defects might occur and the diagnosis of combined immunodeficiency is often considered in these cases. Those with T cell defects are more susceptible to opportunistic infections, and might present with autoimmunity and malignancies. To improve accuracy of patient diagnosis and prognosis, Kamae and collaborators49 studied TREC and KREC in a cohort of 40 hypogammaglobulinemic patient, who did not have molecular diagnosis and classified them in four groups according to the presence of normal numbers of TREC, and immunoglobulin kappa-deleting recombination excision circles (KREC): Group A TREC+KREC+ 19 patients, Group B TREC+KREC− 7 patients, Group C TREC−KREC+ 8 patients, and Group D TREC−KREC− 6 patients. Group D most resembles the description of combined immunodeficiency and presented with a high incidence of opportunistic infections, autoimmunity and malignancy. Group C did not fulfill criteria for combined immunodeficiency with B cells, however both Groups C and D are likely to benefit from patient management similar to SCID/CID. Autoimmunity was significant in Group B. The authors proposed to use this classification to consider therapeutic options for individual CVID patients.
Newborn screening of T cell deficiencies
Kwan and collaborators50 reported 2-yer period data from the SCID newborn screening program in California. From approximately 1 million newborns tested, 50 cases with low TREC number were later confirmed to have T cell lymphopenia. Fourteen of these 50 cases were diagnosed as SCID, and received HSCT. One was diagnosed with complete DiGeorge syndrome and received a thymus transplant. Survival of these patients with these severe conditions was remarkable at 93%. The diagnoses of the other 35 patients with T cell lymphopenia were: secondary T cell lymphopenia (n=9), non-immunological genetic syndromes (n=12), extreme prematurity (n= 8), combined immunodeficiencies (n=6). In addition to improvement of survival outcomes, universal newborn screening has helped to establish true incidence of these relatively rare diseases in California, which for SCID appears to be 1/66,250 live births. Of significance, 3 research groups reported patients with SCID who had normal TREC counts at birth. La Marca et al.51 reported 3 patients with delayed-onset ADA-SCID, with diagnosis at 3, 4 and 7 years-old, and studied their newborn dried blood spots. TREC analyses were within normal limits, but levels of adenosine were elevated. This report proposes that newborn screening should include tandem mass spectrometry, considering its low cost. Lev et al.52 reported eight patients with MHC class II deficiency and reported TCR Vβ spectrotyping and TREC quantification as a measure of thymic activity. The analysis of TCR Vβ repertoire appeared normal when performed on total lymphocytes, however clonal expansion was evident when only CD4+ T cells were examined. In contrast, the CD8+ T cell Vβ repertoire distribution was normal. When TREC counts were measured, they were detected in normal range. Similarly, Kao and collaborators53 reported two patients with MHC Class II deficiency that had normal TRECs at birth. The authors emphasize that normal TREC levels would not rule-out MHC Class II deficiency.
Primary Immunodeficiencies: Management
Cole et al54 reported a cohort of 62 patients with chronic granulomatous disease (CGD) identified in the United Kingdom and Ireland, 52 (92%) were male and with X-linked inheritance. The median age at diagnosis was 5.21 years in males and 4.06 in females. Thirty (48%) of patients had received HSCT, 28 of whom were males. The authors compared survival and complications rate with HSCT. Survival was similar in both groups at 90%, patients without HSCT had a rate of severe infections at 0.71 episodes/CGD life year, compared to 0.15 episodes after receiving HSCT. Although the contrast of infectious episodes did not control for disease severity or patient age, the results support considering HSCT early in the management of CGD patients. Koker and colleagues55 presented a series of 89 Turkish patients with diagnosis of CGD. Fifty-five (61.8%) of patients were suspected to have autosomal recessive inheritance. The authors investigated NADPH oxidase activity, as measured by dihydrorhodamine (DHR) assay, and studied correlation with disease severity. The authors emphasized that severity of clinical presentation is associated to residual NADPH but not with the specific gene that is mutated, and should be considered for assessment of prognosis and use of HSCT. Notarangelo commented on the long road to optional management of patients with CGD and predicted better outcomes with improvement in methods of HCST and gene therapy.56 Horn and Cowan57 reviewed the published literature concerning outcomes of HSCT for SCID.
Based on available data for immunoreconstitution and mortality, they report on the most optimal conditioning regimens and graft vs host prophylaxis for the different SCID phenotypes. The authors propose that the current increased number of SCID infants being identified through the newborn screening program should receive the best treatment protocols. The authors also discussed the need to define long term immune reconstitution and potential late effects of transplantation, such as neurological deficits, delayed growth and development. Haddad and coauthors58 reviewed B cell immunity outcomes after HSCT in SCID patients, with regards to the use of conditioning regimens. They concluded that available studies did not allow a definitive answer to this question, however the data suggested that some genetic defects such as IL7R deficiency would result in normal B cell function without B cell chimerism. Potential long-term adverse effects should be considered in the decision of using of conditioning regimens and GvHD prophylaxis to improve B cell chimerism. Along these lines, the neuropsychological evaluation of 21 patients with hemophagocytic lymphohystiocytosis (HLH) who received HSCT was reported by Jackson and colleagues.59 Cohort characteristics included that most patients were diagnosed with perforin deficiency (n=11), and 16 had reduce intensity conditioning. Full-scale intelligence quotient, social communication and quality of life were reduced to 80%, compared with their healthy siblings. The authors acknowledged that these late neurological effects might also result from the HLH, as it is unclear if these deficiencies were present before transplantation.
Lawrence and collaborators60 reported the presence of allergic disorders in 9 (50%) patients of a cohort of 18 patients with ADA-SCID who had received HSCT, gene therapy and/or polyethylenglycol (PEG) ADA. Eight of the 9 patients with allergy symptoms were receiving PEG-ADA, as compared with three of 9 patients not currently receiving PEG-ADA, suggesting that severity of disease and/or management success are determinant factors for allergy.
Al-Mousa et al.61 reported one case of DOCK8 deficiency who received an HLA-matched stem cell transplant without a conditioning regimen. Their patient presented with lymphoid and myeloid chimerism at 12 months. He was able to clear Cryptosporidium infection and viral infections, although he developed chronic GvHD. The authors raised the questions of whether HSCT for DOCK8 deficiency can be successful with limited conditioning for patients in critical condition. Patients with deficiencies in the IL-10 receptor or IL-10 are characterized by refractory inflammatory bowel disease (IBD), presenting in childhood. In a series of 40 children with IBD, Engelhardt and coauthors62 searched for mutations in the IL10 and IL10R genes. Seven (18%) patients were found with deleterious mutations in the IL10R genes, 3 patients with homozygous mutations in IL10RA, one patient with compound heterozygous mutations in IL10RB and 2 patients with a homozygous mutation in IL10RB. Compared with the other 33 patients without mutations in this series, IL-10R deficient patients were younger, although with similar severe course of disease. One of the patients received a HLA-matched HSCT, and was doing well after a year . By contrast, the other six patients continue with colitis requiring multiple immunosuppression regimens and surgeries. Two patients with IL10 deficiency and inflammatory bowel disease also received HSCT and resolved their colitis. The authors recommend looking for defects of IL-10/IL-10R in pediatric patients with IBD, because HSCT may potentially be curative.
Three articles favor the use of HSCT for treatment of severe immunodeficiencies while the patient was active with chronic viral infections. Kim et al.63 reported clearance of bacterial and viral infections in ZAP70 deficiency using a HLA matched donor. Slatter et al.64 reported that four of 5 patients cleared CMV, RSV and adenoviral infections after receiving HSCT. Toyoda et al.65 used a cord blood transplant with myeloablative conditioning, rituximab and antiviral medications in a 18 month-old boy with WAS who was suffering from refractory thrombocytopenic purpura (TTP) and chronic CMV infection. The patient was immunoreconstituted without TTP recurrence.
Therapeutic use of immunoglobulins
Gelfand and coauthors66 reviewed indications for the use of IgG replacement therapy in antibody deficiency conditions and discussed scenarios in which the use of this therapy might not be justified, such as for patients with IgG subclass deficiency not associated with increased frequency of infections. They emphasized the need of establishing objective criteria to determine whether a patient benefits from IgG replacement therapy, chief among them a validated functional assay of antibody avidity. Agrawal and Cunningham-Rundles67 developed and proposed a scoring system for establishing need of IgG replacement, based on a review of 65 patients with who were evaluated for suspected antibody deficiency. Serum immunoglobulin levels and specific antibody responses as well as clinical history were considered.
A pharmaco-economic comparison of the subcutaneous administration of IgG products at home to the intravenous administration of IgG at a hospital was conducted in a Canadian center, resulting in less expenditure when using the subcutaneous route.68 It is still unclear if similar results would occur in different communities.
While screening for mRNA levels of the receptor FcγRIIa in CVID patients, van der Hedjen and collaborators69 found a novel FcγRIIa isoform that expressed an additional exon due to a point mutation. Neutrophils expressing this isoform showed increased elastase release in response to dimeric IgG, while responses to the stimulant fMLP were not different, suggesting specificity for IgG. This isoform was found in 3 of 8 CVID patients who developed anaphylaxis with IVIG infusions, but in none of 45 CVID patients who received IVIG without side effects. Two of the 3 patients had been shown to have anti-IgA antibodies. The allele was also found in 2.4% of 208 patients with Kawasaki Disease, however they not report side effects. The authors suggest that the novel isoform might increase the risk of anaphylaxis in CVID patient, and possibly only in those who present with anti-IgA antibodies.
Other therapeutic interventions of immunological importance
Wildbaum et al.70 described the use of granulocyte-colony stimulating factor (G-CSF) for the control of CMC in a 59 year-old female, with CMC diagnosis for over 30 years, with a heterozygous gain-of-function mutation in STAT1. The patient has received G-CSF weekly for 19 years. While on G-CSF, TH17 cells counts were comparable to healthy controls, however, within 4 weeks of stopping G-CSF, symptoms recurred and TH17 cells decreased considerably.
Autophagy is a cellular mechanism that allows cell survival avoiding apoptosis and is thought to play a significant role in T cell homeostasis. Van Loosdregt et al.71 investigated the modulation of autophagy in human T cells by hydroxychloroquine, an anti-inflammatory drug that is thought to affect lysosomes and inhibits authophagy. At concentrations known to be anti-inflammatory in humans, exposure to hydroxychlorquine increases the number of autophagosomes in CD4+ T cells and induces apoptosis. When CD4+CD45RA+ naïve T cells, and CD4+CD45RO+ memory T cells were analyzed, the effect of this drug was markedly more significant in CD45RO+ T cells. The authors conclude that this mechanism might explain the modulatory effect of hydroxychloroquine in inflammation.
Motivated by reports of antibody deficiency by use of valproic acid, an anti-epiletic medication, Kienzler et al.72 studied B cell differentiation after exposure to valproic acid. Differentiation into memory B cells and plasmablasts was significantly reduced, most likely as a result of inhibition of proliferation. The authors suggest that valproic acid might be used in the treatment of autoimmunity.
Conclusions
Research activity in primary immunodeficiencies continues to bring new information on genes affecting the differentiation and function of the cells of the immune system (Table I). New pathogenic mutations and molecular mechanisms have been reported. With improved access to gene sequencing, different clinical presentations are attributed to gene defects that, in the past, appeared to have a traditional presentation only. Despite the signal advance afforded by genotype analysis, patients with the same genetic mutations are found to express different phenotypes, thus issuing another challenge to the clinical immunologist to look for other factors, both genetic, and non-genetic, to account for this wonderous diversity of gene expression.
Table I.
Selected key advances in basic and clinic immunology in 2013.
Primary immunodeficiencies: Mechanisms of Disease
|
Primary immunodeficiencies: New genes
|
Primary immunodeficiencies: New phenotypes
|
Primary immunodeficiencies: Diagnosis
|
Primary immunodeficiency: Treatment
|
Acknowledgments
We acknowledge the support of NIH grant, AI082978, and the David Fund, the Immunobiology Research Center, and the Jeffrey Modell Diagnostical Research Center of Texas Children’s Hospital.
Abbreviations
- ADA
adenoside deaminase
- CGD
chronic granulomatous disease
- CMC
chronic mucocutanous candidiasis
- CVID
combined variable immunodeficiency
- HIES
hyper IgE syndrome
- HSCT
hematopoietic stem cell transplantation
- IVIG
intravenous immunoglobulins
- KREC
kappa-deleting recombination excision circles
- RAG
recombination activating gene
- SCID
severe combined immunodeficiency
- STAT
signal transduction and activator
- TREC
T cell receptor excision circles
- Tregs
regulatory T cells
- WAS
Wiskott-Aldrich syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
The authors have no conflict of interest with regard to the contents of this article.
References
- 1.Maggina P, Gennery AR. Classification of primary immunodeficiencies: Need for a revised approach? J Allergy Clin Immunol. 2013;131:292–294. doi: 10.1016/j.jaci.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunnignham Rundles C, et al. Primary immunodeficiency diseases, an update of the classification from the international union of immunological societies expert committee for primary immunodeficiecny. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Notarangelo LD. Partial defects of T cell development associated with poor T cell function. J Allergy Clin Immunol. 2013;131:1297–1305. doi: 10.1016/j.jaci.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al Herz W, Al Mousa H. Combined immunodeficiency: the Middle East experience. J Allergy Clin Immunol. 2013;131:658–660. doi: 10.1016/j.jaci.2012.11.033. [DOI] [PubMed] [Google Scholar]
- 5.Orange JS. Natural Killer cell deficiency. J Allergy Clin Immunol. 2013;132:515–525. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol. 2013:536–544. doi: 10.1016/j.jaci.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pieper K, Grimbacher B, Eibel H. B cell biology and development. J Allergy Clin Immunol. 2013;131:959–971. doi: 10.1016/j.jaci.2013.01.046. [DOI] [PubMed] [Google Scholar]
- 8.Mehling M, Brinkmann V, Burgener AV, Gubser P, Luster AD, Kaposs L, Hess C. Homing frequency of human T cells inferred from peripheral blod depletion kinetics after sphingosine-1-phosphate receptor blockade. J Allergy Clin Immunol. 2013;131:1440–1443. doi: 10.1016/j.jaci.2012.12.1520. [DOI] [PubMed] [Google Scholar]
- 9.Hirahara K, Poholek A, Vahedi G, Laurence A, Kanno Y, Milner JD, O'Shea JJ. Mechanisms underlying helper T cell plasticity: Implication for immune-mediated disease. J Allergy Clin Immunol. 2013;131:1276–1287. doi: 10.1016/j.jaci.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimizu M, Kanegane H, Wada T, Motoyoshi M, Morio T, Candotti F, Yachie A. Aberrant glycosylation of IgA in Wiskott-Aldrich syndrome and X-linked thrombocytopenia. J Allergy Clin Immunol. 2013;131:587–589. doi: 10.1016/j.jaci.2012.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang PA, Shaabani N, Borkens S, Honke N, Scheu S, Booth A, et al. Reduced Type I Interferon production by dendritic cells an weakened antiviral immunity in patients with Wiskott-Aldrich Syndrome protein deficiency. J Allergy Clin Immunol. 2013;131:815–824. doi: 10.1016/j.jaci.2012.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe Y, Sasahara Y, Ramesh N, Massad MJ, Looi CY, Kumaki S, et al. T-cell receptor ligantion causes Wiakott Aldrich protein degradation and F-actin assembly downregulation. J Allergy Clin Immunol. 2013;132:648–655. doi: 10.1016/j.jaci.2013.03.046. [DOI] [PubMed] [Google Scholar]
- 13.Mizesko MC, Banerjee PP, Monaco-Shawver L, Mace EM, Bernal WE, Sawalle-Belohradsky J, et al. Defective actin accumulation impairs human natutal killer cell function in patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2013;131:840–848. doi: 10.1016/j.jaci.2012.12.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegel AM, Stone KD, Cruse G, Lawrence MG, Olivera A, Jung M, et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J Allergy Clin Immunol. 2013;132:1388–1396. doi: 10.1016/j.jaci.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ives ML, Ma CS, Palendira U, Chan A, Bustamante J, Boisson-Dupuis S, et al. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper IgE syndrome impair human CD8+ T cell memory formation and function. J Allergy Clin Immunol. 2013;132:400–411. doi: 10.1016/j.jaci.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazerolles F, Picard C, Kracker S, Fischer A, Durandy Blood CD4+CD45RO+ CXCR5+ T cells are decreased but partially functional in signal transducer and activator of transcription 3 deficiency. J Allergy Clin Immunol. 2013;131:1146–1156. doi: 10.1016/j.jaci.2012.12.1519. [DOI] [PubMed] [Google Scholar]
- 17.Poliani PL, Fontana E, Roifman CM, Notarangelo LD. z-chain associated protein of 70 kDa (ZAP70) deficiency in human subjects is associated with abnormalities in thymic stromal cells: implications for T cell tolerance. J Allergy Clin Immunol. 2013;131:597–600. doi: 10.1016/j.jaci.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Perez de Diego R, Mulvey C, Crawford M, Trotter MWB, Lorenzo L, Sancho-Shimizu V, et al. The proteoma of Toll-like receptor 3-stimulated human immortalized fibroblast: implications for susceptibility herpes simplex encephalitis. J Allergy Clin Immunol. 2013;131:1157–1166. doi: 10.1016/j.jaci.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Driessen GJ, Ijspeert H, Weemaes CMR, Haraldsson A, Trip M, Warris A, et al. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T- cell homeostasis and reduced immune repertoire diversity. J Allergy Clin Immunol. 2013;131:1367–1375. doi: 10.1016/j.jaci.2013.01.053. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Gallo M, Radigan L, Almejun MB, Martinez-Pomar N, Matamoros N, Cunningham-Rundles C. TACI mutations and impaired B cell function in subjects with CVID and healthy heterozygotes. J Allergy Clin Immuno. 2013;132:468–476. doi: 10.1016/j.jaci.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chew CYG, Sinha U, Gatenby PA, DEMalmanche T, Adelstein S, Garsia R, et al. Autoimmunity in primary antibody deficiency is associated with proteinthyrosine phosphatase nonrecetor type 22 (PTPN22) J Allergy Clin Immunol. 2013;131:1130–1135. doi: 10.1016/j.jaci.2012.06.023. [DOI] [PubMed] [Google Scholar]
- 22.Kolly L, Busso N, von Scheven-Gete A, Bagnoud N, Moix I, Holzinger D, et al. Periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis syndrome is linked to dysregulated monocyte IL-1b production. J Allergy Clin Immunol. 2013;131:1635–1643. doi: 10.1016/j.jaci.2012.07.043. [DOI] [PubMed] [Google Scholar]
- 23.Parvaneh N, Casanova JL, Notarangelo LD, Conley ME. Primary immunodeficiencies: A rapidly evolving story. J Allergy Clin Immunol. 2013;132:314–323. doi: 10.1016/j.jaci.2012.11.051. [DOI] [PubMed] [Google Scholar]
- 24.Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredijian-Ding I, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol. 2013:1376–1383. doi: 10.1016/j.jaci.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, Cohen S, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. 2013;131:477–485. doi: 10.1016/j.jaci.2012.11.050. [DOI] [PubMed] [Google Scholar]
- 26.Jabara HH, Ohsumi T, Chou J, Masaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. 2013;132:151–158. doi: 10.1016/j.jaci.2013.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen R, Gilliani S, Lanzi G, Mias GI, Lonardi S, Dobbs K, et al. Whole-exome seqeuncing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J Allergy Clin Immunol. 2013;132:656–664. doi: 10.1016/j.jaci.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magerous-Chatinet A, Stolzenberg MC, Lanzarotti N, Neven B, Daussy C, Picard C, et al. Autoimmune lymphoproliferative syndrome caused by a homozygous null FAS ligand (FASLG) mutation. J Allergy Clin Immunol. 2013;131:486–490. doi: 10.1016/j.jaci.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dinwiddie DL, Kingsmore SF, Caracciolo S, Rossi G, Moratto D, Mazza C, et al. Combined DOCK8 and CLECL7A mutations causing immunodeficiency: 3 brothers with diarrhea, eczema and infections. J Allergy Clin Immunol 2013. 2013;131:594–596. doi: 10.1016/j.jaci.2012.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu AP, Sowerwine KJ, Lawrence MG, Davis J, Henderson CJ, Zarember KA, et al. Intermediate phenotypes in patients with autosomal dominant hyper IgE syndrome caused by somatic mosaicism. J Allergy Clin Immunol. 2013;131:1586–1593. doi: 10.1016/j.jaci.2013.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giancane G, Ferrara S, Carsetti R, Papoff P, Iacobini M, Duse M. Anhidrotic ectodermal dysplasia: A new mutation. J Allergy Clin Immunol. 2013;132:145–147. doi: 10.1016/j.jaci.2013.05.034. [DOI] [PubMed] [Google Scholar]
- 32.Abraham RS, Recher M, Gilliani S, Walter JE, Lee YN, Fragoni F, et al. Adult onset manifestation of idiopathic T-cell lymphopenia due to heterozygous RAG1 mutation. J Allergy Clin Immunol. 2013;131:1421–1422. doi: 10.1016/j.jaci.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henderson LA, Fragoni F, Hopkins G, de Boer H, Pai SY, Lee YN, et al. Expanding the spectrum of recombinase activating gene 1 deficiency: A family with early onset autoimmunity. J Allergy Clin Immunol. 2013;132:969–971. doi: 10.1016/j.jaci.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cattaneo F, Recher M, Masneri S, Baxi SN, Fiorini C, Antonelli F, et al. Hypomorphic Janus kinase 3 mutations result in a spectrum of immune defects, including partial maternal T-cell engrafment. J Allergy Clin Immunol. 2013;131:1136–1145. doi: 10.1016/j.jaci.2012.12.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farnault L, Chambost H, Michel G, Thuret I, de Saint Basile G, Fischer A, et al. Persistence of natural killer cells with expansionof a hypofunctional CD56CD16+KIR+NKG2C+ subset in a patient with atypical Janus Kinase 3-deficient severe combined immunodeficiency. J Allergy Clin Immunol. 2013;131:1230–1231. doi: 10.1016/j.jaci.2012.08.047. [DOI] [PubMed] [Google Scholar]
- 36.Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol. 2013;131:1594–103. doi: 10.1182/blood-2012-07-440339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henderson LA, Fragoni F, Hopkins G, AL-Herz W, Weihacht K, Comeau AM. First reported case of Omenn syndrome in a patient with reticular dysgenesis. J Allergy Clin Immunol. 2013;131:1227–1230. doi: 10.1016/j.jaci.2012.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Lin Z, Gao L, Wang A, Wan Z, Chen W, et al. Exome sequencing reveals a signal transducer and activator of transcription 1 (STAT1) mutations in a child with recalcitrant fusariosis. J Allergy Clin Immunol. 2013;131:1242–1243. doi: 10.1016/j.jaci.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 39.Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias D, Paulson ML, et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutaions and disseminated cocidiodomcyosis and histoplasmosis. J Allergy Clin Immunol. 2013;131:1624–1634. doi: 10.1016/j.jaci.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant gain-of-function STAT1 muatioins in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked -like syndrom. J Allergy Clin Immunol. 2013;131:1611–1623. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland S, et al. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B cell survival. J Allergy Clin Immunol. 2013;131:1691–1693. doi: 10.1016/j.jaci.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allenspach EJ, Bellodi C, Jeong D, Kopmar N, Nakamura T, Ochs H, et al. Common variable immunodeficiency as the initial presentation of dyskeratosis congenita. J Allergy Clin Immunol. 2013;132:223–225. doi: 10.1016/j.jaci.2012.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bacon CM, Wilkinson SJ, Spickett GP, Barge D, Lucraft HH, Jackson G, et al. Epstein Barr independent diffuse large B cell lymphoma in DNA ligase 4 deficiency. J Allergy Clin Immunology. 2013;131:1237–1239. doi: 10.1016/j.jaci.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 44.Al-Zahrani D, Al-Ghonaium A, Al-Mousa H, Al-Kassar A, Roifman CM. Skeletal abnormalities and successful hematopoietic stem cell transplantation in patients with reticular dysgenesis. J Allergy Clin Immunol. 132:993–996. doi: 10.1016/j.jaci.2013.04.055. [DOI] [PubMed] [Google Scholar]
- 45.Tassone L, Carvalho ACC, Calabresi A, Tortoli E, Apostoli E, Scomodoro O, et al. Disseminated Mycobacterial genavense infection after immunosuppressive therapy shows underlying new composite heterozygous mutations of B1 unit of IL12 receptor gene. J Allergy Clin Immunol. 2013;131:607–610. doi: 10.1016/j.jaci.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 46.Geeden J, Pfundt R, Meijer J, Verheijen FW, van Kallenburg ABP, Warris A, Marcelis C. Severe phenotype of severe combined immunodeficiency caused by adenosine deaminase deficiency in a patient with a homozygous mutation due to uniparental unisomy. J Allergy Clin Immunol. 2013;132:222–223. doi: 10.1016/j.jaci.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 47.Alsina L, Gonzalez-Roca E, Giner MT, Piquer M, Puga I, Pascal M, et al. Massive parallel sequencing reveals maternal somatic IL2RG mosaicism in an X-linked severe combined immunodeficiency family. J Allergy Clin Immunol. 2013;132:741–743. doi: 10.1016/j.jaci.2013.03.038. [DOI] [PubMed] [Google Scholar]
- 48.Abbott JK, Ochs HD, Gelfand Ew. Coding-region alterations in BTK do not universally cause X-linked agammaglobulinemia. J Allergy Clin Immunol. 2013;132:1246–1248. doi: 10.1016/j.jaci.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 49.Kamae C, Nakagawa N, Saito H, Honma K, Mitsuiki N, Ohara O, et al. Common variable immunodeficiency classification by quantifying T cell receptor and immunoglobulin k-deleting recombination excision circles. J Allergy Clin Immunol. 2013;131:1437–1440. doi: 10.1016/j.jaci.2012.10.059. [DOI] [PubMed] [Google Scholar]
- 50.Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, et al. Newborn screening for severe combined immunodeficiency in California: results of the first two years. J Allergy Clin Immunol. 2013;132:140–150. doi: 10.1016/j.jaci.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.la Marca G, Canessa C, Giocaliere E, Romano F, Duse M, Malvagia S, et al. Tandem mass spectrometry, but not T cell receptor excision circle analysis, identifies newborns with late onset adenosine deaminase deficiency. J Allergy Clin Immunol. 2013;131:1604–1610. doi: 10.1016/j.jaci.2012.08.054. [DOI] [PubMed] [Google Scholar]
- 52.Lev A, Simon AJ, Broides A, Levi J, Garty BZ, Rosenthal E, et al. Thymic function in MHC Class II-deficient patients. J Allergy Clin Immunol. 2013;131:831–839. doi: 10.1016/j.jaci.2012.10.040. [DOI] [PubMed] [Google Scholar]
- 53.Kao CY, Chase J, Lloret MG, Stiehm ER, Moore T, Aguilera MJM, et al. Newborn screening for severe combined immunodeficiency does not identify bare lymphocyte syndrome. J Allergy Clin Immunol. 2013;131:1693–1695. doi: 10.1016/j.jaci.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 54.Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2013;132:1150–1155. doi: 10.1016/j.jaci.2013.05.031. [DOI] [PubMed] [Google Scholar]
- 55.Koker MY, Camcioglu Y, van Leeuwen K, Kilic SS, Barlan I, Yilmaz M, et al. Clinical, functional and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. 2013;132:1156–1163. doi: 10.1016/j.jaci.2013.05.039. [DOI] [PubMed] [Google Scholar]
- 56.Notarangelo LD. The long road to optimal management for chronic granulomatous disease. J Allergy Clin Immuol. 2013;132:1164–1165. doi: 10.1016/j.jaci.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 57.Horn B, Cowan MJ. Unresolved issues in hematopoietic stem cell transplantation for severe combined immunodeficiency: Need for safer conditioning and reduced late effects. J Allergy Clin Immunol. 2013;131:1306–1311. doi: 10.1016/j.jaci.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haddad E, Leroy S, Buckley RH. B-cell reconstitution for SCID: Should a conditioning regimen be used in SCID treatment? J Allergy Clin Immunol. 2013;131:994–1000. doi: 10.1016/j.jaci.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson J, Titman P, Butler S, Bond K, Rao A, Veys P, et al. Cognitive and psychosocial function post hematopoietic stem cell transplantation in hemophagocytic lymphohystiocytosis. J Allergy Clin Immunol. 2013;132:889–995. doi: 10.1016/j.jaci.2013.05.046. [DOI] [PubMed] [Google Scholar]
- 60.Lawrence MG, Barber JS, Sokolic RA, Garabedian EK, Desai A, O'Brien M, et al. Elevated IgE and atopy in patients treated for early onset ADA-SCID. J Allergy Clin Immunol. 2013;132:1444–1446. doi: 10.1016/j.jaci.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al-Mousa H, Hawwari A, Alsum Z. in DOCK8 deficiency donor cell engraftment post-genoidentical hematopoietic stem cell transplantation is possible without conditioning. J Allergy Clin Immunol. 2013;131:1244–1245. doi: 10.1016/j.jaci.2012.12.663. [DOI] [PubMed] [Google Scholar]
- 62.Engelhardt KR, Shah N, Faizura-Yeop I, Kocacik Uygun DF, Frede N, Muise AM, et al. Clinical outcome in IL-10 and IL10 receptor deficient patients with and without hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2013;131:825–830. doi: 10.1016/j.jaci.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 63.Kim VHD, Murguia L, Schechter T, Gruneaum E, Roifman CM. Emergency treatment for z chain-associated protein of 70 kDa (ZAP70) deficiency. J Allergy Clin Immunol. 2013;131:1233–1235. doi: 10.1016/j.jaci.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 64.Slatter M, Nademi Z, Patel S, Barge D, Valappil M, Brigham K, et al. Haploidentical hematopoitic stem cell transplantation can lead to viral clearance in severe combined immunodeficiency. J Allergy Clin Immunol. 2013;131:1705–1709. doi: 10.1016/j.jaci.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 65.Toyoda H, Azuma E, Kawasaki Y, Iwasa T, Ohashi H, Otsuki S, et al. Cord blood transplantation combined with rituximab for Wiskott-Aldrich syndrome with autoimmune thrombotic thrombocytopenic purpura. J Allergy Clin Immunol. 2013;132:226–227. doi: 10.1016/j.jaci.2013.01.040. [DOI] [PubMed] [Google Scholar]
- 66.Gelfand EW, Ochs HD, Shearer WT. Controversies in IgG replacement therapy in patients with antibody deficiency disease. J Allergy Clin Immunol. 2013;131:1001–1005. doi: 10.1016/j.jaci.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 67.Agarwal S, Cunningham-Rundles C. Treatment of hypogammaglobulinemia in adults: a scoring system to guide decisions of immunoglobulin replacement. J Allergy Clin Immunol. 2013;131:1699–1701. doi: 10.1016/j.jaci.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ducruet T, Levasseur MC, Des Roches A, Kafal A, Dicaire R, Haddad E. Pharmaco-economic advantages of subcutanous versus intravenous immunoglobulin treatment in a Canadian pediatric center. J Allergy Clin Immunol. 2013;131:585–587. doi: 10.1016/j.jaci.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 69.van der Heidjen J, Geissler J, van Mirre E, van Deuren M, va der Meer JWM, Salama A, et al. A novel spliced variant of FcgRIIa: a risk factor for anaphylaxis in patients with hypogammaglobulinemia. J Allergy Clin Immunol. 2013;131:1408–1416. doi: 10.1016/j.jaci.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 70.Wildbaum G, Shahar E, Katz R, Karin N, Etzioni A, Pollack S. Continuous G-CSF therapy for isolated chronic mucocutanous candidiasis: complete clinical remission with restoration of IL-17 secretion. J Allergy Clin Immunol. 2013;132:761–763. doi: 10.1016/j.jaci.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 71.van Loosdregt J, Spreafico R, Rossetti M, Prakken BJ, Lotz M, Albani S. Hydroxychloroquine preferentially induces apoptosis of CD45RO+ effector T cells by inhibiting autophagy: a possible mechanism for therapeutic modulation of T cells. J Allergy Clin Immunol. 2013;131:1443–1447. doi: 10.1016/j.jaci.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kienzler AK, Rizzi M, Reith M, Nutt SL, Eibel H. Inhibition of human B cell development into plasmablasts by histone deacetylase inhibitor valproic acid. J Allergy Clin Immunol. 2013;131:1695–1699. doi: 10.1016/j.jaci.2013.01.018. [DOI] [PubMed] [Google Scholar]