

Summary
Fragile X Syndrome (FraX) is a broad-spectrum neurological disorder with symptoms ranging from hyperexcitability to mental retardation and autism. Loss of the fragile X mental retardation 1 (fmr1) gene product, the mRNA-binding translational regulator FMRP, causes structural over-elaboration of dendritic and axonal processes as well as functional alterations in synaptic plasticity at maturity. It is unclear, however, whether FraX is primarily a disease of development, a disease of plasticity or both; a distinction vital for engineering intervention strategies. To address this critical issue, we have used the Drosophila FraX model to investigate the developmental roles of Drosophila FMRP (dFMRP). dFMRP expression and regulation of chickadee/profilin coincides with a transient window of late brain development. During this time, dFMRP is positively regulated by sensory input activity, and required to limit axon growth and for efficient activity-dependent pruning of axon branches in the Mushroom Body learning/memory center. These results demonstrate that dFMRP has a primary role in activity-dependent neural circuit refinement in late brain development.
Keywords: Fragile X Syndrome, mental retardation, autism, neural development, translation, synaptogenesis, synaptic pruning
Introduction
Fragile X Syndrome (FraX) is a commonly inherited mental retardation disorder causing hyperactivity, hypersensitivity to sensory stimuli, epileptic seizures and autism (Belmonte and Bourgeron, 2006; Boccia and Roberts, 2000; Freund and Reiss, 1991; Sabaratnam et al., 2001). The fragile X mental retardation protein (FMRP) is an mRNA-binding protein proposed to function in mRNA trafficking, stability and translational regulation (Ashley et al., 1993; De Diego Otero et al., 2002; Feng et al., 1997; Laggerbauer et al., 2001; Weiler et al., 1997; Weiler et al., 2004; Xu et al., 2004; Zalfa et al., 2007; Zalfa et al., 2003; Zhang et al., 2007). Although FMRP potentially binds 4% of brain mRNAs, only a handful of targets have been validated (Brown et al., 2001; Hayashi et al., 2007; Zhang and Broadie, 2005). Neural activity from sensory experience and metabotropic glutamate receptor signaling increases FMRP expression and function (Gabel et al., 2004; Hou et al., 2006; Irwin et al., 2005; Irwin et al., 2000; Restivo et al., 2005; Todd and Mack, 2000; Todd et al., 2003; Valentine et al., 2000; Weiler et al., 1997). FMRP may repress translation via association with polyribosomes (Aschrafi et al., 2005; Khandjian et al., 2004; Stefani et al., 2004), transported to sites of local synaptic translation in response to neurotransmission (Ferrari et al., 2007; Greenough et al., 1985; Ostroff et al., 2002).
Activity plays critical roles in sculpting neural circuits during development and later mediating plasticity (Desai et al., 2002; Zito and Svoboda, 2002). FMRP may perform a common function in regulating activity-dependent protein synthesis in both settings. In FraX patients, mutant mice and Drosophila, dendritic arbors are overgrown with immature dendritic spines suggesting a failure of synapse maturation (Comery et al., 1997; Galvez et al., 2003; Galvez and Greenough, 2005; Galvez et al., 2005; Grossman et al., 2006; Irwin et al., 2002; Irwin et al., 2001; Ivanco and Greenough, 2002; McKinney et al., 2005; Nimchinsky et al., 2001; Pan et al., 2004; Rudelli et al., 1985). Dendritic defects appear most evident during early postnatal development and abrogate with maturation (Galvez and Greenough, 2005; Nimchinsky et al., 2001). Similarly, mutant neurons exhibit axonal over-branching in mice and Drosophila indicating a similar requirement presynaptically (Antar et al., 2006; Pan et al., 2004). FMRP is required for plasticity in mature synapses; long-term depression (LTD) is enhanced (Hou et al., 2006; Huber et al., 2002; Koekkoek et al., 2005) and potentiation (LTP) is reduced (Larson et al., 2005; Li et al., 2002; Zhao et al., 2005). These data suggest two roles for FMRP: during development to regulate the structuring of neural circuits and during maturity to regulate maintained plasticity.
Drosophila is well suited for the dissection of developmental processes, however the well-characterized Drosophila FraX model has yet to be exploited for this purpose. Therefore, we investigated developmental roles of dFMRP in the Drosophila brain, specifically activity-dependent structural changes driven by sensory input. We find that dFMRP expression is maximal during late-stage periods of axon pruning, which requires both dFMRP and sensory input activity. These results reveal a prominent role for dFMRP in activity-dependent neural circuit refinement.
Materials and Methods
Fly stocks and genetics
The genetic wildtype strain was w1118. The dfmr1 null allele was w; dfmr150M/TM6GFP. The control strain for MARCM analyses was obtained by crossing heatshock-FLP, mCD8-GFP; FRT82B, Tubulin P-Gal80/Sb; Gal4-OK107 with y, w; FRT82B/Sb. The control for sensory stimulation experiments was crossed to y, w; FRT82B, Sb/TM6. The mutant MARCM chromosome was FRT82B, dfmr150M/TM6GFP and overexpression was FRT82B, dfmr150M,UAS-dfmr1/TM6GFP. Standard techniques produced two recombinant channelrhodopsin-2 lines: FRT82b,CHR2/TM6 and FRT82b, dfmr150M, CHR2/TM6GFP. Sensory mutants used were or83b-2 (provided by Dr. Leslie Vosshall, Rockefeller) and ninaE (provided by Dr. Bih-Hwa Shieh, Vanderbilt).
Protein and RNA extraction
Fly heads were frozen on dry ice and stored at −80°C. Protein/RNA was extracted from the same samples of 10–25 pooled heads using Trizol (Invitrogen, Carlsbad, CA). Protein pellets were resuspended in 8 M Urea, 1% SDS supplemented with 1X Complete Protease Inhibitors (Roche, Indianapolis, IN) incubated at 50°C for 1 hour with intermittent vortexing. Protein concentration was determined using a MicroBCA Assay (Pierce, Rockford, IL). RNA pellets were resuspended in DEPC treated water, and concentration determined by absorbance at 260 nm.
Western blotting
Single heads were homogenized in 1X Nupage LDS Sample Buffer (Invitrogen, Carlsbad, CA) with 55 mM DTT. Debris was pelleted by centrifugation at 16,000 × g at 25°C and samples boiled for 10 minutes. Extracts were loaded onto a 4–12% Bis-Tris gel and electrophoresed at 200V in 1X MOPS or 1XMES buffer (Invitrogen, Carlsbad, CA). Protein was transferred to nitrocellulose in 1X Nupage transfer buffer (Invitrogen, Carlsbad, CA) plus 10% methanol at 100V for 1 hour. The membrane was blocked for 1 hour in Odyssey Blocking Buffer (Li-Cor, Lincoln, NE) and probed for 12–16 hours at 4°C with the following antibodies: dFMRP: 6A15 (Sigma, St. Louis, MO) 1:5000 (for developmental blots) or 1:500 (for sensory-deprivation blots); chickadee/profilin (Chi1J, Developmental studies Hybridoma Bank, Iowa) 1:10; α-tubulin (Sigma, St. Louis, MO) 1:400,000. Membranes were washed 3X with buffer (25 mM Tris pH8.0, 150 mM sodium chloride, 0.05% Ige-PAL-CA630). The secondary antibody, anti-mouse IgG IR800 (Rockland, Gilbertsville, PA) was diluted 1:10,000 in Odyssey Blocking Buffer and applied for 1 hour at 25°C. The blot was washed 3X with buffer and then scanned on the Odyssey Infrared Imaging System.
Quantitative RT-PCR
Extracted RNA (2μg) treated with DNase I Turbo (Ambion, Austin, TX) was made into cDNA using random hexamer primers and Superscript II RNase H-Reverse Transcriptase (Invitrogen, Carlsbad, CA). Quantitative PCR of cDNA (1μl) was done using SYBR Green JumpStart Taq Ready Mix (Sigma, St. Louis, MO). The following primers were used at 0.5 μM concentrations each per reaction: gapdh2: 5′-CCTTGCAAGCAAGCCGATAG-3′, 5′-CGACATGGTTAACTTTTTGT-3′; dfmr1: 5′-GTTCGGCTCGACAATGGCGC-3′, 5′-GCGACAGCTGTCACCTGGCC-3′; chickadee: 5′-CGCAGTCCAGTGGCTTTGAG-3′, 5′CGCTGATCAGTTTGGAGAGC-3′. Cycling parameters were 95°C-3 minutes, (95°C-10 seconds, 60°C-30 seconds, 72°C- 30 seconds) x 40 cycles (Bio-Rad iQ5 Thermal Cycler). Each experiment consisted of 3 biological replicates for each time point plated in duplicate.
Immunocytochemistry
Brains were dissected in 1XPBS and fixed in 4% paraformaldehyde + 4% sucrose in 1X PBS for 40 minutes at 25°C. Brains were washed 3X with buffer (1XPBS, 1% BSA, 0.1% Triton X-100) for 30 minutes and incubated with the following primary antibodies at 4°C for 12–16 hours: dFMRP: 6A15 (Sigma, St. Louis, MO) 1:250, mouse CD8 (Caltag, Burlingame, CA) 1:50, FasII (Developmental studies hybridoma bank 1D4, Iowa City, IA) 1:10. Secondary antibodies (anti-mouse-IgG Cy3, anti-rat-IgG-Cy5 (Jackson Immunoresearch, West Grove, PA), anti-mouse-IgG AlexaFluor 488 (Molecular Probes, Eugene, OR)) diluted 1:400 were applied for 2–3 hours at 25°C. Brains were washed 3X for 1 hour before mounting in Vectashield (Vector Labs, Burlingame, CA) and imaging on a Zeiss Meta 510 confocal microscope. Images were collected at identical settings and presented as maximum Z-projections. As previously (Pan et al., 2004), MARCM branch parameters were determined with LSM software on 3D confocal Z-stacks of each neuron.
Results
dFMRP developmentally regulates brain RNA and protein levels
FMRP negatively regulates protein translation in mature brain, but little is known about developmental requirements (Khandjian et al., 2004; Laggerbauer et al., 2001; Li et al., 2001; Qin et al., 2005; Sung et al., 2003). Research on Drosophila brain development has focused on the first 48 hours during pupal metamorphosis (Awasaki and Ito, 2004; Awasaki et al., 2006; Brown et al., 2006; Lee et al., 1999; Marin et al., 2005; Watts et al., 2004; Williams and Truman, 2005; Zheng et al., 2003), but little is known of latter half of brain development. Presumed roles for activity mediate late use-dependent refinement of neural circuits (Boothe et al., 1979; Desai et al., 2002; Fox and Wong, 2005; Hinds and Hinds, 1976; Huttenlocher, 1979; Lund et al., 1977; Pan et al., 2004; Stern et al., 2001; Turrigiano and Nelson, 2004). To begin to assay temporal requirements for dFMRP during brain development versus maturity, we analyzed developmental time points beginning 60 hours after puparium formation (APF) and extending into the mature adult (9 days post-eclosion).
Null dfmr1 mutants (dfmr150M) were compared to controls (w1118) at mid-pupal day 3 (P3; 60–72 hours APF), mid pupal day 4 (P4; 80–90 hours APF), immediately post-eclosion (0–7 hours after eclosion (AE)) and at 1 day (1d), 4 days (4d) and 9 days (9d) in the adult. Total RNA was quantified as μg per head (Fig. 1A). The total amount of RNA was higher in both wildtype and mutants during stages of pupal brain development than at maturity. In dfmr1 null brain, there was a significant increase in RNA only during a restricted window of late pupal development (Fig. 1A). There was a 38% (P3) and 51% (P4) increase in total RNA in the dfmr1 null compared to wildtype (P3: WT, 0.58±0.14μg, dfmr1, 0.79±0.08μg, P=0.025, N=6; P4: WT, 0.47±0.17μg, dfmr1, 0.71±0.11μg, P= 0.016, N=6). Conversely, there were no significant differences in RNA levels throughout all adult time points (Fig. 1A). Thus, dFMRP functions to negatively regulate RNA levels during a restricted window of late pupal brain development.
Figure 1. dFMRP regulates protein and RNA levels during brain maturation.

Total RNA (A) and protein (B) extracted at indicated developmental time points from dfmr1 null (dfmr150M) and control (w1118). Bars show the mean μg per head ± standard deviation. Significance: 0.05>p>0.01 (*), 0.01<p<0.001 (**). Stages: P3 (60–70 hrs APF), P4 (88–96 hrs APF), 0–7 hrs AE, 1d (21–24 hrs AE), 4d (96–112 hrs AE), 9d (216–232 hrs AE).
As with RNA levels, total protein extracted from wildtype and mutants was higher in the developing brain compared to the mature brain. There was a 2-fold decrease in protein (μg/head) over the 12-day assay period (Fig. 1B). The dfmr1 null brain had significantly elevated protein during a restricted window of development, with elevated protein levels persisting into the early-use period following eclosion (Fig. 1B). There were significant protein increases of 42% (P3), 31% (P4) and 44% (0–7 hours AE) in dfmr1 compared to wildtype (P3: WT, 16.7±2.5μg, dfmr1, 23.73±5.8μg, P= 0.01, N=6; P4: WT, 16.0±3.4μg, dfmr1, 20.9±3.0μg, P=0.04, N=6; 0–7 hours: WT, 10.5±3.1μg, dfmr1, 15.2±2.1μg, P=0.001, N=12). There were no significant differences in adult animals for several days following this early requirement (Fig. 1B). However, a new period of requirement in the mature brain occurred at nine days, when the dfmr1 null contained 43% more protein than wildtype (9d: WT, 7.68±0.4μg, dfmr1, 11.0±2.0μg, P= 0.004, N=6). These results suggest that there is a transient window of dFMRP requirement during late pupal development extending into the early-use period of the young adult, followed by a separable requirement much later in the mature brain.
dFMRP expression and function is developmentally regulated
The restricted window of RNA/protein upregulation during late dfmr1 brain development/early adult use, suggested that dFMRP expression itself might be similarly developmentally regulated. FMRP is expressed at higher levels during development in mammals (Khandjian et al., 1995; Lu et al., 2004; Singh et al., 2007; Wang et al., 2004), but there is no indication of whether developmental regulation occurs at the level of transcription or translation. FMRP represses its own translation (Ashley et al., 1993; Brown et al., 1998; Ceman et al., 1999; Schaeffer et al., 2001; Sung et al., 2000) adding a complicating factor to understanding the mechanism of regulation. To address this issue, dfmr1 mRNA and dFMRP protein brain levels were assayed from pupal development to maturity.
The dfmr1 mRNA level was measured by quantitative RT-PCR with the same time points as above but narrowing the eclosion time point to 0–3 hours AE and including 7 days AE to better define the late expression profile. The dfmr1 mRNA level was normalized to a housekeeping gene, GAPDH2, and reported as a fold change relative to the first time point (Fig. 2A). The expression of dfmr1 mRNA follows two distinct patterns. First, there is high abundance during pupal brain development, which then falls rapidly upon eclosion and remains low for the first day of adult life (Fig. 2A). This pattern is consistent with the reduction in overall RNA abundance during the development to adult transition, as shown in Figure 1A. The abundance of dfmr1 mRNA, however, increases 50% between days 1 and 4 (P=0.009). In the adult (>7 days post-eclosion), dfmr1 message remains elevated to levels similar to those at pupation (Fig. 2A). This second period of dfmr1 mRNA elevation is divergent from the total RNA profile, which declines into maturity (Fig. 1A). Thus, there are two distinct phases of dfmr1 transcription, with one peak during late brain maturation and a second comparable plateau in the fully mature brain.
Figure 2. dFMRP protein and mRNA are differentially developmentally regulated.

A) Quantitative RT-PCR of dfmr1 mRNA levels normalized to GAPDH2 and reported as fold changes relative to first time point. Bars show mean ± standard error. B) Western blot analysis of dFMRP protein. Each lane represents a single head at indicated stages (2 heads per stage). α-tubulin is the loading control. C) Immunocytochemistry of dFMRP in control (w1118) brain. CB: central brain, OL: optic lobe. Scale bar=100 μm. Stages: P3 (60–70 hrs APF), P4 (88–96 hrs APF), E (0–3 hrs AE), 1d (21–24 hrs AE), 4d (96–112 hrs AE), 7d (168–184 hrs AE), 9d (216–232 hrs AE).
The dFMRP protein level in the brain was measured by both immunoblot and immunocytochemistry. Western blot analyses were performed on heads from single animals, in duplicate, from seven developmental time points (Fig. 2B). As predicted from the dfmr1 mRNA levels, dFMRP protein expression is maximal during late stages of brain development and decreases during the first day post-eclosion (Fig. 2B). At maturity (>1 day), dFMRP protein was difficult to detect on western blots and, surprisingly, no longer correlated with dfmr1 mRNA levels (compare Figs. 2A and B). While dfmr1 mRNA was high after 4 days AE, dFMRP protein levels remained minimal. This expression profile was also evident by brain immunocytochemistry (Fig. 2C). During late pupal brain development, dFMRP is expressed at high levels throughout the entire brain, primarily in neuronal soma, whereas in the mature brain dFMRP expression is strongly reduced except in limited central brain regions. These data show that dfmr1 mRNA and dFMRP protein levels correlate closely during brain development, but that dfmr1 transcription and translation become uncoupled in the mature brain.
To test whether dFMRP function is similarly regulated in the same developmental pattern, we examined the dFMRP target chickadee/profiling (Reeve et al., 2005). Actin-binding profilin is upregulated in the absence of dFMRP, but nothing is known about developmental regulation. We performed quantitative RT-PCR for profilin mRNA on dfmr1 and control brain extracts from development through maturity (Fig. 3A). In wildtype, profilin mRNA nearly doubles during late pupal brain development and then falls precipitously immediately after eclosion, and remain low in the adult brain (Fig. 3A). In dfmr1 mutants, profilin mRNA levels follow a similar profile with two important differences: first, the pupal day 4 spike is ~3 times the level of mRNA at pupal day 3 and 46% increased over controls, and secondly, in aged adults (9 days) of age, profilin mRNA levels are actually decreased (43%) compared to controls (Fig. 3A). For profilin protein levels, maximum brain expression is normally during pupal day 4, rapidly falling at eclosion (Fig 3B), closely consistent with the mRNA levels. In dfmr1 null brains, in contrast, profilin protein is maintained at aberrantly high levels following eclosion and through the early use period in the young adult (Fig. 3B). No protein changes are apparent at 9 days when profilin mRNA levels increase in the mutant. These findings are totally consistent with distinct developmentally controlled roles for dFMRP function during late-brain development/early-use refinement periods and at maturity.
Figure 3. The dFMRP target chickadee/profilin is developmentally regulated.

A) Quantitative RT-PCR of chickadee/profilin mRNA normalized to GAPDH2 and reported as fold changes relative to first time point. Bars show mean ± standard error. B) Western blot analysis of chickadee/profilin from single control and dfmr1 (1 head per lane) at the developmental time points shown. α-tubulin is the loading control.
dFMRP regulates a late development period of axonal pruning
The Mushroom Body (MB) is a primary learning/memory center in Drosophila brain and therefore the focus of behavioral, structural and functional studies (Margulies et al., 2005; Zars et al., 2000). We have shown previously that dfmr1 null MB neurons exhibit increased axonal growth and over-branching (Pan et al., 2004). However, these analyses were done in unstaged brains and the cause of defects was not determined. In light of our finding that dFMRP expression and function is differentially regulated during development vs. maturity, we reexamined MB neuron axon morphogenesis throughout development. The Mosaic Analysis with a Repressible Cell Marker (MARCM) genetic clonal technique (Lee and Luo, 2001) was used to label single homozygous mutant MB gamma neurons (Fig. 4A). This method allows for the direct visualization of individual neuron structure in the intact brain, and also permits direct analysis of the cell autonomous function of dFMRP in that single neuron.
Figure 4. Single cell MARCM clonal analysis of MB gamma neuron development.

A) Representative image of Fasciclin II (FasII, red) labeled Mushroom Body containing a single MARCM gamma neuron clone (green). The white box highlights the area of axonal projection. B) Developmental profile of axon projections of single cell MARCM gamma neuron clones. Boxed insets are magnifications of areas of small (<5μm) presynaptic branches (arrows), which are subject to pruning. Scale bar=10μm. Stages: P3 (60–70 hrs APF), P4 (88–96 hrs APF), 0–3 hrs AE, 4d (96–112 hrs AE).
MARCM analyses were performed on single control and dfmr1 null MB neurons with MB axonal lobes defined with anti-Fasciclin II (FASII, red, Fig. 4A). The gamma neuron class was selected for analysis due to its well-defined morphology and simple single axon projection (Fig. 4A, green). Single neuron structure was analyzed at four time points: P3, P4, 0–3 hours AE and 4 days AE (Fig. 4B). Just prior to eclosion, both control and dfmr1 neurons undergo a period of increased axonal growth as the total length of branches increases significantly (Fig. 4B). While there were no differences in branch number or axon length at pupal day 3, by pupal day 4 dfmr1 null axons have grown 25% longer than control axons (P=0.02), although overall branch number remains comparable (Fig. 4B). However, dfmr1 neurons at P4 contain 31% more large branches (>10μm) and 26% fewer small branches (<5μm) than control neurons (P=0.004 and P=0.026, respectively). Thus, dFMRP negatively regulates MB axon branch growth specifically during late stages of pupal brain development.
Eclosion heralds the onset of use-dependent activity in brain circuits. In control neurons at eclosion, there were rapid decreases in both overall branch length and branch number relative to P4, consistent with a pruning mechanism (Fig. 4B). The specific branches pruned were short processes (<5μm) (Fig. 4B, inset arrows). In contrast, dfmr1 null neurons show no significant decrease in axon branches during this normal pruning period. As a result, dfmr1 neurons have 24% more and 30% longer axonal branches than controls immediately following eclosion (P=0.02 and P=0.003, respectively; Fig 4B). By 4 days post-eclosion, when dFMRP protein is minimally expressed (Fig. 2B), both branch number and length become static. Conversely, pruning absent in dfmr1 neurons post-eclosion, now manifested by 4 days AE (Fig. 4B). Once again, primarily branches <5μm were pruned by this mechanism (Fig. 4B, inset). Interestingly, the excessive branching in dfmr1 neurons present in young animals is absent in mature adults (branch number and length: P=0.17, 0.84, respectively). In fact, over-pruning is evidenced by a 35% decrease in small (<5μm) axon branches in dfmr1 neurons compared to controls at maturity (P=0.01; Fig. 4B).
We previously showed that dFMRP overexpression of dFMRP causes under-branching of gamma neurons, the inverse of the dfmr1 null phenotype (Pan et al., 2004). Here, we examine the developmental profile in single cell MARCM analysis clones overexpressing (OE) dFMRP. At P3, underbranching is already apparent with 47% fewer branches (P=0.002), which averaged 61% shorter (P<0.001) than controls (Fig 5A). The axon growth that occurs normally during P4, fails in dFMRP OE neurons as branch number and length remain statistically unchanged (Fig 5B; number: P3, 5.5±3.6 (N=21); P4, 4.1±1.8 (N=20); length: P3, 25.5±11.6μm (N=21); P4, 20.5 ±8.7μm (N=20)). This undergrowth persists into maturity at 4 days post-eclosion and is worsened by aberrant excessive pruning immediately after eclosion. Axon branch number in OE neurons decreases ~30% (P=0.03) upon eclosion (Fig 5B; P4, 4.1±1.8 (N=20); 0–3hrs AE, 2.9±2.4 (N=17)). The only branches available for pruning are the short filipodial-like branches persistent throughout late pupation (Fig 5A, insets). Thus, dFMRP overexpression inhibits axonal elongation and accelerates post-eclosion process refinement.
Figure 5. dFMRP overexpression abrogates normal MB neuronal development.

A) Developmental profile of axon projections of single cell MARCM gamma neuron clones overexpressing dFMRP. Boxed insets highlight presynaptic branches that are pruned. Scale bar=10 μm. Stages: P3 (60–70 hrs APF), P4 (88–96 hrs APF), 0–3 hrs AE, 4d (96–112 hrs AE). B) Quantitation of total axon branch number of dFMRP overexpressing MARCM clones. Each point is data from a single-cell MARCM clone. Horizontal lines represent the mean for each data set. Significance: 0.01<p<0.05 (*).
Activity-dependent regulation of dFMRP expression and function
The pruning of small axonal branches in MB neurons occurs concomitantly with the onset of use, consistent with a period of refinement to remove weak or improperly formed synaptic connections. The observation that this pruning mechanism is delayed beyond the early-use period in dfmr1 null neurons suggests that activity may regulate dFMRP function. To begin to test this hypothesis, we performed sensory-deprivation experiments to block external stimulation that should be required for the use-dependent testing of synaptic connectivity. 48 hour old, individual pupae were isolated in 1.5mL tubes filled with 1mL of food to prevent social stimulation and limit exploration/movement. Tubes were maintained in total darkness to prevent visual stimulation and kept in isolation boxes to mitigate auditory stimulation. Animals were allowed to develop under these conditions (sensory-deprived; SD) until 4 days AE. Controls developed in standard fly vials with ~30 other animals and maintained in 12:12 L/D conditions until 4 days AE.
FMRP expression has been reported to be elevated in response to sensory and neuronal stimulation (Gabel et al., 2004; Hou et al., 2006; Irwin et al., 2005; Irwin et al., 2000; Todd and Mack, 2000; Todd et al., 2003; Valentine et al., 2000; Weiler et al., 1997). Therefore, we first tested whether dFMRP expression was affected by sensory-deprivation. The abundance of dfmr1 mRNA was assayed by quantitative RT-PCR (Fig. 6A). The dfmr1 mRNA level was significantly reduced (P=0.02) by ~20% in SD animals, indicating that sensory input positively regulates dfmr1. Western analyses on single heads showed dFMRP protein levels were also significantly reduced (P=0.04; Fig. 6B) by~20% in SD animals (seven pairs of control and SD heads, 3 trials; Fig. 6C). Thus, sensory input activity positively regulates dFMRP protein levels.
Figure 6. Sensory input deprivation reduces dFMRP expression.
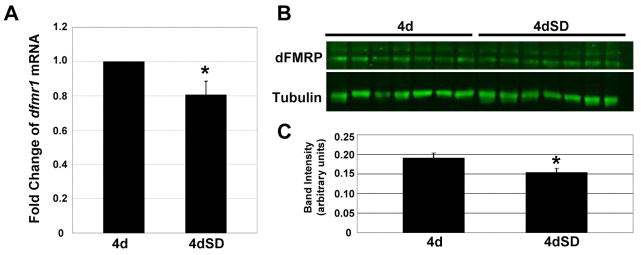
A) Quantitative RT-PCR of dfmr1 mRNA from normally-reared and sensory-deprived (SD) animals at 4 days (96–112 hrs AE). Values are normalized to GAPDH2. Bars show the mean ± standard error (N=4). Significance indicated 0.05>p>0.01 (*). B) Western blot for dFMRP of normally-reared and SD animals at 4 days (96–112 hrs AE), with one head per lane. B) Quantitation of dFMRP western blot normalized to α-tubulin loading control. Bars show the mean ± standard error. Significance: 0.05>p>0.01 (*).
To refine this analyses to specific sensory modalities, we next assayed dFMRP expression in two sensory transduction mutants: or83b, which eliminates a widely expressed odorant co-receptor required for olfaction of a broad spectrum of odors, and ninaE, which encodes rhodopsin required for visual phototransduction (Larsson et al., 2004; O’Tousa et al., 1985). In these mutants, there is a comparable ~20% reduction in dFMRP expression by brain western analysis (Fig. 7A, B), demonstrating that both vision and olfaction positively regulate dFMRP. Importantly, these sensory mutants concomitantly display a significant, opposing increase in the dFMRP target chickadee/profilin (Figs. 7A, B). Together, these results demonstrate that neural activity driven by sensory input is a positive regulator of dFMRP expression and function.
Figure 7. Olfactory and vision mutants reduce dFMRP expression and function.

A) Western blot analysis of dFMRP and chickadee/profilin in control, odorant receptor mutant or83b, and rhodopsin mutant ninaE. Single heads from <16 hour old animals loaded per lane. α-tubulin is the loading control. B) Quantitation of dFMRP and chickadee/profilin western analysis. Bars show the mean ± standard error (N=6). Significance: 0.05>p>0.01 (*).
We next assayed the effect of sensory input activity on MB axon pruning (Fig. 8). Single MB gamma neuron MARCM clones were analyzed in animals grown until 4 days AE in normal vs. sensory-deprived (SD) conditions. SD animals displayed a significant 34% (P=0.03) increase in axon branch number in control neurons, but a highly significant 86% (P=0.001) increase in dfmr1 null neurons (Fig. 8A). Axon branch length did not increase in control neurons, but significantly (P<0.001) increased by 47% in dfmr1 null neurons. As a consequence, after 4 days of sensory-deprivation dfmr1 axon branches were 30% longer than controls (P=0.002; Fig. 5A). The failure to prune was most apparent in dfmr1 neurons in branches less than 5μm (P<0.001), but also apparent in longer branches binned between 5–10μm (P<0.001). Pruning normally occurs within 3 hours post-eclosion. Since sensory-deprivation inhibits pruning, we wanted to determine whether pruning could be restored with a subsequent activity period of the same duration. To test this possibility, 4 day SD animals were acutely stimulated with normal rearing conditions for 3 hours immediately prior to analysis. Neurons from these animals were identical to SD animals (Fig. 8A), with 24% longer dfmr1 axon branches compared to controls (P=0.002). Thus, there is a restricted developmental window in which dFMRP mediates use-dependent pruning of axonal branches.
Figure 8. Sensory-deprivation modifies dFMRP-dependent axon pruning.

A) Representative images of single cell MARCM gamma neuron axon projections at 4 days post-eclosion. Animals raised in standard conditions (top row), sensory-deprived (middle row) and following 3 hours of normal sensory stimulation (bottom row). Scale bar=10μm. B) Diagram of dFMRP-dependent changes in MB axonal projections. dFMRP protein and mRNA are expressed maximally during late pupal development and the early-use period of the newly-eclosed animal. Activity-dependent pruning in MB axons occurs during this window, dependent on dFMRP. At maturity, transcriptional and translational regulation of dfmr1 becomes uncoupled as mRNA levels inversely correlate with protein levels.
Neuronal activation enhances dFMRP-dependent axon pruning
Since sensory-deprivation blocks pruning, inducing neuronal activity may increase pruning. To test this hypothesis, we generated MARCM clones expressing Chlamydomonas reinhardtii light-gated channelrhodopsin-2 (CHR2) (Nagel et al., 2003). In the presence of cofactor all-trans retinal, CHR2 channels open in response to blue light (Schroll et al., 2006). We generated recombinant MARCM CHR2 animals in the control or in the dfmr1 null mutant. Flies were fed all-trans retinal or an ethanol vehicle throughout development. Immediately upon eclosion (<12hrs AE), both genotypes were subjected to 1Hz blue light (470nm) pulses for 6 hours. Brains were dissected and single-cell gamma neuron MARCM clones analyzed (Fig. 9A).
Figure 9. CHR2-induced neuronal activation drives dFMRP-dependent pruning.

A) Single cell MARCM clones expressing channelrhodopsin-2 (CHR2) in control or in dfmr1 null backgrounds, with animals grown on food containing all-trans retinal (ATR) or ethanol (ETOH vehicle). Post-eclosion animals (<12 hours after eclosion) were stimulated with 470 nm light at 1Hz pulses for 6 hours. Scale bar=10μm. Quantified total axon branch number (B) and <5μm branch number (C) of CHR2-expressing MARCM clones after blue light stimulation. Horizontal bars are the mean for each data set. Significance: 0.01<P<0.05 (*).
Neuronal activation of MB neurons in CHR2-expressing clones resulted in significant 21% reduction (p=0.02) in the total number of axonal branches in animals fed the all-trans retinal cofactor (experimental) compared to vehicle-fed controls (Fig. 9B). Importantly, small axon branches (<5μm) that are not pruned during sensory-deprivation are reduced 27% by neuronal activation (Fig. 9C). Induced pruning was totally dependent on dFMRP, as no effect was observed in dfrm1 null neurons expressing CHR2 (Fig. 9). Thus, pruning of axon branches during the early use-refinement phase requires both neuronal electrical activaty and dFMRP function.
Two-phase dFMRP requirement: axonogenesis and activity-dependent pruning
Quantification of neuronal architecture reveals two phases of dFMRP regulation in the MB; axon growth during late-stage pupal development and activity-dependent axon pruning during the early-use following eclosion. The cumulative length of axon branches is significant increased in dfmr1 null neurons during P3/P4 (WT: P3, 66.1±30.2μm (N=17), P4, 86.5±20.6μm (N=18), P=0.03; dfmr1: P3, 82.8±38.3μm (N=19), P4, 108.1±30.7μm (N=19), P=0.02; Fig. 10A). In contrast, the number of axon branches is not significantly during this development period (WT: P3, 10.5±4.5 (N=17), P4, 11.4±2.8 (N=18), P=0.5; dmfr1: P3, 12.2±6.3 (N=19), P4, 10.5±3.2 (N=19), P=0.4; Fig. 10B). Thus, dFMRP regulates axon growth but not branching during late-stage brain development.
Figure 10. Developmental changes in axon branch length and number in single cell Mushroom Body clones in control and dfmr1 null mutants.

Total axon branch length (A) and total axon branch number (B) of gamma neurons from the indicated genotypes and stages. Each point is data from a single-cell MARCM clone. Horizontal lines represent the mean for each data set. Stages: P3 (60–70 hours APF), P4 (88–96 hours APF), E (0–3 hours AE), 4D (96–112 hours AE), sensory input deprived (SD, 96–112 hours AE), 3 hours sensory stimulation (3H Stim, sensory-deprived for 4 days followed by 3 hours of sensory input stimulation). Significance: 0.01<P<0.05 (*), 0.001<P<0.01 (**), P<0.001 (***).
At eclosion, MB neurons normally decrease both axon branch length and number (length: 0–3 hrs AE, 72.1±19.8μm (N=21), P=0.02 relative to P4 (Fig. 10A); number: 0–3 hrs AE 8.8±2.9 (N=21), P=0.007 relative to P4; Fig. 10B). In contrast, dfmr1 neurons display no such pruning at eclosion (length: 0–3 hrs AE, 93.7±21.9 μm (N=19), P=0.11 relative to P4 (Fig. 10A); number: 0–3 hrs AE, 10.9±2.8 (N=19), P=0.48 relative to P4; Fig. 10B). Instead, pruning manifests in a delayed fashion in dfmr1 neurons. By 4 days post-eclosion, dfmr1 neurons display fewer axon branches (length: 72.9±29.7 μm (N=22), P=0.01 relative to eclosion (Fig. 10A); number: 7.2±3.1 (N=22), P=0.002 relative to eclosion; Fig. 10B). Indeed, over-pruning in dfmr1 null neurons was evident in the reduction of small processes (<5μm) compared to controls (control: 5.4±2.7 (N=24), dfmr1: 3.5±1.9 (N=22), P=0.01). Thus, there is an early-use period of pruning whose timing is dependent on dFMRP.
Sensory-deprivation (SD) revealed that dFMRP function is activity-dependent. In control SD neurons, there was a significant increase in axon branch number (4d control: 9.0±3.3 (N=24); 4d SD: 12.1±3.8 (N=20), P=0.028; Fig. 10B), with a non-significant tendency towards greater length (4d control: 68.9±27.8μm (N=24); 4d SD: 82.7±21.2μm (N=20), P=0.17). In dfmr1 SD neurons, there was a significantly more pronounced increase in branch number (4d: 7.23±3.1 (N=22); 4d SD: 13.4±3.5 (N=24), p<0.001; Fig. 10B), and branch length was also significantly increased (4d: 72.9±29.7μm (N=22); 4d SD: 107.3±27.8μm (N=24), P<0.001; Fig. 10A). Acute stimulation following SD rearing was unable to induce pruning in either dfmr1 or control neurons (Fig. 10). Thus, sensory input activity strongly influences both the timing and extent of the dFMRP-dependent pruning of axonal processes.
Discussion
Remarkably, it remains unknown whether Fragile X Syndrome (FraX) is a disease of development or a disease of plasticity, although these two possibilities give rise to entirely different strategies for intervention and treatment. Most recent FraX research has focused on acute defects in synaptic plasticity, with only minimal attention to developmental dysfunction. A key concept is that a restricted function during development may well be reflected in impaired plasticity at maturity, and thus the timing of dysfunction does not necessarily correspond to the period of functional requirement. While mammalian studies have indicated peak FMRP levels during early postnatal development (Khandjian et al., 1995; Lu et al., 2004; Singh et al., 2007; Wang et al., 2004), very little work has characterized the temporal requirements of FMRP. We have therefore sought to employ the Drosophila FraX model to analyze dFMRP-dependent processes in neuronal development. Our initial findings highlighted roles for dFMRP in 1) late stages of brain development and 2) very early-use circuit refinement, and we therefore focused on these mechanisms.
In the absence of dFMRP, elevated levels of both total RNA and total protein are evident during a restricted period of late pupal brain development, with the protein increase persisting into a post-eclosion, early-use refinement period (Fig. 1). These increases are transient and disappear in mature brain thereby defining a limited developmental window of dFMRP function. The increase in protein is predicted since FMRP/dFMRP negatively regulates translation (Khandjian et al., 2004; Laggerbauer et al., 2001; Li et al., 2001; Qin et al., 2005; Sung et al., 2003). The elevated RNA is more surprising. FMRP/dFMRP can both negatively and positively regulate mRNA stability (Xu et al., 2004; Zalfa et al., 2007; Zhang et al., 2007), and, therefore, dFMRP may have a developmentally-restricted role primarily as a negative regulator of mRNA stability. Alternatively, the RNA increase may be caused by elevated transcription, via an uncharacterized direct or indirect transcriptional inhibition function of dFMRP. Since the increase in total protein/RNA is not biased towards selected dFMRP targets, these results suggest globally upregulated transcription/translation in the dfmr1 mutant brain during a restricted window of very late maturation and early-use refinement.
During brain development, dfmr1 mRNA and dFMRP protein levels tightly correlate with the above changes but, surprisingly, dfmr1 mRNA levels actually inversely correlate with dFMRP protein levels in the mature brain (Fig. 2). By 4 days AE, dfmr1 mRNA levels rise to levels nearly as high as those present during development, but dFMRP protein is maintained at a basal level in the mature brain. This change strongly suggests a distinct switch in dFMRP regulation, in which transcription and translation become uncoupled. Since dFMRP/FMRP represses the translation of its own mRNA (Ashley et al., 1993; Brown et al., 1998; Ceman et al., 1999; Schaeffer et al., 2001; Sung et al., 2000), it is tempting to speculate that this negative feedback mechanism specifically regulates dFMRP in the mature brain. FMRP modulates synaptic plasticity at maturity, as evidenced by region-specific decreased LTP and enhanced LTD in fmr1 KO mice (Hou et al., 2006; Huber et al., 2002; Koekkoek et al., 2005; Larson et al., 2005; Li et al., 2002; Wilson and Cox, 2007; Zhao et al., 2005). Consistent with such a mature function, elevated total protein levels are once again evident in fully mature dfmr1 null brain (Fig. 1). A similar increase in cerebral protein synthesis occurs in adult fmr1 KO mice (Qin et al., 2005). Together, these data suggest that a switch in dFMRP/FMRP regulation defines separate windows of function in development versus maturity.
It was critical to determine whether dFMRP function correlated to its developmental expression profile. A known dFMRP target is chickadee/profilin; dFMRP binds chickadee mRNA and negatively regulates translation (Reeve et al., 2005). Importantly, the dynamics of chickadee misregulation in the dfmr1 null brain indicate the dFMRP functional requirement mirrors its development expression profile (Fig. 3). Chickadee expression normally sharply peaks in very late-stage brain development (P4), and it is during this development window, and shortly following, that dramatic overexpression is manifest in the dfmr1 null brain. As for general brain profiles, an increase in chickadee transcript parallels the increase in protein, suggesting that dFMRP regulation may be at the level of the mRNA, for example mRNA stability. dFMRP reportedly interacts with miRNA machinery to control mRNA levels of the sodium channel pickpocket1 (Xu et al., 2004). A similar mechanism for chickadee regulation would be consistent with our results. Interestingly, the increase in chickadee/profilin protein levels coincides with this period of use-dependent neural circuit refinement at eclosion. At least one dFMRP/FMRP target mRNA, MAP1b/futsch, is regulated specifically at postnatal day 10 in fmr1 KO mice (Lu et al., 2004). These new insights suggest it will be vital to ascertain the developmental expression of all putative FMRP targets in the context of these distinct temporal windows of regulation, and in order to validate in vivo functions.
During the peak period of dFMRP expression, there are two phases of dFMRP-dependent axon maturation. During late pupal development, dFMRP inhibits axon elongation, with dfmr1 null neurons exhibiting branches 25% longer than controls (Fig. 4). This function is restricted to very late stages (P4), with no differences in branch length or number earlier (P3). Immediately upon eclosion, dFMRP is required for use-dependent pruning, causing a decrease in both axon branch length and number (Fig. 4). Pruning is most evident in the smallest presynaptic branches (<5μm) and occurs quickly (hours) following the onset of adult activity. Targeted overexpression of dFMRP causes inverse defects in both phases of dFMRP requirement (Fig. 5). Axon undergrowth is apparent early (P3) and axons fail to grow later (P4). Axon branches present in these neurons are short, filipodial-like structures and, at eclosion, there is excessive pruning to result in ~30% fewer branches than in P4 and ~3 times fewer branches than in controls (Fig. 5). Thus, both axonogenesis and axon branch pruning are bidirectional modified by inverse changes in dFMRP expression.
Blocking sensory input activity maintains dFMRP in its early development regulative state, with a correlative reduction in both dfmr1 mRNA and dFMRP protein (Fig. 6). Both olfactory (or83b) and phototransduction mutants (ninaE) (Larsson et al., 2004; O’Tousa et al., 1985) similar suppress dFMRP level, indicating that these two primary modes of brain sensory input both positively drive dFMRP expression (Fig. 7). Similarly, mammalian FMRP expression is elevated following activity stimulation by both environmental enrichment and mGluR signaling activation (Gabel et al., 2004; Hou et al., 2006; Irwin et al., 2005; Todd and Mack, 2000; Todd et al., 2003; Valentine et al., 2000; Weiler et al., 1997). Blocking mGluR activity in Drosophila and mice can rescue some dfmr1 defects, including impaired learning and memory (McBride et al., 2005; Yan et al., 2005; Pan et al. 2007). From these similar findings, it is enticing to suggest that FMRP/dFMRP may function downstream of mGluR signaling activity, perhaps differentially in development versus maturity. Importantly, both or83b and ninaE sensory mutants cause elevation of chickadee/profilin in the same brains in which dFMRP is suppressed (Fig. 7). This finding is consistent with activity-dependent regulation of dFMRP to regulate chickadee/profilin expression.
This study shows for the first time that Drosophila neurons undergo activity-dependent pruning coincident with the onset of use. This pruning is primarily restricted to final terminal axonal process. In the absence of dFMRP, pruning does not occur during the normal developmental window (Fig. 8). Indeed, blocking sensory input activity leads to further increases in the axon branch number and length in dfmr1 null neurons. Moreover, at maturity, sensory stimulation following sensory-deprivation does not induce pruning, likely because the dFMRP level has fallen too low. We hypothesize that there is a threshold of dFMRP required for efficient activity-dependent pruning during the early-use period, which is normally defined by the window of high dFMRP expression. Reinstated sensory stimulation following sensory-deprivation does cause a significant dFMRP-dependent increase in the number of long axon branches (>10μm; Fig. 8). These data are consistent with the need for high dFMRP expression to both limit axonal growth and mediate early-use refinement of circuits. Importantly, we confirmed that neuronal activation bidirectionally drives the pruning process using targeted expression of the exogenous light-gated channelrhodopsin-2 channel (Schroll et al., 2006). Light-driven activation of CHR2 channels induces pruning of the same small (<5μm) axonal processes that aberrantly persist in the dfmr1 null brain (Fig. 9). As predicted, the induced pruning process fails to occur in the absence of dFMRP.
Delayed pruning eventually occurs in dfmr1 null neurons to ultimately rescue the overbranching defect present in younger animals. A similar transient elongation of dendritic spines occurs in young postnatal fmr1 KO mice, although a secondary overgrowth phenotype may appear months later in adult animals (Galvez and Greenough, 2005; Nimchinsky et al., 2001). In Drosophila, the delayed axon pruning in dfmr1 null neurons actually goes too far, resulting in reduced neuronal complexity in mature adult animals (Fig. 10). The small presynaptic branches (<5μm) are reduced 35% in dfmr1 null neurons compared to controls at 4 days. Since pruning normally occurs very rapidly (<3 hours after eclosion), coincident with initial use, it is likely that the pruning process is strictly controlled for that developmental time. By delaying pruning in the absence of dFMRP, it appears that other factors that buffer the extent of process elimination fail to provide adequate regulation of the mechanism. Indeed, this mitigation may be a function of dFMRP itself, since dFMRP levels drop drastically immediately following the normal pruning window. FMRP potentially regulates many proteins involved in a diverse set of functions (Brown et al., 2001; Miyashiro et al., 2003; Zhang et al., 2005). Understanding the developmental regulation of proteins that associate with FMRP and FMRP target mRNAs will be crucial to unraveling the underlying pruning mechanisms of activity-dependent neural circuit refinement.
Acknowledgments
We thank Dr. Luyuan Pan in the Broadie Lab for guidance on the MARCM technique. We are grateful to members of Broadie Lab for discussion, especially Dr. Cheryl Gatto and Emma Rushton for comments on the manuscript. This work was supported by a FRAXA fellowship to C.R.T and NIH grant GM54544 to K.B.
References
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Aschrafi A, Cunningham BA, Edelman GM, Vanderklish PW. The fragile X mental retardation protein and group I metabotropic glutamate receptors regulate levels of mRNA granules in brain. Proc Natl Acad Sci U S A. 2005;102:2180–5. doi: 10.1073/pnas.0409803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley CT, Jr, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–6. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- Awasaki T, Ito K. Engulfing action of glial cells is required for programmed axon pruning during Drosophila metamorphosis. Curr Biol. 2004;14:668–77. doi: 10.1016/j.cub.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Awasaki T, Tatsumi R, Takahashi K, Arai K, Nakanishi Y, Ueda R, Ito K. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–67. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–7. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9:1221–5. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- Boccia ML, Roberts JE. Behavior and autonomic nervous system function assessed via heart period measures: the case of hyperarousal in boys with fragile X syndrome. Behav Res Methods Instrum Comput. 2000;32:5–10. doi: 10.3758/bf03200783. [DOI] [PubMed] [Google Scholar]
- Boothe RG, Greenough WT, Lund JS, Wrege K. A quantitative investigation of spine and dendrite development of neurons in visual cortex (area 17) of Macaca nemestrina monkeys. J Comp Neurol. 1979;186:473–89. doi: 10.1002/cne.901860310. [DOI] [PubMed] [Google Scholar]
- Brown HL, Cherbas L, Cherbas P, Truman JW. Use of time-lapse imaging and dominant negative receptors to dissect the steroid receptor control of neuronal remodeling in Drosophila. Development. 2006;133:275–85. doi: 10.1242/dev.02191. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–87. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Brown V, Small K, Lakkis L, Feng Y, Gunter C, Wilkinson KD, Warren ST. Purified recombinant Fmrp exhibits selective RNA binding as an intrinsic property of the fragile X mental retardation protein. J Biol Chem. 1998;273:15521–7. doi: 10.1074/jbc.273.25.15521. [DOI] [PubMed] [Google Scholar]
- Ceman S, Brown V, Warren ST. Isolation of an FMRP-associated messenger ribonucleoprotein particle and identification of nucleolin and the fragile X-related proteins as components of the complex. Mol Cell Biol. 1999;19:7925–32. doi: 10.1128/mcb.19.12.7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–4. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Diego Otero Y, Severijnen LA, van Cappellen G, Schrier M, Oostra B, Willemsen R. Transport of fragile X mental retardation protein via granules in neurites of PC12 cells. Mol Cell Biol. 2002;22:8332–41. doi: 10.1128/MCB.22.23.8332-8341.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–9. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17:1539–47. doi: 10.1523/JNEUROSCI.17-05-01539.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–4. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, Bagni C. The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol Cell Neurosci. 2007;34:343–54. doi: 10.1016/j.mcn.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Fox K, Wong RO. A comparison of experience-dependent plasticity in the visual and somatosensory systems. Neuron. 2005;48:465–77. doi: 10.1016/j.neuron.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Freund LS, Reiss AL. Cognitive profiles associated with the fra(X) syndrome in males and females. Am J Med Genet. 1991;38:542–7. doi: 10.1002/ajmg.1320380409. [DOI] [PubMed] [Google Scholar]
- Gabel LA, Won S, Kawai H, McKinney M, Tartakoff AM, Fallon JR. Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J Neurosci. 2004;24:10579–83. doi: 10.1523/JNEUROSCI.2185-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez R, Gopal AR, Greenough WT. Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 2003;971:83–9. doi: 10.1016/s0006-8993(03)02363-1. [DOI] [PubMed] [Google Scholar]
- Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A. 2005;135:155–60. doi: 10.1002/ajmg.a.30709. [DOI] [PubMed] [Google Scholar]
- Galvez R, Smith RL, Greenough WT. Olfactory bulb mitral cell dendritic pruning abnormalities in a mouse model of the Fragile-X mental retardation syndrome: further support for FMRP’s involvement in dendritic development. Brain Res Dev Brain Res. 2005;157:214–6. doi: 10.1016/j.devbrainres.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Greenough WT, Hwang HM, Gorman C. Evidence for active synapse formation or altered postsynaptic metabolism in visual cortex of rats reared in complex environments. Proc Natl Acad Sci U S A. 1985;82:4549–52. doi: 10.1073/pnas.82.13.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Elisseou NM, McKinney BC, Greenough WT. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158–64. doi: 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci U S A. 2007;104:11489–94. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds JW, Hinds PL. Synapse formation in the mouse olfactory bulb. I. Quantitative studies. J Comp Neurol. 1976;169:15–40. doi: 10.1002/cne.901690103. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–54. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–50. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Synaptic density in human frontal cortex - developmental changes and effects of aging. Brain Res. 1979;163:195–205. doi: 10.1016/0006-8993(79)90349-4. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Christmon CA, Grossman AW, Galvez R, Kim SH, DeGrush BJ, Weiler IJ, Greenough WT. Fragile X mental retardation protein levels increase following complex environment exposure in rat brain regions undergoing active synaptogenesis. Neurobiol Learn Mem. 2005;83:180–7. doi: 10.1016/j.nlm.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ, Crisostomo RA, Larsen BP, Mehta A, Alcantara CJ, et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet. 2002;111:140–6. doi: 10.1002/ajmg.10500. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–7. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Swain RA, Christmon CA, Chakravarti A, Weiler IJ, Greenough WT. Evidence for altered Fragile-X mental retardation protein expression in response to behavioral stimulation. Neurobiol Learn Mem. 2000;73:87–93. doi: 10.1006/nlme.1999.3914. [DOI] [PubMed] [Google Scholar]
- Ivanco TL, Greenough WT. Altered mossy fiber distributions in adult Fmr1 (FVB) knockout mice. Hippocampus. 2002;12:47–54. doi: 10.1002/hipo.10004. [DOI] [PubMed] [Google Scholar]
- Khandjian EW, Fortin A, Thibodeau A, Tremblay S, Cote F, Devys D, Mandel JL, Rousseau F. A heterogeneous set of FMR1 proteins is widely distributed in mouse tissues and is modulated in cell culture. Hum Mol Genet. 1995;4:783–9. doi: 10.1093/hmg/4.5.783. [DOI] [PubMed] [Google Scholar]
- Khandjian EW, Huot ME, Tremblay S, Davidovic L, Mazroui R, Bardoni B. Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc Natl Acad Sci U S A. 2004;101:13357–62. doi: 10.1073/pnas.0405398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, VanderWerf F, Bakker CE, et al. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005;47:339–52. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–38. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Larson J, Jessen RE, Kim D, Fine AK, du Hoffmann J. Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci. 2005;25:9460–9. doi: 10.1523/JNEUROSCI.2638-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson MC, Domingos AI, Jones WD, Chiappe ME, Amrein H, Vosshall LB. Or83b encodes a broadly expressed odorant receptor essential for Drosophila olfaction. Neuron. 2004;43:703–14. doi: 10.1016/j.neuron.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Lee T, Lee A, Luo L. Development of the Drosophila mushroom bodies: sequential generation of three distinct types of neurons from a neuroblast. Development. 1999;126:4065–76. doi: 10.1242/dev.126.18.4065. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–4. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–51. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–83. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wang H, Liang Z, Ku L, O’Donnell WT, Li W, Warren ST, Feng Y. The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci U S A. 2004;101:15201–6. doi: 10.1073/pnas.0404995101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JS, Boothe RG, Lund RD. Development of neurons in the visual cortex (area 17) of the monkey (Macaca nemestrina): a Golgi study from fetal day 127 to postnatal maturity. J Comp Neurol. 1977;176:149–88. doi: 10.1002/cne.901760203. [DOI] [PubMed] [Google Scholar]
- Margulies C, Tully T, Dubnau J. Deconstructing memory in Drosophila. Curr Biol. 2005;15:R700–13. doi: 10.1016/j.cub.2005.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin EC, Watts RJ, Tanaka NK, Ito K, Luo L. Developmentally programmed remodeling of the Drosophila olfactory circuit. Development. 2005;132:725–37. doi: 10.1242/dev.01614. [DOI] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–64. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- McKinney BC, Grossman AW, Elisseou NM, Greenough WT. Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am J Med Genet B Neuropsychiatr Genet. 2005;136:98–102. doi: 10.1002/ajmg.b.30183. [DOI] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417–31. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–5. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–46. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Tousa JE, Baehr W, Martin RL, Hirsh J, Pak WL, Applebury ML. The Drosophila ninaE gene encodes an opsin. Cell. 1985;40:839–50. doi: 10.1016/0092-8674(85)90343-5. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–45. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14:1863–70. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005;25:5087–95. doi: 10.1523/JNEUROSCI.0093-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SP, Bassetto L, Genova GK, Kleyner Y, Leyssen M, Jackson FR, Hassan BA. The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol. 2005;15:1156–63. doi: 10.1016/j.cub.2005.05.050. [DOI] [PubMed] [Google Scholar]
- Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, Bagni C, Ammassari-Teule M. Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102:11557–62. doi: 10.1073/pnas.0504984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol (Berl) 1985;67:289–95. doi: 10.1007/BF00687814. [DOI] [PubMed] [Google Scholar]
- Sabaratnam M, Vroegop PG, Gangadharan SK. Epilepsy and EEG findings in 18 males with fragile X syndrome. Seizure. 2001;10:60–3. doi: 10.1053/seiz.2000.0492. [DOI] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, Moine H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. Embo J. 2001;20:4803–13. doi: 10.1093/emboj/20.17.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroll C, Riemensperger T, Bucher D, Ehmer J, Voller T, Erbguth K, Gerber B, Hendel T, Nagel G, Buchner E, et al. Light-induced activation of distinct modulatory neurons triggers appetitive or aversive learning in Drosophila larvae. Curr Biol. 2006;16:1741–7. doi: 10.1016/j.cub.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Singh K, Gaur P, Prasad S. Fragile x mental retardation (Fmr-1) gene expression is down regulated in brain of mice during aging. Mol Biol Rep. 2007;34:173–81. doi: 10.1007/s11033-006-9032-8. [DOI] [PubMed] [Google Scholar]
- Stefani G, Fraser CE, Darnell JC, Darnell RB. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci. 2004;24:7272–6. doi: 10.1523/JNEUROSCI.2306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Maravall M, Svoboda K. Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron. 2001;31:305–15. doi: 10.1016/s0896-6273(01)00360-9. [DOI] [PubMed] [Google Scholar]
- Sung YJ, Conti J, Currie JR, Brown WT, Denman RB. RNAs that interact with the fragile X syndrome RNA binding protein FMRP. Biochem Biophys Res Commun. 2000;275:973–80. doi: 10.1006/bbrc.2000.3405. [DOI] [PubMed] [Google Scholar]
- Sung YJ, Dolzhanskaya N, Nolin SL, Brown T, Currie JR, Denman RB. The fragile X mental retardation protein FMRP binds elongation factor 1A mRNA and negatively regulates its translation in vivo. J Biol Chem. 2003;278:15669–78. doi: 10.1074/jbc.M211117200. [DOI] [PubMed] [Google Scholar]
- Todd PK, Mack KJ. Sensory stimulation increases cortical expression of the fragile X mental retardation protein in vivo. Brain Res Mol Brain Res. 2000;80:17–25. doi: 10.1016/s0169-328x(00)00098-x. [DOI] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A. 2003;100:14374–8. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Valentine G, Chakravarty S, Sarvey J, Bramham C, Herkenham M. Fragile X (fmr1) mRNA expression is differentially regulated in two adult models of activity-dependent gene expression. Brain Res Mol Brain Res. 2000;75:337–41. doi: 10.1016/s0169-328x(99)00310-1. [DOI] [PubMed] [Google Scholar]
- Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT, Oostra BA. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature. 1993;363:722–4. doi: 10.1038/363722a0. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Wang H, Ku L, Osterhout DJ, Li W, Ahmadian A, Liang Z, Feng Y. Developmentally-programmed FMRP expression in oligodendrocytes: a potential role of FMRP in regulating translation in oligodendroglia progenitors. Hum Mol Genet. 2004;13:79–89. doi: 10.1093/hmg/ddh009. [DOI] [PubMed] [Google Scholar]
- Watts RJ, Schuldiner O, Perrino J, Larsen C, Luo L. Glia engulf degenerating axons during developmental axon pruning. Curr Biol. 2004;14:678–84. doi: 10.1016/j.cub.2004.03.035. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, et al. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci U S A. 2004;101:17504–9. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Truman JW. Cellular mechanisms of dendrite pruning in Drosophila: insights from in vivo time-lapse of remodeling dendritic arborizing sensory neurons. Development. 2005;132:3631–42. doi: 10.1242/dev.01928. [DOI] [PubMed] [Google Scholar]
- Wilson BM, Cox CL. Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci U S A. 2007;104:2454–9. doi: 10.1073/pnas.0610875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Bogert BA, Li W, Su K, Lee A, Gao FB. The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr Biol. 2004;14:1025–34. doi: 10.1016/j.cub.2004.05.055. [DOI] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–66. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, et al. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10:578–87. doi: 10.1038/nn1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–27. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- Zars T, Fischer M, Schulz R, Heisenberg M. Localization of a short-term memory in Drosophila. Science. 2000;288:672–5. doi: 10.1126/science.288.5466.672. [DOI] [PubMed] [Google Scholar]
- Zhang M, Wang Q, Huang Y. Fragile X mental retardation protein FMRP and the RNA export factor NXF2 associate with and destabilize Nxf1 mRNA in neuronal cells. Proc Natl Acad Sci U S A. 2007;104:10057–62. doi: 10.1073/pnas.0700169104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Broadie K. Fathoming fragile X in fruit flies. Trends Genet. 2005;21:37–45. doi: 10.1016/j.tig.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Friedman DB, Wang Z, Woodruff E, 3rd, Pan L, O’Donnell J, Broadie K. Protein expression profiling of the drosophila fragile X mutant brain reveals up-regulation of monoamine synthesis. Mol Cell Proteomics. 2005;4:278–90. doi: 10.1074/mcp.M400174-MCP200. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25:7385–92. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Wang J, Haerry TE, Wu AY, Martin J, O’Connor MB, Lee CH, Lee T. TGF-beta signaling activates steroid hormone receptor expression during neuronal remodeling in the Drosophila brain. Cell. 2003;112:303–15. doi: 10.1016/s0092-8674(03)00072-2. [DOI] [PubMed] [Google Scholar]
- Zito K, Svoboda K. Activity-dependent synaptogenesis in the adult Mammalian cortex. Neuron. 2002;35:1015–7. doi: 10.1016/s0896-6273(02)00903-0. [DOI] [PubMed] [Google Scholar]