

Abstract
Fragile X syndrome (FXS), caused by loss of FMR1 gene function, is the most common heritable cause of intellectual disability and autism spectrum disorders. The FMR1 protein (FMRP) translational regulator mediates activity-dependent control of synapses. In addition to the metabotropic glutamate receptor (mGluR) hyperexcitation FXS theory, the GABA theory postulates that hypoinhibition is causative for disease state symptoms. Here, we use the Drosophila FXS model to assay central brain GABAergic circuitry, especially within the Mushroom Body (MB) learning center. All 3 GABAA receptor (GABAAR) subunits are reportedly downregulated in dfmr1 null brains. We demonstrate parallel downregulation of glutamic acid decarboxylase (GAD), the rate-limiting GABA synthesis enzyme, although GABAergic cell numbers appear unaffected. Mosaic analysis with a repressible cell marker (MARCM) single-cell clonal studies show that dfmr1 null GABAergic neurons innervating the MB calyx display altered architectural development, with early underdevelopment followed by later overelaboration. In addition, a new class of extra-calyx terminating GABAergic neurons is shown to include MB intrinsic α/β Kenyon Cells (KCs), revealing a novel level of MB inhibitory regulation. Functionally, dfmr1 null GABAergic neurons exhibit elevated calcium signaling and altered kinetics in response to acute depolarization. To test the role of these GABAergic changes, we attempted to pharmacologically restore GABAergic signaling and assay effects on the compromised MB-dependent olfactory learning in dfmr1 mutants, but found no improvement. Our results show that GABAergic circuit structure and function are impaired in the FXS disease state, but that correction of hypoinhibition alone is not sufficient to rescue a behavioral learning impairment.
Keywords: Fragile X Mental Retardation Protein (FMRP), Mushroom Body, glutamic acid decarboxylase, synapse, calcium signaling, associative learning
Introduction
GABAergic dysfunction is implicated in a range of neurodevelopmental disorders, including autism, epilepsy, Rett syndrome, and Fragile X syndrome (FXS) (D'Hulst and Kooy, 2007; 2009; Gatto and Broadie, 2010; Paluszkiewicz et al., 2010; Pizzarelli et al., 2011; Coghlan et al., 2012). In mouse and Drosophila FXS disease models, GABAergic deficiencies include depressed glutamic acid decarboxylase (gad) mRNA (D'Hulst et al., 2009) and GABAA receptor (GABAAR) mRNA/protein levels (Idrissi et al., 2005; D'Hulst et al., 2006; Gantois et al., 2006; Curia et al., 2009; Adusei et al., 2010). GABAergic functional defects also occur in fragile X mental retardation 1 (FMR1) knockout mice. In the hippocampus, there is decreased GABAergic tonic inhibition (Curia et al., 2009). In the amygdala, both tonic and phasic inhibitory currents are reduced (Olmos-Serrano et al., 2010). Striatal spiny neurons display increased spontaneous inhibitory current frequency, with reduced paired-pulse ratio of evoked inhibitory currents (Centonze et al., 2008), consistent with reported fronto-striatal circuit disruption in clinical FXS (Menon et al., 2004). Together, these findings indicate depressed GABAergic function in the FXS disease state (Paluszkiewicz et al., 2010), suggesting GABAergic therapeutic treatment potential. The first such trial was conducted in the Drosophila FXS model, with a chemical screen demonstrating that feeding GABA or GABA reuptake inhibitors prevents a host of Drosophila FMR1 (dfmr1) null phenotypes (Chang et al., 2008).
With only 3,000 GABAergic neurons in the genetically-tractable Drosophila brain (Buchner et al., 1988; Chiang et al., 2011), this disease model provides an accessible system for testing GABAergic circuit impairments. A key focus is the central brain Mushroom Body (MB) olfactory learning and memory center, in which the resistance to dieldrin (RDL) GABAAR in MB-intrinsic Kenyon Cell (KC) neurons is involved in synaptic integration within the MB calyx (Su and O'Dowd, 2003; Enell et al., 2007; Liu et al., 2007; Leiss et al., 2009). MB-targeted RDL knockdown elevates calcium responses to odor presentation and increases olfactory learning acquisition, although not memory stability (Liu et al., 2007). Importantly, we have shown that dfmr1 null mutants display altered KC architecture (Pan et al., 2004; Tessier and Broadie, 2008; Siller and Broadie, 2011) and striking olfactory learning and memory impairments (Bolduc et al., 2008; Coffee et al., 2012). We therefore hypothesized that loss of Drosophila Fragile X mental retardation protein (dFMRP) may cause GABAergic circuitry defects in the MB underlying disruption of learning and memory behaviors. In this study, we assay GABAergic system expression, structure, and function in the well-characterized Drosophila FXS model (Repicky and Broadie, 2008; Coffee et al., 2010; Gatto and Broadie, 2011; Siller and Broadie, 2011; Tessier and Broadie, 2011). We report GAD depression with normal GABAergic neuron numbers, developmental stage-specific MB GABAergic neuron structural defects, a novel subclass of MB-intrinsic GABAergic KCs, and functional alterations in GABAergic neuron calcium signaling dynamics. However, efforts to pharmacologically augment GABAergic signaling, as done previously (Chang et al., 2008), failed to restore MB-dependent learning defects in dfmr1 mutants. These findings reveal MB GABAergic circuitry defects in the Drosophila FXS disease model, but suggest that restoring inhibitory balance alone is insufficient to correct learning.
Materials and Methods
Drosophila Genetics
Drosophila stocks were maintained on standard cornmeal/agar/molasses food in humidity-controlled incubators with a 12 hour light:dark cycle at 25°C. Two independent dfmr1 null alleles (dfmr150M (Zhang et al., 2001) and dfmr12 (Dockendorff et al., 2002)) were compared to w1118 genetic background control. The A9.9 GAD1-Gal4/CyO driver line (Gero Misenböck, University of Oxford, Oxford, UK (Ng et al., 2002)) was used for GABAergic neuron labeling and transgene expression. The OK107-Gal4 driver line was used for MB KC labeling and transgene expression. Transgenes included: UAS-mCD8∷GFP, -DenMark, -dBrainbow, -GFP.nls (Bloomington Drosophila Stock Center, Bloomington, IN), and the UAS-GCaMP1.3 Ca2+ reporter (Nakai et al., 2001). For comparisons of control and dfmr1, standard genetic techniques were used to introduce all transgenic elements into both backgrounds. For pharmacological modulation of GABAergic signaling, animals were raised throughout development and into adulthood on standard food supplemented with either 40μM GABA or 40μM nipecotic acid (NipA) (Chang et al., 2008). As previously, heated molten food was cooled to 50°C before drugs were added and then thoroughly mixed to achieve even distribution.
Drosophila Brainbow and MARCM
Drosophila Brainbow (dBrainbow) was used to clonally subdivide the GAD-Gal4 GABAergic neuron population (Hampel et al., 2011). For Cre-mediated recombination, y1w67c23 P{Crey}1b; snaSco/CyO; Dr1/TM3, Sb1 (Bloomington) was crossed to GAD-Gal4/CyO and GAD-Gal4; dfmr150M/TM6. F1 offspring harboring recombinase and driver were then crossed to w1118; P{UAS-Brainbow}attP40 (Bloomington) and P{UAS-Brainbow}attP40/CyO; dfmr150M/TM6, respectively. Experimental animals were females of the genotypes: y1w67c23 P{Crey}1b/w1118; GAD-Gal4/P{UAS-Brainbow}attP40 and y1w67c23 P{Crey}1b/+; GAD-Gal4/P{UAS-Brainbow}attP40; dfmr150M. Mosaic analysis with a repressible cell marker (MARCM) clonal analyses were then employed to further dissect the GABAergic neuron population (Lee and Luo, 2001; Wu and Luo, 2006). GAL80 inhibits GAL4 transcription factor activity, and following temperature-sensitive flippase (FLP) recombinase/FLP recombination target (FRT)-mediated mitotic recombination, the GAL80 transgene is removed from one of the daughter cells providing for expression of a GAL4-driven reporter gene specifically in this daughter cell and its progeny. For control and dfmr1 clone induction, UAS-mCD8∷GFP, hsFLP; FRT82b, tubGal80 was crossed to either GAD-Gal4; FRT82b or GAD-Gal4; FRT82b, dfmr150M/TM6. FLP/FRT-mediated recombination was then induced with 37°C heat-shock (HS) at indicated times; 1 hour HS at 2 days after egg lay (AEL) was used for most examinations. Clones generated were of the following genotypes: UAS-mCD8∷GFP, hsFLP/+; GAD-Gal4/+; FRT82b and UAS-mCD8∷GFP, hsFLP/+; GAD-Gal4/+; FRT82b, dfmr150M. Structural analyses were done using the ImageJ/FIJI (NIH, Bethesda, MD, http://rsb.info.nih.gov/ij/) Simple Neurite Tracer segmentation tool to generate overlaid, skeletonized outlines for quantification (Longair et al., 2011).
Immunocytochemistry
Studies were performed essentially as previously described (Gatto and Broadie, 2008; 2009; 2011). Briefly, staged brains were dissected in phosphate buffered saline (PBS) and then fixed for 30 minutes with 4% paraformaldehyde/4% sucrose in PBS (pH 7.4) at room temperature or with ice-cold Bouin's fixative (GAD only (Kolodziejczyk et al., 2008)). Preparations were rinsed with PBS, then blocked and permeablized with 0.2% triton X-100 in PBS (PBST) supplemented with 1% bovine serum albumin (BSA) and 0.5% normal goat serum (NGS) for 1 hour at room temperature. Primary and secondary antibodies were diluted in PBST with 0.2% BSA and 0.1% NGS and incubated overnight at 4°C and 2–4 hours at room temperature, respectively. Antibodies used included: anti-FasII (1:10; 1D4, mouse, Developmental Studies Hybridoma Bank (DSHB), University of Iowa), anti-GAD (1:500; 819, rabbit (Featherstone et al., 2000)), anti-GFP (1:2,000; ab290, rabbit, AbCAM Inc., Cambridge, MA), anti-GFP-FITC (1:200; ab6662, goat, AbCAM), anti-myc (1:50; 9E10, mouse, DSHB), anti-HA (1:100; 3F10, rat, Roche Applied Science, Indianapolis, IN), anti-RFP (1:2,000; 600-401-379, rabbit, Rockland Immunochemicals Inc., Gilbersville, PA), anti-ChAT (1:100; ChAT4B1, mouse, DSHB), anti-dFMRP (1:500; 6A15, mouse, Sigma, St. Louis, MO), and anti-GABA (1:500; A2052, rabbit, Sigma). Alexa-568/633-phalloidin (1:25; Invitrogen-Molecular Probes, Carlsbad, CA) was used for F-actin visualization. Alexa-Fluor secondaries (1:250; Invitrogen-Molecular Probes) were used throughout. All specimens were mounted in Fluoromount-G (Electron Microscopy Sciences, Hatfield, PA), and fluorescent images were collected using a ZEISS LSM 510 META laser scanning confocal microscope.
Neuronal Cell Counts
To examine the cell number and organization of GABAergic neurons, a nucleus-targeted UAS-GFP.nls (Bloomington) transgene driven by GAD-Gal4 was introduced into both control and dfmr1 null backgrounds. Brains from 1 day old adult animals were dissected and processed following the immunocytochemical methods described above. Confocal imaging was done of whole brains, with a particular focus on the posterior aspect of the MB calyx region. Maximum intensity projections were generated from Z-stacks to quantify GFP-positive nuclei within the area adjacent to the F-actin delineated MB calyx. From the center of the calyx, a 50μm diameter circle was drawn and all marked nuclear profiles in this brain region counted in control and dfmr1 null animals.
MB Structural Assays
Staged brains from 1 day old adults were dissected, fixed, and labeled with anti-FasII (DSHB), as described above, in order to examine MB structural phenotypes. Controls and dfmr1 null animals were examined with and without constant 40μM GABA feeding (Chang et al., 2008). Confocal Z-stacks were obtained of the entire collection of MB axonal lobes and scored for neuronal structural abnormalities, including overgrowth of the β lobes across the brain midline and other overt lobe development phenotypes.
Western Blotting
Studies were performed as previously described (Gatto and Broadie, 2008; 2009; 2011). Briefly, heads from staged animals were homogenized and boiled in NuPage sample buffer (Invitrogen, Carlsbad, CA) supplemented with 40mM DTT. Total protein from 3 heads per sample was loaded onto 4–12% Bis-Tris gels and electrophoresed in NuPage MES Buffer (Invitrogen) for 1 hour at 200 V. Transfer to nitrocellulose was performed for 1 hour at 100 V in NuPage transfer buffer (Invitrogen) with 10% MeOH. Processing was completed using the Odyssey near infrared fluorescence detection system (Li-COR, Lincoln, NE). Antibodies used included: anti-dFMRP (1:3,000; 6A15), anti-α-tubulin (1:100,000; B512, mouse, Sigma), anti-GAD (1:1,000; 819), Alexa-goat-anti-rabbit-680 (1:10,000; Invitrogen–Molecular Probes), and IR-goat-anti-mouse-800 (1:10,000; Rockland). GAD levels were quantified by measuring immunoreactive band intensities compared to the α-tubulin loading control. The dfmr1 null was normalized to w1118 genetic background control to report percent expression levels.
GCaMP Calcium Imaging
Studies were done as previously described (Gatto and Broadie, 2011; Tessier and Broadie, 2011). Briefly, to examine GABAergic neurons, brains from staged animals (UAS-GCaMP/+; GAD-Gal4/+ or UAS-GCaMP/+; GAD-Gal4/+; dfmr150M) were dissected in room temperature physiological saline containing 128mM NaCl, 2mM KCl, 4mM MgCl2, 35.5mM sucrose, 5mM HEPES, and 1.5mM CaCl2, pH 7.2. Isolated brains were immediately imaged under a 40X water-immersion objective with maximal pinhole aperture on a ZEISS LSM 510 META laser scanning confocal microscope (zoom 2, scan speed 10, 256 × 256 pixels). Preparations were scanned for 1 minute to ascertain baseline fluorescence and then acutely depolarized with 70mM KCl. Calcium transients were monitored for at least 1 additional minute. ImageJ/FIJI (NIH) was used for image registration and determination of fluorescence intensity values for regions of interest. The baseline fluorescence level was defined as the average value during the initial period from 40–50 seconds of acquisition and used to normalize the data set.
Olfactory Associative Learning
Studies were done as previously described (Coffee et al., 2012). Briefly, stocks were maintained on 12 hour light:dark cycle (4 AM lights on, 4 PM lights off) and tested at Zeitgeber time 12–14 at 4–7 days post-eclosion (Lyons and Roman, 2009). All procedures were carried out in the dark at 23°C and 85–95% humidity. 75–100 flies were acclimated for 2 minutes in a cylindrical tube lined with parallel copper wire for electric shock reinforcement coincident with an olfactory cue. The sequentially introduced odors of 10−3 3-Octanol (OCT) and 1.5 × 10−3 4-methylcyclohexanol (MCH) were presented for one minute. For the conditioned stimulus (CS+), a train of 10 electrical shocks were given at 80 V every 5 seconds for 2.5 seconds. Animals were then exposed to 1 minute intervals of odorless airflow, unshocked odor (control stimulus, CS−), and again odorless airflow. After 2 minutes acclimation, animals were given a T-maze choice of converging air currents carrying CS+ and CS− odors for 2 minutes. Animals were then trapped in choice tubes, anesthetized, and counted. The final learning index (LI) was computed as follows: LI=[(CS−)−(CS+)]/[(CS−)+(CS+)] (Tully and Quinn, 1985).
Statistics
Analyses were performed using InStat 3 and Prism 6.02 (GraphPad Software, Inc., La Jolla, CA). Unpaired, two-tailed t-tests with Welch correction were applied for normal data sets with unequal standard deviations. When data were not normally distributed, pair-wise comparisons were made with non-parametric Mann-Whitney tests. For parametric multiple comparisons, one-way analysis of variance (ANOVA) was performed with Tukey-Kramer multiple comparison post-tests. For nonparametric multiple comparisons, a Kruskal-Wallis test was applied with Dunn's multiple comparison tests. For line comparisons in calcium dynamics studies, linear regressions were fit to rise and decay to enable two-tailed calculations in slope comparisons. Significance in figures is presented as p<0.05 (*), p<0.01 (**), and p<0.001 (***).
Results
Null dfmr1 brains show GAD loss in normally arrayed GABAergic circuitry
To begin assaying GABAergic circuitry in the Drosophila FXS model, we first tested brain expression of the rate-limiting GABA synthesizing enzyme GAD in developmental studies (Fig. 1). In control (w1118), GAD is concentrated in several distinct brain regions. Viewed from the anterior aspect, strong GAD expression is present throughout the ventral subesophageal ganglion and in neural cell bodies surrounding the antennal lobes (Fig. 1A). When examined from the posterior, GAD-positive neuronal soma clusters are bilaterally distributed at the upper border of the central brain and optic lobes situated between the lateral horn and lobula plate, and in another distinct cluster near the dorsal midline (Fig. 1B). Importantly, punctate GAD bilateral foci occur within the region of the MB calyx. In dfmr1 nulls, GAD is expressed in similar patterns, but with reduced intensities. Specifically, MB calyx-associated GAD is reduced and also more diffuse in mutants (Fig. 1B). Quantified anti-GAD fluorescence levels were significantly lower in null dfmr1 brains compared to controls, at both day 1 (control: 1±0.09, N=14 vs. dfmr1: 0.74±0.06, N=14; p=0.0274) and day 5 (control: 1±0.1, N=15 vs. dfmr1: 0.75±0.07, N=17; p=0.0452) post-eclosion (Fig. 1C). This GAD reduction was confirmed by Western blot (Fig. 1D). Controls display the expected dual dFMRP bands (Tessier and Broadie, 2008; Gatto and Broadie, 2011) and several GAD bands representing the major GAD 65/67kDa isoforms (Idrissi et al., 2005). Two independent dfmr1 null alleles, dfmr150M and dfmr12, show complete loss of dFMRP and strong GAD reduction (Fig. 1D). Western blot quantification showed GAD expression to be 64±6% and 61±0.2% of control levels in dfmr150M and dfmr12, respectively (N=3 for each genotype; p=0.0552). Taken together, these results show reduced GAD levels in the dfmr1 null brain, presumably causing GABAergic hypoinhibition in the FXS disease state.
Figure 1. Null dfmr1 mutant brains display depressed GAD protein levels.

A) Adult Drosophila control (w1118) and dfmr1 null (dfmr150M) brains labeled with anti-GAD (green) and anti-FasII (red) imaged from anterior (A). Optic and antennal lobes (OL and AL, respectively) and MB α, β, and γ lobes are indicated. A secondary antibody control shows no labeling (goat-anti-rabbit 488 (GAR488, green) and goat-anti-mouse 568 (GAM568, red)). B) Brains labeled with anti-GAD (green) imaged from the posterior (P). The calyx (C), optic lobe (OL), and esophageal foramen (EF) are labeled for spatial orientation. The dashed rectangular area, including the right-side MB calyx region, is shown at higher magnification on the right. C) GAD immunolabeling intensity quantification at day 1 and 5 post-eclosion normalized to w1118 control. Error bars represent s.e.m. Significance is shown at p<0.05 (*). D) Representative anti-GAD Western blot of isolated adult brains at 1 day post-eclosion. Top row shows dFMRP loss in two independent null alleles (dfmr150M, dfmr12) compared to control (w1118). Middle row shows GAD reduction in both mutants. α-tubulin serves as the loading control.
The loss of GAD could be due to either reduced GABAergic neuron numbers or specific GAD downregulation in normally arrayed cells. To test for potential differences in GABAergic neuron organization, we first used the UAS/GAL4 system (Duffy, 2002) with the A9.9 GAD-Gal4 line (3.1 kb of upstream sequence from the gad1 start codon (Ng et al., 2002)) driving UAS-mCD8::GFP to label neuronal membranes (Fig. 2A). We assayed dFMRP expression relative to this marked GABAergic neuron population. Examined from both anterior and posterior directions in higher magnification single optical slices, dFMRP co-labeled with GFP-positive GABAergic neurons (Fig. 2A). dFMRP (red) is more ubiquitously expressed, as expected from known expression in other neuronal classes, with relative exclusion from areas devoid of neuronal somas, such as the MB calyx neuropil. This apparent process exclusion is a consequence of the significantly higher dFMRP protein expression levels within the neuronal soma cytoplasm. Closer inspection reveals dFMRP puncta within the MB neuropil calyx region (data not shown). Importantly, GABAergic neurons (green) adjacent to the MB calyx region display robust dFMRP expression (Fig. 2A, bottom row). Comparing control to dfmr1 null brains, there was no discernible difference in the extent or spatial distribution of GAD-Gal4-driven UAS-mCD8::GFP (data not shown), suggesting the formation of grossly normal GABAergic neuronal circuitry throughout the brain in the absence of dFMRP.
Figure 2. Brain GABAergic neurons express dFMRP.

A) Brain immunocytochemistry to visualize GAD-Gal4-driven UAS-mCD8::GFP (green) at the membrane and cytoplasmic anti-dFMRP labeling (red) from anterior (A) and posterior (P) perspectives (y1w*/+; GAD-Gal4/UAS-mCD8::GFP.L.LL5). Optic and antennal lobes (OL and AL, respectively) and MB calyx (C, arrowhead) are indicated for reference. Higher magnification views of single optical slices show antennal lobe (A, boxed) and calyx regions (P, boxed). B) Brain immunocytochemistry to visualize GAD-Gal4-driven nuclear UAS-GFP.nls (NLS-GFP, green) and anti-dFMRP labeling (red) from both anterior (A) and posterior (P) (w1118/+; GAD-Gal4/UAS-GFP.nls.14). The dashed box denotes the MB calyx region shown at higher magnification in the second row. GABAergic neurons in all brain areas express dFMRP. Arrowheads denote neurons displaying GAD-Gal4-driven UAS-GFP.nls with associated dFMRP. Arrows denote dFMRP-expressing neurons not within the GAD-Gal4 population.
To further assay dFMRP expression within GABAergic neurons, anti-dFMRP labeling was assessed relative to GAD-Gal4-driven expression of a nuclear GFP transgene (UAS-GFP.nls (Neufeld et al, 1998); Fig. 2B). dFMRP is clearly apparent in all GABAergic cell bodies. In higher magnification singular optical sections, cytosolic dFMRP can be seen surrounding the GFP-positive nuclei (Fig. 2B, bottom row). Given the ubiquitous expression of dFMRP in all neurons, not all dFMRP-expressing neurons are GAD-Gal4 GABAergic neurons, as expected (Fig. 2B). The clear localization of dFMRP in Drosophila GABAergic neurons agrees with mouse studies showing FMRP expression in GAD65/67-positive neurons (Olmos-Serrano et al., 2010).
The relative numbers of GABAergic neurons in control and dfmr1 null brains were next assessed. The GAD-Gal4 line shows good overlap with anti-GABA immunostaining (Hamasaka et al., 2005), as well as anti-GAD and anti-vGAT visualization (Enell et al., 2007), and consistent cellular distribution with gad1 in situ hybridization (Okada et al., 2009). We similarly observe anti-GAD labeling overlapping the GAD-Gal4 neuron population (Fig. 3A), and again used GAD-Gal4 to drive nuclear UAS-GFP.nls transgene expression in control and dfmr1 null brains (Fig. 3B). The overall pattern of distribution and density of GABAergic neurons in control and dfmr1 null brains appeared comparable, with no detectable differences (Fig. 3B). To quantify GABAergic cell numbers, maximum intensity projections were generated of the GFP-positive nuclei surrounding the MB calyx target region (Fig. 3B, right column). The number of GABAergic nuclei was counted within a 50μm diameter region centered on the MB calyx (Fig. 3C, top). No significant difference in cell number was observed between control and dfmr1 null (control: 150±5, N=12 vs. dfmr1: 142±5 N=12; p=0.3118; Fig. 3C, bottom). Thus, the decreased GAD levels observed in the dfmr1 null brain cannot be attributed to a loss of GABAergic neurons.
Figure 3. GABAergic neuron numbers are unaltered in dfmr1 null brains.

A) Immunocytochemistry to visualize GAD-Gal4-driven UAS-mCD8::GFP (green) at the membrane relative to anti-GAD (red) immunoreactivity in 1 day old control brains. The MB calyx (C) is labeled. B) Day 1 adult brain immunocytochemistry to visualize GADGal4-driven nuclear UAS-GFP.nls (NLS-GFP, green) and F-actin (red) from the posterior in both controls and dfmr1 nulls. The dashed square box denotes the MB calyx region shown at higher magnification, with GFP-positive nuclei shown relative to the actin-enriched MB calyx. C) For nuclei count quantification, a 50μm diameter circle was centered on the MB calyx, with maximum intensity projections from confocal Z-stacks (top). There is no significant difference (n.s.) between control and dfmr1 nulls.
To sub-divide the complex GAD-Gal4 GABAergic pattern, we next employed dBrainbow technology (Hampel et al., 2011), which uses Cre-recombinase-mediated, stochastic, and mutually exclusive selection of 1 of 3 tandemly arrayed UAS-fluorescent markers for labeling (Fig. 4). The GAD-Gal4-driven UAS-dBrainbow cassette was studied with immunolabeling for anti-GFP (green), anti-myc (red), and anti-HA (blue) to assay clonal GABAergic neuron lineages. As active neuronal migration outside the optic lobes does not occur in Drosophila, static clonal units are built locally via neuroblast proliferation (Ito et al., 2013; Yu et al., 2013), with the central brain region being generated from a pool of approximately 100 neuroblasts (Ito and Hotta, 1992; Urbach and Technau, 2003). Thus, the clearly separate marked dBrainbow domains represent separate GABAergic lineages (Fig. 4A). With recombination taking place early in neuroblast division, anterior dBrainbow tracing (Fig. 4A, A) indicates that GABAergic neurons arise from at least 25 distinct lineages (31±2 clonal groups, excluding optic lobes and subesophageal ganglion regions; N=6 brains). More specifically, GABAergic cell bodies adjacent to the MB calyx are present in several domains marked by different dBrainbow colors (Fig. 4A, P), indicating that they arise from several distinct lineages (10±1 clonal groups; N=16 calyces). Control and dfmr1 null brains did not display any discernible differences in GABAergic lineage size or spatial organization, imaged from either the anterior or posterior orientations (Fig. 4A). More explicitly, GABAergic cell numbers and spatial distribution in the MB calyx region do not detectably change in dfmr1 null compared to control brains (Fig. 4B). Despite the stochastic nature of the dBrainbow labeling technique, high magnification images reveal comparable GAD-Gal4 neuronal lineages in controls and dfmr1 nulls, based on clonal size and distribution. We conclude that dfmr1 null brains exhibit specific loss of GAD expression within grossly normal GABAergic circuitry, with no detectable differences in cell number or brain region innervation patterns.
Figure 4. Brainbow indicates normal GABAergic cell populations in dfmr1 nulls.

A) To subdivide the GAD-Gal4 GABAergic neuron population, dBrainbow Cre-mediated recombination events drive specific fluorophore choices; EGFP-V5 (green), mKO2-myc (red), and EBFP2-HA (blue). Anterior (A) and posterior (P) views are presented for a comprehensive low magnification comparison of GABAergic neuron lineages in control and dfmr1 null brains. The optic (OL) and antennal (AL) lobes, MB calyx (C), and esophageal foramen (EF) are indicated for orientation. B) Higher magnification GADGal4-mediated UAS-dBrainbow images comparing control (top) and dfmr1 null (bottom) GABAergic neuron lineages. The boxed areas (1–3) within the first column denote the specific regions shown at higher magnification to the right. Posterior views that include the MB calyx region are shown in all cases.
Gross GABAergic innervation of the MB calyx is unaltered in dfmr1 null brains
Our next goal was to specifically assay GABAergic innervation of the MB calyx, a key brain region underlying learning and memory behavior output (Jefferis et al., 2002; Davis, 2005; Margulies et al., 2005; Keene and Waddell, 2007; Tanaka et al., 2008; Waddell, 2010; Davis, 2011; Kahsai and Zars, 2011). Recent work has detailed inhibitory influence provided to the MB by the anterior paired lateral (APL) neurons (Liu and Davis, 2009; Pitman et al., 2011). Singular neurons in each hemisphere have been shown to mediate ipsilateral MB contact projecting to the lobes, peduncle, and calyx (Liu and Davis, 2009). However, we observed a much broader cohort of MB-infiltrating GABAergic neurons to be considered herein.
The MB calyx contains actin-rich KC dendritic claws that encircle the incoming presynaptic projection neurons (PNs) to form hundreds of discrete synaptic microglomeruli, which are also sites of GABAergic input (Fig. 5) (Leiss et al., 2009). This calyx region was demarcated with MB-specific OK107-Gal4-driven expression of UAS-DenMark, an ICAM5-mCherry fusion that selectively labels the dendritic compartment (Nicolaï et al., 2010). By then co-labeling with anti-Fasciclin II (FasII, green) to mark MB axonal lobes (Crittenden et al., 1998), the DenMark (red) calyx labeling was readily distinguished (Fig. 5A). In parallel, relative to GABAergic processes labeled with GAD-Gal4-driven UAS-mCD8::GFP (green), the MB calyx was marked using phalloidin (red) to label the F-actin concentration in the KC dendritic claws (Fig. 5B). The local actin enrichment in the calyx can be seen bilaterally on either side of the protocerebral bridge. The cholinergic PN terminals residing at the center of each microglomerular unit were also marked by immunolabeling for choline acetyltransferase (Fig. 5B, anti-ChAT, blue). Together, these labeling approaches were used to assay GABAergic MB calyx innervation in dfmr1 nulls compared to genetic background controls.
Figure 5. GABAergic processes normally innervate the MB calyx in dfmr1 nulls.

A) OK107-Gal4-driven UAS-DenMark (ICAM5::mCherry fusion, red) marks the MB calyx dendritic region, with KCs labeled with anti-FasII (green). Brains are imaged from posterior for improved MB calyx resolution. MB calyx (C), MB α and β lobes, optic lobes (OL), and ellipsoid body (EB, arrowhead) are labeled for orientation. B) Control and dfmr1 null (dfmr150M) brains show GAD-Gal4-driven UAS-mCD8::GFP (green) relative to F-actin (phalloidin, red) and choline acetyltransferase (anti-ChAT, blue). Optic lobes (OL), MB calyx (C, arrow), esophageal foramen (EF), and protocerebral bridge (PCB, arrowhead) are labeled for reference. Higher magnification views depict MB calyces (arrows) in both genotypes. The convergence of presynaptic PN terminals (ChAT), postsynaptic KC dendritic claws (F-actin), and GABAergic interneuron processes (GFP) is evident in single microglomeruli (arrowheads).
Examining the GAD-Gal4 labeled innervation of the MB calyx revealed little discernible difference between dfmr1 null mutants and controls (Fig. 5B). GABAergic processes densely innervate the MB calyx in both genotypes, providing strong inhibitory synaptic input (Fig. 5B). Multiple quantification approaches revealed no significant differences in either the number or density of GABA-innervated microglomeruli in dfmr1 null mutants compared to controls (data not shown). The relative association of GABAergic process involvement at the microcircuit level appeared the same in both genotypes. At low magnification (Fig. 5B, control and dfmr1 - top rows), the calyx regions can be clearly detected with respect to their GABAergic innervation (anti-GFP, green, C, arrow), KC dendritic actin enrichment (phalloidin, red), and PN boutons (anti-ChAT, blue). The higher magnification images (Fig. 5B, control and dfmr1 - bottom rows) reveal KC dendritic claws (F-actin, red) coalescing to encircle PN boutons (anti-ChAT, blue) often with associated GABAergic processes (anti-GFP, green). The only quantified difference that could be defined in dfmr1 nulls was found when measuring the cross-sectional area of synaptic microglomeruli with GABAergic process innervation (Fig. 5B, arrowheads). Null dfmr1 microglomeruli display a highly significant decrease in the maximal area (control: 5.81±0.24 μm2, N=53 vs. dfmr1: 4.64±0.21 μm2, N=66; p=0.0003). This suggests that dfmr1 influences the contact area between GABAergic inputs and KC dendritic claws within the MB calyx. Overall, however, GABAergic process organization in the MB calyx appears to be largely unaffected in dfmr1 null mutants. To dissect possible GABAergic circuitry differences, it was clear that a higher level of cellular resolution would be required.
Generation of dfmr1 null MARCM clones from birth-dated GABAergic lineages
To assess the fine subcellular architecture of individual GABAergic neurons innervating the MB calyx, high resolution, single-cell labeling was done using GAD-Gal4-driven MARCM (Lee and Luo, 2001; Pan et al., 2004; Wu and Luo, 2006; Tessier and Broadie, 2008; Siller and Broadie, 2011). This clonal technique involves GAL80-mediated UAS/GAL4 repression with FLP/FRT systems for the generation of GFP-labeled control or homozygous dfmr1 null neurons in an otherwise wild-type brain (Fig. 6). This approach allows the definition of cell-autonomous effects of dFMRP via its removal from individual, identified GABAergic neurons. Co-labeling for the anti-GFP clonal marker (green) and anti-dFMRP (red) demonstrated the efficient generation of dfmr1 null mutant neurons (Fig. 6A). In control clones, dFMRP is readily evident within the GABAergic neuronal cell bodies targeted. However, in dfmr1 clones, there is a complete loss of dFMRP within the GFP-positive neurons, with retained immunolabeling in the surrounding cells (Fig. 6A). Thus, we are able to specifically eliminate dFMRP within marked subsets of the GABAergic neuron population upon MARCM induction.
Figure 6. MARCM dfmr1 GABAergic neuron clones in developmental windows.

A) GAD-Gal4-mediated MARCM clones of control and dfmr1 null neurons with heat-shock (HS) induction on the day of egg lay (+0d AEL). The clonal marker (anti-GFP, green) is shown relative to anti-dFMRP (red). Insets depict higher magnification views of indicated cells (arrows). B) Developmental time course of GAD-Gal4-mediated MARCM for both control and dfmr1 null. The optic lobe (OL), MB calyx (C), and esophageal foramen (EF) are labeled for spatial orientation. HS inductions at the indicated developmental windows are presented: embryonic (emb), second instar (2iL), third instar (3iL), pupal days 1 and 3 (P1, P3), and adult (ad). The best induction paradigm for isolated MB-extrinsic GABAergic neurons with calyx-associated processes is +2d AEL. Higher magnification images of the MB calyx region to highlight these birth-dated cells are shown in both control and dfmr1 null brains.
Although GABAergic signaling components have been identified and well-localized in the Drosophila brain (Kuppers et al., 2003; Enell et al., 2007; Liu et al., 2007; Kolodziejczyk et al., 2008), little has been reported about the development of GABAergic neurons. We therefore first devised a series of HS induction paradigms to determine the most appropriate developmental window in which to generate clones of GABAergic neurons innervating the MB calyx (Fig. 6B). We tested MARCM induction at progressive 24 hour time points ranging from embryos at staged periods AEL, to larvae from all three instars, through adults immediately post-eclosion (day 0). MARCM clone patterns were examined in both control and dfmr1 nulls to ensure that the inductive capacity was not altered in the mutants (Fig. 6B). Viewing from the posterior to best image the MB calyx region, we found very little auto-induction in the absence of HS (Fig. 6B, NO HS). Each sequential time point examined revealed GAD-Gal4 neuronal subsets to be marked based on relative birth dates. GABAergic neuron clone generation was readily achieved with HS applied from early embryonic stages (0d AEL) through pupal day 3 (8d AEL), showing that GABAergic neurons are born throughout the entire neuroblast proliferative period (Fig. 6B). The most widespread GABAergic neuron labeling was accomplished with HS from 2–5 days AEL corresponding to the 2nd through wandering 3rd instar developmental windows. The best inductive period allowing sparse labeling of individual GABAergic cell bodies adjacent to and innervating the MB calyx was 2 days AEL in a 2nd instar larval period (Fig. 6B, HS + 2d AEL (2iL)). Higher magnification images of this time point show the GABAergic cells surrounding the MB calyx, which are targeted below (Fig. 6B, upper right corner images for both control and dfmr1 null). Later time points of pupal induction were not as successful in generating MARCM clones, showing that the majority of GABAergic neurons are born during postembryonic larval stages. However, induction at 6 days AEL during pupal day 1 labeled a distinct concentration of late-born GABAergic neurons at the immediate periphery of the MB calyx (Fig. 6B, HS + 6d AEL (P1)), which will also be addressed in detail below. HS induction in newly eclosed adults provided no labeling of new MARCM clones over the background control (Fig. 6B, HS + 10d AEL (ad)), indicating that all GABAergic neurons are born in pre-adult developmental stages and are post-mitotic by eclosion. Having defined the profile of GAD-Gal4 GABAergic neuron birthdates and mitotic potential with MARCM labeling, we could then proceed in examining isolated single-cell clones for dfmr1 phenotypes in GABAergic neuron fine architecture.
Developmental changes in GABAergic neuron architecture in dfmr1 null mutants
To test potential roles for dfmr1 in MB GABAergic neuron architecture, single-cell MARCM labeling was done (Fig. 7A). Individual cell clones were visualized for GAD-Gal4-driven UAS-mCD8∷GFP (anti-GFP, green) relative to the actin-rich MB calyx (phalloidin, C, red) and PN terminal position (anti-ChAT, blue). Importantly, the generation and retention of dfmr1 null clones alone revealed that dfmr1 is not essential for GABAergic neuron survival in a cell-autonomous manner. Two classes of MB GABAergic neurons were immediately obvious, with intra- vs. extra-calyx termination (Fig. 7A). We first focused on the intra-calyx terminating neurons that provide MB calyx microglomerular microcircuit inhibitory input. To quantitatively measure GABAergic neuron architecture, we used the ImageJ/FIJI Neurite Tracer segmentation tool (Fig. 7B) (Longair et al., 2011). Overlaid, skeletonized outlines were made with the mapping origin initiated at the centroid of the neuronal cell body. Traces were progressively extended to encompass the entire collection of neuronal branches emanating from the major primary process (Fig. 7B). As a first assessment, total process length was compared in MARCM clone control and dfmr1 null neurons at both days 1 and 5 post-eclosion. Cumulative length comparisons showed no significant differences between the genotypes at either time point (day 1: control, 75±5 μm (N=50 single-cell clones); dfmr1, 67±3 μm (N=50); p=0.5980. day 5: control, 83±4 μm (N=50); dfmr1, 90±4 μm (N=50); p=0.1487; Fig. 7C). However, dfmr1 mutant neurons showed a differential developmental trajectory in process elaboration, with a highly significant (p<0.001) increase in process length between days 1 and 5, which did not occur in control neurons (Fig. 7C). These results suggest that dFMRP serves to potentially constrain MB innervating GABAergic neuron outgrowth during post-eclosion development.
Figure 7. GABAergic single neuron MARCM for developmental structure studies.
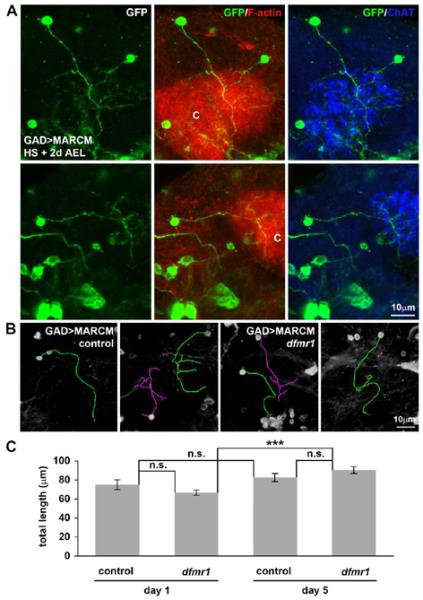
A) Examples of identifiable single-cell GAD-Gal4-mediated MARCM (anti-GFP, green) of neurons innervating the labeled MB calyx (C) (568-phalloidin revealing F-actin, red; anti-ChAT, blue). B) Control and dfmr1 null neuron traces done using the ImageJ/FIJI Neurite Tracer segmentation tool. Overlaid, skeletonized outlines used for quantification are shown (purple, green). The different colors employed indicate separate individual neurons that were sequentially traced within a given Z-series of images. Mapping originated at the centroid of the cell body encompassing all neuronal processes. C) Quantification of total process length of intra-calyx terminating GABAergic neurons for control and dfmr1 null at day 1 and day 5 post-eclosion. Null dfmr1 neurons exhibit significant (p<0.001, ***) elaboration from day 1 to 5, but there are no significant differences (n.s.) between genotypes at either time point. Error bars represent s.e.m.
For more detailed analyses, MB-innervating GABAergic neurons were classified based on process number; simple (primary process only), moderate (1–3 additional processes), and complex (≥4 additional processes). For all three classes, the criterion for analyses was clearly defined and separable marked processes showing no overlap with other MARCM-labeled cells. Any cells that failed to meet these criteria were excluded from the analysis. For these three structural classes, developmental age comparisons were made on days 1 and 5 post-eclosion between control and dfmr1 null neurons (N=50 neurons of each genotype at each developmental stage; Fig. 8). At least 10 individual MARCM clone cells of each GABAergic cell class were examined for each genotype at each time point, with the exception only of the simple neuron class in dfmr1 null mutants, which were rare at both time points. On day 1 post-eclosion, the labeled, identifiable and singularly traceable GABAergic controls considered were comprised of 42% simple, 30% moderate, and 28% complex neurons, whereas dfmr1 nulls were 22% simple, 54% moderate, and 24% complex neurons. By day 5, controls were 20% simple, 30% moderate, and 50% complex, and dfmr1 nulls were 10% simple, 36% moderate, and 54% complex (Fig. 8). Within these GABAergic neuron classes, total process length was under-elaborated in dfmr1 null complex neurons compared to controls at day 1 (control: 118±10 μm (N=14); dfmr1, 83±6 μm (N=12); p=0.0063; Fig. 8A). Moreover, dfmr1 null simple neurons exhibited increased length compared to controls by day 5 (control: 51±5 μm (N=10); dfmr1: 65±4 μm (N=5); p=0.0498; Fig. 8A). A developmental trajectory influence was prominent in dfmr1 null GABAergic neurons, with an increased total process length between day 1 and day 5 across all three structure categories (simple, moderate, and complex; p=0.0132, p=0.0280, and p=0.0181, respectively; Fig. 8A). Moreover, dfmr1 null complex neurons appear to harbor an initial extension failure and thus are likely deficient in the subsequent pruning of processes compared to controls (day 1 vs. day 5: controls, 118±10 μm (day 1, N=14) vs. 110±5 μm (day 5, N=25); dfmr1, 83±6 μm (day 1, N=12) vs. 103±5 μm, (day 5, N=27); Fig. 8A). Taken together, these results show that dFMRP regulates both the early establishment and late refinement of the MB-innervating GABAergic neurons, with differential roles based on neuronal complexity and development stage of the circuitry.
Figure 8. GABAergic dfmr1 neurons display differential developmental growth.

A) Intra-calyx terminating GABAergic neurons identified via GAD-Gal4-mediated MARCM classified by branching complexity; single primary process (simple), 1–3 accessory processes (moderate), and >4 accessory processes (complex). Quantification of total process length comparing control and dfmr1 null single-cell clones at day 1 and 5 post-eclosion. B) Quantification of process length by type in moderate complexity GABAergic neurons, showing primary, secondary, tertiary, and cell body processes in control and dfmr1 null single-cell clones at day 1 and 5 post-eclosion. In controls, accessory cell body-associated processes were never observed at day 1, and in both genotypes, tertiary processes were rarely detected at either day 1 or day 5. C) Quantification of process length for complex neurons, including quarternary processes. Throughout, error bars represent s.e.m. Significance is indicated as p<0.05 (*), p<0.01 (**), and p<0.001 (***).
In a final series of analyses, GABAergic cell body-associated, primary, secondary, tertiary, and quarternary processes were compared in the moderate and complex neuron categories (Fig. 8B, C). In moderate neurons, no differences occurred between control and dfmr1 nulls at a given time (day 1 or day 5) or across developmental time points (day 1 vs. day 5) (Fig. 8B). In sharp contrast, complex GABAergic neurons were significantly different between genotypes both at a given stage and in comparing developmental differences (Fig. 8C). At day 1 post-eclosion, both primary and secondary branches of control GABAergic neurons were longer than the processes in dfmr1 null neurons (primary: control, 72±6 μm (N=14) vs. dfmr1, 57±5 μm (N=12); p=0.0441; secondary: control, 12±1 μm (N=47) vs. dfmr1, 8±1 μm (N=31); p=0.0261). In addition to changes in process length, the prevalence of secondary and tertiary branches was increased in control complex neurons at day 1 compared to dfmr1 mutants. Controls averaged 3.4±0.3 secondary branches (N=14) vs. 2.6±0.2 in dfmr1 nulls (N=12; p=0.0486), and controls displayed 1.6±0.5 tertiary branches (N=14) vs. 0.3±0.1 in dfmr1 nulls (N=12; p=0.0087). Across developmental time, increased primary process length occurred in dfmr1 null GABAergic neurons (57±5 μm (day 1, N=12) vs. 68±3 μm (day 5, N=27); p=0.0400; Fig. 8C), whereas only slight changes in secondary and tertiary branches of control neurons occur between days 1 and 5, decreasing (p=0.0464) and increasing (p=0.0401), respectively. These results show that dFMRP has the strongest influence on MB-innervating GABAergic neurons with the greatest structural complexity, with important roles in regulating connectivity differences during post-eclosion development.
A novel class of MB intrinsic GABAergic neurons
A second class of late-born GABAergic neurons we identified appears to be intrinsic to the MB circuit, with a primary process that passes through the calyx with subsequent extra-calyx termination in the MB axonal lobes (Fig. 9A). Surrounding the MB circuit, it has previously been shown that extrinsic GABAergic APL neurons innervate the MB proper to influence both canonical (Liu and Davis, 2009) and reversal learning (Wu et al., 2012). Additionally, as discussed above, MB calyx microglomeruli are innervated by GABAergic processes initiated from MB-extrinsic GABAergic neurons with cell bodies adjacent to the calyx (Leiss et al., 2009), and MB KCs express GABAARs to receive and integrate these inhibitory cues (Enell et al., 2007; Liu et al., 2007). However, MB-intrinsic GABAergic neurons have not previously been reported in any published study.
Figure 9. A novel class of intrinsic Mushroom Body GABAergic neurons.

A) GAD-Gal4-mediated MARCM with HS induction at + 6d AEL showing clonal marker (anti-GFP, green) relative to anti-ChAT (blue). Cell bodies (CB, arrows), dendrites (D), and initial axonal process extensions are evident within the MB calyx (middle panel). Axonal extensions beyond the calyx (right panel) show clear divergence into MB α and β lobes. Inset shows the overlay of the separable neuron compartments comprising the complex α/β KCs. B) MB-specific OK107-Gal4-driven UAS-mCD8∷GFP (green) to delineate MB KCs. Cell bodies identified in inset (arrow) shown at higher magnification co-labeled with anti-GABA (red). A GABAergic neuron in the MB population is highlighted (arrow). C) OK107-Gal4-driven UAS-mCD8∷GFP relative to co-labeling with anti-GAD (red). Two GABAergic neurons in the MB population are highlighted (arrow).
In our MARCM studies, we identified a novel class of GABAergic neurons that extend through and well beyond the MB calyx, bearing a striking resemblance to α/β KCs (Fig. 9A). Unlike the early-born MB-extrinsic intra-calyx terminating GABAergic neurons characterized above, these late-born MB-intrinsic neurons were preferentially labeled using MARCM with early pupal HS induction (Fig. 6B; HS + 6d AEL (P1)). Their cell bodies maintain typical positioning seen in KCs (Fig. 9A; CB, arrows), and they make clearly defined dendritic claws within the calyx (Fig. 9A; D) associated with the cholinergic terminals of the presynaptic PNs (Fig. 9A; anti-ChAT, blue). Moreover, the processes that extend through the calyx remain in the MB proper and bifurcate into the α and β axonal lobes (Fig. 9A, right panel). To confirm the identity of these KCs as GABAergic neurons, the MB-specific OK107-Gal4 was used to drive expression of membrane-tethered mCD8∷GFP (green) coupled to immunolabeling for both GABA (Fig. 9B, red) and GAD (Fig. 9C, red). A subgroup of these neurons clearly labeled with both GABAergic markers, identifying them as MB-intrinsic GABAergic α/β KCs. These results reveal that subsets of MB KCs are themselves GABAergic, conferring a previously unrecognized level of MB inhibitory control. Moreover, as we have previously described, dfmr1 null KCs exhibit overelaborated axonal and dendritic processes (Pan et al., 2004; Tessier and Broadie, 2008); therefore, it is probable that these novel GABAergic MB KCs display similar architectural phenotypes in the dfmr1 null.
Altered GABAergic neuron calcium dynamics in dfmr1 null mutants
Having shown that dfmr1 null GABAergic neurons display structural alterations, we next tested their functional capacity to ascertain if the mutant GABAergic neurons may likewise present compromised signaling. Calcium influx is the key response to neuronal activity, signaling both short- and long-term responses. In the MB circuit, calcium transients are elicited upon odor presentation (Wang et al., 2004) and serve as cellular learning and memory traces under various stimulation paradigms (Yu et al., 2006; Wang et al., 2008). In the Drosophila FXS model, altered calcium dynamics have been previously reported in both PDF-tritocerebral and KC neurons (Gatto and Broadie, 2011; Tessier and Broadie, 2011). In particular, dfmr1 null KC neurons displayed elevated amplitudes of depolarization-induced calcium influx transients and retarded rise to peak times, resulting in an extended calcium signal time course (Tessier and Broadie, 2011). To test MB-innervating GABAergic neuron signaling function, we assayed calcium dynamics incurred upon acute depolarizing stimulation using a transgenic GCaMP calcium reporter. This reporter incorporates a calmodulin sensor tethered to GFP, with calcium influx altering GFP conformation to increase fluorescence output (Nakai et al., 2001; Akerboom et al., 2009). GAD-Gal4-driven UAS-GCaMP was assayed in control and dfmr1 null GABAergic neurons. A summary of these data is shown in Figure 10.
Figure 10. GABAergic dfmr1 neurons display altered calcium signaling dynamics.

A) Representative still frames of MB-calyx (C) adjacent GABAergic neurons with GAD-Gal4-driven UAS-GCaMP fluorescent calcium reporter before, during, and after depolarization. Control and dfmr1 nulls are shown, rainbow heat-map pseudocolored for fluorescence intensity. Cooler colors (blue end of the spectrum) represent lower intensities, whereas warmer colors (red end of the spectrum) represent higher intensities. B) Quantification of fluorescence intensity relative to the baseline prior to KCl depolarizing stimulation (t=0). Control (blue) and dfmr1 null (red) show comparable slopes in the rise kinetics to maximum, but mutants exhibit retardation in time to maximum, elevated maximal transient amplitude, and delayed but more rapid decay kinetics. Error bars represent s.e.m.
MB-innervating GABAergic neurons were first imaged to establish baseline GCaMP reporter fluorescence (Fig. 10A, pre-stimulation) and then acutely depolarized with 70mM KCl application (Fig. 10B, t=0) while continuously monitoring calcium transient development and dissipation (Fig. 10A, transient maximum and post-transient). Fluorescence intensity was normalized to baseline and reported as the fluorescence change over time. The rise time slope, from baseline to transient maximum, was indistinguishable between control and dfmr1 null neurons; however, the dfmr1 nulls achieved an elevated response peak compared to control (Fig. 10B). Moreover, there was significantly altered calcium dissolution kinetics between control and dfmr1 null GABAergic neurons, as evidenced by comparing the decay slopes (control: −0.4795 ± 0.0048 (R2: 0.9872), N=35 vs. dfmr1: −0.9737 ± 0.0117 (R2: 0.9815), N=37; p<0.0001; Fig. 10B). Thus, depolarization-induced calcium signaling dynamics in MB-innervating GABAergic neurons are altered by the loss of dFMRP, suggesting potential compensatory efforts within the dfmr1 null MB GABAergic circuitry.
Pharmaceutical GABAergic modulation in MB-dependent olfactory learning
To assess the contribution of dfmr1 null mutant GABAergic neuron dysregulation to MB-dependent behavioral output, we assayed classical Pavlovian olfactory learning using the standard T-maze training paradigm (Quinn et al., 1974; Tully and Quinn, 1985). We have previously shown that dfmr1 null mutants exhibit strongly compromised MB-dependent learning and memory (Bolduc et al., 2008; Coffee et al., 2012). Moreover, the RDL GABAAR has been shown to influence MB-dependent memory in both overexpression and knockdown studies, to impair and enhance memory acquisition, respectively (Liu et al., 2007). We therefore hypothesized that dFMRP and RDL functionally interface in the manifestation of MB-dependent learning and memory behavior. Importantly, previous studies have shown that GABAergic modulation via feeding of GABA or the GABA reuptake inhibitor NipA can strongly restore dfmr1 null mutant behavioral defects, including learning during naïve courtship (Chang et al., 2008). We therefore hypothesized the MB-dependent olfactory associative learning should be similarly restored by these GABAergic pharmaceutical treatments.
To ensure our feeding paradigm was sufficient to replicate the dfmr1 rescue previously reported (Chang et al., 2008), we examined MB neuron structural phenotypes employing anti-FasII to visualize the MB axonal lobes (Fig. 11A). MB β lobe crossing and fusion across the midline results from axons failing to terminate at the midline in dfmr1 null mutants (Michel et al., 2004; Pan et al., 2004), and this phenotype has been shown to be significantly rescued by feeding 40μM GABA (Chang et al., 2008). In 1 day old adults, β lobe crossing rarely occurred in controls (2/55 brains, 4% aberrant, 96% normal) but was 10 times more common in dfmr1 nulls (26/65 brains, 40% aberrant; Fig. 11A). When fed throughout development and into adulthood with GABA (40μM), MB β lobe crossing remained rare in controls (6/56 brains, 11% aberrant, 89% normal), and was reduced by ~50% in dfmr1 nulls (13/54 brains, 24% aberrant), consistent with previous reports (Chang et al., 2008). A second striking abnormality in dfmr1 nulls was the absence or loss of MB lobes, most often the α lobe (Fig. 11A). This phenotype was very rarely observed in controls (1/65 brains, 1.5%), but was nearly 20 times more frequent in dfmr1 nulls (19/65 brains, 29%). With GABA feeding, controls maintained the same lobe loss frequency (1/56 brains, 2%), whereas GABA-treated dfmr1 nulls again showed nearly 50% restoration (9/56 brains, 16%; Fig. 11A). Taken together, these results indicated successful GABA dosage and delivery based on effectiveness in alleviating dfmr1 null phenotypes in the MB olfactory learning center.
Figure 11. Null dfmr1 learning defect not altered by GABAergic modulation.

A) Adult day 1 Drosophila control (w1118) and dfmr1 null (dfmr150M) brains labeled with anti-FasII (green). Shown are examples of the variable MB lobe phenotypes observed. Within the MB region, α, β, and γ lobes are highlighted. Neuronal abnormalities including β lobe overextension/fusion (asterisk) and absent/missing MB lobes (arrows) are presented. Quantification demonstrates the percentage of brains examined per genotype and treatment group exhibiting each illustrated phenotype. Animals were supplied with either standard food harboring an ethanol vehicle control or 40μM GABA. B) MB-dependent associative learning assessed with the T-maze olfactory training paradigm, with quantification of population LI. Control (w1118) and dfmr1 null (dfmr150M) are shown raised on standard food or with constitutive feeding of GABA or NipA. Controls show no significant differences (n.s.) under these conditions. A comparable dfmr1 null mutant learning defect occurs under all 3 conditions (n.s.). Error bars represent s.e.m. Significance is indicated as p<0.05 (*), p<0.01 (**) and p<0.001 (***).
To then assay learning, two equally aversive odor stimuli were sequentially presented, coupling introduction of the CS+ odor with an electric shock (80 V, 2.5 s, 5 s intervals, 10x) compared to a CS− odor, and then testing choice between shocked and non-shocked odors. Controls and dfmr1 null mutants were compared for associative learning performance, with and without drug feeding throughout life. These data are summarized in Figure 11B. We first confirmed the strong learning deficit in dfmr1 null mutants. Using standard training and testing conditions, controls displayed a significantly higher LI compared to dfmr1 nulls (control LI=0.46±0.04 vs. dfmr1 LI=0.25±0.03; N=7 independent trials per genotype, p=0.001; Fig. 11B). To then attempt GABAergic modulation, GABA or NipA were fed throughout development and into adulthood, as above, at the same therapeutic dosage (40μM) used successfully in previous studies (Chang et al., 2008). However, neither treatment provided any benefit, and learning performance in both control and dfmr1 null mutants remained entirely unaltered (Fig. 11B). In quantifying LI values, GABA (control+GABA: LI=0.42±0.02, N=5 vs. dfmr1+GABA: LI=0.24±0.01, N=7; p=0.0005) and NipA (control+NipA: LI=0.44±0.06, N=5 vs. dfmr1+NipA: LI=0.19±0.04, N=7; p=0.0108) treatments caused no change in the compromised learning evidenced in dfmr1 null mutants. These results show that GABAergic intervention alone at this level is insufficient to remediate dfmr1 null defects in MB-dependent learning.
Discussion
The lion's share of FXS neuronal studies have focused on glutamatergic hyperexcitation, but there is clear evidence that GABAergic hypoinhibition may also be important in both Drosophila and mouse disease models (D'Hulst and Kooy, 2007; Paluszkiewicz et al., 2010; Pizzarelli et al., 2011; Coghlan et al., 2012). In the current study, we probed GABAergic circuitry in the Drosophila brain, especially in relation to the MB olfactory learning and memory center, and assessed GABAergic modulation as an FXS intervention strategy. In dfmr1 null mutants, we found depressed GAD expression, altered GABAergic neuron architecture and developmental refinement, and altered GABAergic neuron calcium signaling function within the MB circuit, consistent with GABAergic dysfunction contributing to compromised MB-dependent learning in the Drosophila FXS model (Bolduc et al., 2008; 2010; Coffee et al., 2012). However, pharmacological attempts to alleviate GABAergic signaling defects, modeled on previously reported successful intervention strategies feeding GABA and a GABA reuptake inhibitor, NipA (Chang et al., 2008), did not improve the behavioral output in dfmr1 null mutants. Taken together, this study reveals GABAergic impairments in the FXS disease state, but does not show that GABAergic hypoinhibition is a strong determinant of the learning component of FXS cognitive compromise.
Our results support the reported loss of gad mRNA in the Drosophila FXS model (D'Hulst et al., 2009) and reduced GAD protein in the amygdala basolateral nucleus in the mouse FXS model (Olmos-Serrano et al., 2010). Importantly, FMRP has been suggested to bind gad mRNA directly based on cross-linking immunoprecipitation (HITS-CLIP) (Darnell et al., 2011). However, given FMRP is best defined as a translational repressor (e.g. MAP1B/futsch (Zhang et al., 2001), profilin/chickadee (Reeve et al., 2005; Tessier and Broadie, 2008), etc.), this direct interaction would predict GAD elevation, not the reported loss in the FXS disease state. Alternatively, FMRP may confer transcript stability (Zalfa et al., 2007), such that in its absence gad mRNA would be subject to degradation, causing subsequent loss of the GAD protein. During development, loss of GABA production may have consequences unrelated to its mature inhibitory function, as GABA can serve as an excitatory neurotransmitter in immature stages owing to the high intracellular chloride concentration maintained by Na+/K+/Cl− co-transporter NKCC1 (Yamada et al., 2004; Akerman and Cline, 2007). In Drosophila, however, early GABA roles in synaptogenesis, circuit establishment, and transmission are unclear, as detectable levels of GABA are reported only late in development, since regulatory mechanisms reportedly suppress GAD activity during earlier maturation phases (Kuppers et al., 2003). Developmental roles for GABA will be an area of future investigation in our Drosophila FXS model.
GAD-positive neurons have been reported to innervate the Drosophila MB calyx, with GAD-Gal4-driven synaptobrevin-GFP punctae adjacent to the inner rim of the actin-rich microglomerular rings, establishing GABAergic synaptic input on KCs and/or PNs (Leiss et al., 2009). This GABAergic innervation appears grossly normal in dfmr1 mutants, with a normal array of cell bodies and projection pathways. dBrainbow subdivision (Hampel et al., 2011) of the complex GABAergic circuitry suggests that GABAergic lineages are also normal in dfmr1 nulls. Therefore, there is no evidence that FMRP is required for the generation, placement, or maintenance of GABAergic neurons in the Drosophila brain. However, MARCM clonal analyses employed for single-cell resolution to test cell-autonomous FMRP requirements (Lee and Luo, 2001; Wu and Luo, 2006) show that dfmr1 null MB-innervating GABAergic neurons display early undergrowth in complex cells followed by later overgrowth in simple cells. Thus, the developmental trajectory of GABAergic innervation is dependent upon FMRP. The GABAergic neuron undergrowth opposes the overgrowth seen in excitatory KCs at the same period of MB development (Pan et al., 2004; Tessier and Broadie, 2008; Siller and Broadie, 2011), consistent with theories of GABAergic circuit hypoinhibition in the presence of hyperexcitation in the FXS disease state. In MB-extrinsic GABAergic neurons, FMRP may enhance the expression of targets involved in synapse assembly/maintenance early in development and repress them later, while the converse occurs in MB-intrinsic excitatory KCs. The cell-type-specific translation roles for FMRP in these inhibitory vs. excitatory neurons will be an area of future investigation in our Drosophila FXS model.
In addition to MB-extrinsic GABAergic neurons, we discovered that a subset of MB-intrinsic KCs is also GABAergic, as confirmed by co-labeling for both GAD and GABA. These newly-defined GABAergic neurons provide a potentially self-regulating inhibitory component within the MB circuit proper, which has not previously been recognized. This late-born class of MB-intrinsic GABAergic neurons can be MARCM marked with HS induction applied as late as pupal day 4 (9d AEL), giving them a birthdate easily distinguished from the early-born MB-extrinsic GABAergic neurons. Importantly, late-born MB core neurons play a permissive role in long-term memory formation, able to both facilitate and limit memory consolidation (Huang et al., 2012). These core KCs transiently express glutamate during early MB development (Sinakevitch et al., 2010), but then transition to a different neurotransmitter output. In future work, we plan to determine if the developmental switch in the neurotransmitter employed in these cells involves a potential acquisition of GABAergic function relevant to MB phenotypes in our Drosophila FXS model.
Beyond their architecture, dfmr1 GABAergic neurons display altered calcium signaling dynamics in response to depolarization, showing elevated and prolonged responses. This signaling change may serve as a compensatory mechanism, as GAD activity can be upregulated by increasing intracellular calcium (Kuppers et al., 2003). In Drosophila embryos, negative regulation of GAD can be overridden by agents that elevate free calcium (i.e., thapsigargin and monensin) (Kuppers et al., 2003). It is unclear whether a similar calcium-dependent modulation of GAD might occur in the brain, but this possibility suggests a prospective means to circumvent GAD depression in the FXS disease state. Elevated calcium signaling could also drive direct enhancement of GABA release. In the mouse FXS model striatal region, spontaneous miniature inhibitory synaptic event frequency is elevated, presumably from increased GABA release, even with a lower density of GABAergic synapses (Centonze et al., 2008). In contrast, GABAergic tonic inhibition is decreased in pyramidal cells, although phasic currents remain unchanged (Curia et al., 2009), implicating GABA receptor not neurotransmitter insufficiencies. However, the mouse amygdala exhibits decreased frequency/amplitude of both tonic and phasic inhibitory currents, linked to decreased GABAergic synapse number and reduced GAD65/67 levels (Olmos-Serrano et al., 2010). These findings reveal brain-region-specific changes in the mouse FXS model. Our future Drosophila work will test GABAergic function in MB-extrinsic vs. –intrinsic neurons, as well as upstream GABAergic neurons in the antennal lobe.
How might we correct GABAergic circuit dysfunction in the FXS disease state? Previous work reports that feeding with GABA or the GABA reuptake inhibitor NipA is remarkably effective in correcting a range of dfmr1 null mutant phenotypes, including glutamate toxicity, Futsch/MAP1B over-expression, MB growth, and courtship behavioral memory impairments (Chang et al., 2008). Subsequent studies in the mouse FXS model likewise indicate correction of mutant defects using GABAAR-targeted reagents. For example, the GABAAR agonist gaboxadol/THIP restores disinhibition-related principal neuron excitability deficits in the amygdala and significantly attenuates hyperactivity and reduced prepulse inhibition, although it fails to reverse deficits in cued fear or startle response (Olmos-Serrano et al., 2011). Moreover, GABAAR modulation via benzodiazepine diazepam or neuroactive steroid alphaxalone rescues audiogenic seizures in the mouse FXS model (Heulens et al., 2012). Clinically, an open-label FXS trial employing riluzole, hypothesized to have an inhibitory effect on glutamate release, block excitotoxic effects of glutamate, and potentiate postsynaptic GABAAR function, showed behavioral improvement assessed via the ADHD Rating Scale-IV, with significant correction of the ERK activation FXS biomarker in all subjects (Erickson et al., 2011). Together, these studies suggest that elevating GABAergic function should be an effective strategy for treating the FXS disease state.
Nevertheless, GABAergic augmentation failed to provide any improvement in MB-dependent olfactory learning defects in our Drosophila FXS model, despite partial correction of MB structural defects (Chang et al., 2008). MB over-expression of the RDL GABAAR impairs olfactory learning (Liu et al., 2007), suggesting that elevated GABAergic signaling could be counterproductive; however, given the RDL reduction in dfmr1 mutants (D'Hulst et al., 2006), the intervention was aimed only at restoration. Interestingly, RDL knockdown fails to enhance learning in cAMP signaling pathway mutants, such as rutabaga and NF1 (Liu et al., 2009), suggesting that RDL works upstream of cAMP signaling driving learning. This connection is particularly relevant to FXS, as cAMP is reduced in patient platelets, human neural progenitor cells, and brains in both mouse and Drosophila disease models (Berry-Kravis and Sklena, 1993; Kelley et al., 2007). Moreover, FMR1 overexpression in the HN2 mammalian cell line and dfmr1 overexpression in the null mutant both increase cAMP production (Berry-Kravis and Ciurlionis, 1998; Kelley et al., 2007). Finally, recent electrophysiological studies in Drosophila primary neuronal cultures demonstrate that application of an adenylate cyclase activator suppresses inhibitory GABAergic postsynaptic currents via tempering of GABAAR receptor sensitivity (Ganguly and Lee, 2013). Taken together, these findings suggest a cascade interaction between FMRP, GABAARs, and cAMP signaling during learning. In future work, we plan to coordinately target this pathway at multiple levels to test for restoration of learning in our Drosophila FXS disease model.
Highlights
GAD expression is reduced in dfmr1 nulls, especially within the MB calyx region.
dfmr1 null GABAergic cell numbers and gross organization appear normal.
MARCM reveals GABAergic neuron structural and developmental changes in dfmr1 nulls.
Novel MB-intrinsic GABAergic neurons identified.
GABAergic modulation in dfmr1 nulls fails to rescue olfactory learning defects.
Acknowledgements
We are grateful to Terry Page for expert guidance in olfactory learning experiments. We thank generous donors of key reagents: dfmr12 from Thomas Dockendorff (University of Tennessee, Knoxville, Tennessee), GAD-Gal4 from Gero Misenböck (University of Oxford, Oxford, UK), and anti-GAD from F. Rob Jackson (Tufts University, Boston, MA). We also thank Broadie Lab members, especially Caleb Doll, Neil Dani, William Parkinson, and Emma Rushton for insightful discussions on the study and critical feedback during manuscript preparation, He Zhu for assistance with time-lapse data management, and Eriny Hanna for continued experimental interest. This work was supported by NIH R01 MH084989 to K.B.
Abbreviations
- 2iL
2nd instar larval
- AEL
after egg lay
- ANOVA
analysis of variance
- APL
anterior paired lateral
- BSA
bovine serum albumin
- ChAT
choline acetyltransferase
- CS+
conditioned stimulus
- CS-
control stimulus
- dBrainbow
Drosophila Brainbow
- dfmr1
Drosophila FMR1
- dFMRP
Drosophila fragile X mental retardation protein
- DSHB
Developmental Studies Hybridoma Bank
- FasII
Fasciclin II
- FLP
flippase
- FMR1
fragile X mental retardation 1
- FMRP
fragile X mental retardation protein
- FRT
FLP recombination target
- FXS
Fragile X syndrome
- GABAAR
GABAA receptor
- GAD
glutamic acid decarboxylase
- HITS-CLIP
high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation
- HS
heat-shock
- KC
Kenyon Cell
- LI
learning index
- MARCM
mosaic analysis with a repressible cell marker
- MB
Mushroom Body
- MCH
4-methylcyclohexanol
- mGluR
metabotropic glutamate receptor
- NGS
normal goat serum
- NipA
nipecotic acid
- OCT
3-Octanol
- P1
pupal day 1
- PBS
phosphate buffered saline
- PBST
0.2% triton X-100 in PBS
- PN
projection neuron
- RDL
resistance to dieldrin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest The authors declare the absence of any commercial or financial relationships that could be possibly construed as a potential conflict of interest.
References
- Adusei DC, Pacey LK, Chen D, Hampson DR. Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology. 2010;59:167–171. doi: 10.1016/j.neuropharm.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Akerboom J, Rivera JD, Guilbe MM, Malave EC, Hernandez HH, Tian L, Hires SA, Marvin JS, Looger LL, Schreiter ER. Crystal structures of the GCaMP calcium sensor reveal the mechanism of fluorescence signal change and aid rational design. J Biol Chem. 2009;284:6455–64. doi: 10.1074/jbc.M807657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman CJ, Cline HT. Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 2007;30:382–9. doi: 10.1016/j.tins.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Ciurlionis R. Overexpression of fragile X gene (FMR-1) transcripts increases cAMP production in neural cells. J Neurosci Res. 1998;51:41–8. doi: 10.1002/(SICI)1097-4547(19980101)51:1<41::AID-JNR4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Sklena P. Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. Am J Med Genet. 1993;45:81–7. doi: 10.1002/ajmg.1320450120. [DOI] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11:1143–5. doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Rosenfelt C, Cox H, Tully T. Fragile x mental retardation 1 and filamin a interact genetically in Drosophila long-term memory. Front Neural Circuits. 2010;3:22. doi: 10.3389/neuro.04.022.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner E, Bader R, Buchner S, Cox J, Emson PC, Flory E, Heizmann CW, Hemm S, Hofbauer A, Oertel WH. Cell-specific immuno-probes for the brain of normal and mutant Drosophila melanogaster. I. Wildtype visual system. Cell Tissue Res. 1988;253:357–70. doi: 10.1007/BF00222292. [DOI] [PubMed] [Google Scholar]
- Centonze D, Rossi S, Mercaldo V, Napoli I, Ciotti MT, de Chiara V, Musella A, Prosperetti C, Calabresi P, Bernardi G, Bagni C. Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol Psychiatry. 2008;63:963–73. doi: 10.1016/j.biopsych.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, Warren ST. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4:256–63. doi: 10.1038/nchembio.78. [DOI] [PubMed] [Google Scholar]
- Chiang AS, Lin CY, Chuang CC, Chang HM, Hsieh CH, Yeh CW, Shih CT, Wu JJ, Wang GT, Chen YC, Wu CC, Chen GY, Ching YT, Lee PC, Lin CY, Lin HH, Wu CC, Hsu HW, Huang YA, Chen JY, Chiang HJ, Lu CF, Ni RF, Yeh CY, Hwang JK. Three-dimensional reconstruction of brain-wide wiring networks in Drosophila at single-cell resolution. Curr Biol. 2011;21:1–11. doi: 10.1016/j.cub.2010.11.056. [DOI] [PubMed] [Google Scholar]
- Coffee RL, Jr, Williamson AJ, Adkins CM, Gray MC, Page TL, Broadie K. In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum Mol Genet. 2012;21:1–16. doi: 10.1093/hmg/ddr527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee RL, Tessier CR, Woodruff EA, Broadie K. Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis Model Mech. 2010;3:471–485. doi: 10.1242/dmm.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci Biobehav Rev. 2012;36:2044–2055. doi: 10.1016/j.neubiorev.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden JR, Skoulakis EM, Han KA, Kalderon D, Davis RL. Tripartite mushroom body architecture revealed by antigenic markers. Learn Mem. 1998;5:38–51. [PMC free article] [PubMed] [Google Scholar]
- Curia G, Papouin T, Seguela P, Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex. 2009;19:1515–20. doi: 10.1093/cercor/bhn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hulst C, De Geest N, Reeve SP, van den Van Dam D, La De Deyn de PP, Hassan BA, Kooy RF. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121:1–8. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- D'Hulst C, Heulens I, Brouwer JR, Willemsen R, de Geest N, Reeve SP, de Deyn PP, Hassan BA, Kooy RF. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS) Brain Res. 2009;1253:176–83. doi: 10.1016/j.brainres.2008.11.075. [DOI] [PubMed] [Google Scholar]
- D'Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30:425–31. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- D'Hulst C, Kooy RF. Fragile X syndrome: from molecular genetics to therapy. J Med Genet. 2009;46:577–84. doi: 10.1136/jmg.2008.064667. [DOI] [PubMed] [Google Scholar]
- Darnell JC, van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–61. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL. Olfactory memory formation in Drosophila: from molecular to systems neuroscience. Annu Rev Neurosci. 2005;28:275–302. doi: 10.1146/annurev.neuro.28.061604.135651. [DOI] [PubMed] [Google Scholar]
- Davis RL. Traces of Drosophila memory. Neuron. 2011;70(1):8–19. doi: 10.1016/j.neuron.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–84. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- Duffy JB. GAL4 system in Drosophila: a fly geneticist's Swiss army knife. Genesis. 2002;34:1–15. doi: 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- Enell L, Hamasaka Y, Kolodziejczyk A, Nassel DR. gamma-Aminobutyric acid (GABA) signaling components in Drosophila: immunocytochemical localization of GABA(B) receptors in relation to the GABA(A) receptor subunit RDL and a vesicular GABA transporter. J Comp Neurol. 2007;505:18–31. doi: 10.1002/cne.21472. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Weng N, Weiler IJ, Greenough WT, Stigler KA, Wink LK, McDougle CJ. Open-label riluzole in fragile X syndrome. Brain Res. 2011;1380:264–70. doi: 10.1016/j.brainres.2010.10.108. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Rushton EM, Hilderbrand-Chae M, Phillips AM, Jackson FR, Broadie K. Presynaptic glutamic acid decarboxylase is required for induction of the postsynaptic receptor field at a glutamatergic synapse. Neuron. 2000;27:71–84. doi: 10.1016/s0896-6273(00)00010-6. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Lee D. Suppression of inhibitory GABAergic transmission by cAMP signaling pathway: alterations in learning and memory mutants. Eur J Neurosci. 2013;37:1383–93. doi: 10.1111/ejn.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantois I, Vandesompele J, Speleman F, Reyniers E, D'Hooge R, Severijnen LA, Willemsen R, Tassone F, Kooy RF. Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis. 2006;21:346–57. doi: 10.1016/j.nbd.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development. 2008;135:2637–48. doi: 10.1242/dev.022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. Temporal requirements of the fragile x mental retardation protein in modulating circadian clock circuit synaptic architecture. Front Neural Circuits. 2009;3:8. doi: 10.3389/neuro.04.008.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front Syn Neurosci. 2010;2 doi: 10.3389/fnsyn.2010.00004. doi: 10.3389/fnsyn.2010.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. Fragile X mental retardation protein is required for programmed cell death and clearance of developmentally-transient peptidergic neurons. Dev Biol. 2011;356:291–307. doi: 10.1016/j.ydbio.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaka Y, Wegener C, Nässel DR. GABA modulates Drosophila circadian clock neurons via GABAB receptors and decreases in calcium. J Neurobiol. 2005;65(3):225–40. doi: 10.1002/neu.20184. [DOI] [PubMed] [Google Scholar]
- Hampel S, Chung P, McKellar CE, Hall D, Looger LL, Simpson JH. Drosophila Brainbow: a recombinase-based fluorescence labeling technique to subdivide neural expression patterns. Nat Methods. 2011;8:253–9. doi: 10.1038/nmeth.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heulens I, D'Hulst C, van Dam D, de Deyn PP, Kooy RF. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res. 2012;229:244–9. doi: 10.1016/j.bbr.2012.01.031. [DOI] [PubMed] [Google Scholar]
- Huang C, Zheng X, Zhao H, Li M, Wang P, Xie Z, Wang L, Zhong Y. A permissive role of mushroom body α/β core neurons in long-term memory consolidation in Drosophila. Curr Biol. 2012;22:1981–9. doi: 10.1016/j.cub.2012.08.048. [DOI] [PubMed] [Google Scholar]
- Idrissi el A, Ding XH, Scalia J, Trenkner E, Brown WT, Dobkin C. Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett. 2005;377:141–6. doi: 10.1016/j.neulet.2004.11.087. [DOI] [PubMed] [Google Scholar]
- Ito K, Hotta Y. Proliferation pattern of postembryonic neuroblasts in the brain of Drosophila melanogaster. Dev Biol. 1992;149:134–48. doi: 10.1016/0012-1606(92)90270-q. [DOI] [PubMed] [Google Scholar]
- Ito M, Masuda N, Shinomiya K, Endo K, Ito K. Systematic Analysis of Neural Projections Reveals Clonal Composition of the Drosophila Brain. Curr Biol. 2013;23:644–655. doi: 10.1016/j.cub.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Jefferis GS, Marin EC, Watts RJ, Luo L. Development of neuronal connectivity in Drosophila antennal lobes and mushroom bodies. Curr Opin Neurobiol. 2002;12(1):80–6. doi: 10.1016/s0959-4388(02)00293-3. [DOI] [PubMed] [Google Scholar]
- Kahsai L, Zars T. Learning and memory in Drosophila: behavior, genetics, and neural systems. Int Rev Neurobiol. 2011;99:139–67. doi: 10.1016/B978-0-12-387003-2.00006-9. [DOI] [PubMed] [Google Scholar]
- Keene AC, Waddell S. Drosophila olfactory memory: single genes to complex neural circuits. Nat Rev Neurosci. 2007;8(5):341–54. doi: 10.1038/nrn2098. [DOI] [PubMed] [Google Scholar]
- Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JC, Bhattacharyya A. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS ONE. 2007;2:e931. doi: 10.1371/journal.pone.0000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk A, Sun X, Meinertzhagen IA, Nassel DR. Glutamate, GABA and acetylcholine signaling components in the lamina of the Drosophila visual system. PLoS ONE. 2008;3:e2110. doi: 10.1371/journal.pone.0002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppers B, Sanchez-Soriano N, Letzkus J, Technau GM, Prokop A. In developing Drosophila neurones the production of gamma-amino butyric acid is tightly regulated downstream of glutamate decarboxylase translation and can be influenced by calcium. J Neurochem. 2003;84:939–51. doi: 10.1046/j.1471-4159.2003.01554.x. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–4. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Leiss F, Groh C, Butcher NJ, Meinertzhagen IA, Tavosanis G. Synaptic organization in the adult Drosophila mushroom body calyx. J Comp Neurol. 2009;517:808–24. doi: 10.1002/cne.22184. [DOI] [PubMed] [Google Scholar]
- Liu X, Buchanan ME, Han KA, Davis RL. The GABAA receptor RDL suppresses the conditioned stimulus pathway for olfactory learning. J Neurosci. 2009;29:1573–9. doi: 10.1523/JNEUROSCI.4763-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Davis RL. The GABAergic anterior paired lateral neuron suppresses and is suppressed by olfactory learning. Nat Neurosci. 2009;12:53–9. doi: 10.1038/nn.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Krause WC, Davis RL. GABAA receptor RDL inhibits Drosophila olfactory associative learning. Neuron. 2007;56:1090–102. doi: 10.1016/j.neuron.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA, Armstrong JD. Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics. 2011;27:2453–4. doi: 10.1093/bioinformatics/btr390. [DOI] [PubMed] [Google Scholar]
- Lyons LC, Roman G. Circadian modulation of short-term memory in Drosophila. Learning & memory (Cold Spring Harbor, N.Y.) 2009;16:1–10. doi: 10.1101/lm.1146009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies C, Tully T, Dubnau J. Deconstructing memory in Drosophila. Curr Biol. 2005;15(17):R700–13. doi: 10.1016/j.cub.2005.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon V, Leroux J, White CD, Reiss AL. Frontostriatal deficits in fragile X syndrome: relation to FMR1 gene expression. Proc Natl Acad Sci U S A. 2004;101:3615–20. doi: 10.1073/pnas.0304544101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel CI, Kraft R, Restifo LL. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J Neurosci. 2004;24(25):5798–809. doi: 10.1523/JNEUROSCI.1102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K, Ohkura KM. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:1–5. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93(7):1183–93. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Ng M, Roorda RD, Lima SQ, Zemelman BV, Morcillo P, Miesenbock G. Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron. 2002;36:463–74. doi: 10.1016/s0896-6273(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Nicolaï LJ, Ramaekers A, Raemaekers T, Drozdzecki A, Mauss AS, Yan J, Landgraf M, Annaert W, Hassan BA, Nicolai LJ, Ramaekers A, Raemaekers T, Drozdzecki A, Mauss AS, Yan J, Landgraf M, Annaert W, Hassan BA. Genetically encoded dendritic marker sheds light on neuronal connectivity in Drosophila. Proc Natl Acad Sci USA. 2010;107:1–6. doi: 10.1073/pnas.1010198107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada R, Awasaki T, Ito K. Gamma-aminobutyric acid (GABA)-mediated neural connections in the Drosophila antennal lobe. J Comp Neurol. 2009;514(1):74–91. doi: 10.1002/cne.21971. [DOI] [PubMed] [Google Scholar]
- Olmos-Serrano JL, Corbin JG, Burns MP. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev Neurosci. 2011;33:395–403. doi: 10.1159/000332884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30:9929–38. doi: 10.1523/JNEUROSCI.1714-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluszkiewicz SM, Martin BS, Huntsman MM. Fragile X syndrome: the GABAergic system and circuit dysfunction. Dev Neurosci. 2010;33:349–64. doi: 10.1159/000329420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14:1863–70. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- Pitman JL, Huetteroth W, Burke CJ, Krashes MJ, Lai SL, Lee T, Waddell S. A pair of inhibitory neurons are required to sustain labile memory in the Drosophila mushroom body. Curr Biol. 2011;21(10):855–61. doi: 10.1016/j.cub.2011.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzarelli R, Pizzarelli R, Cherubini E, Cherubini E. Alterations of GABAergic Signaling in Autism Spectrum Disorders. Neural Plast. 2011;2011:1–12. doi: 10.1155/2011/297153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn W, Harris W, Benzer S. Conditioned behavior in Drosophila melanogaster. Proc Natl Acad Sci USA. 1974;71:708–12. doi: 10.1073/pnas.71.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SP, Bassetto L, Genova GK, Kleyner Y, Leyssen M, Jackson FR, Hassan BA. The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol. 2005;15:1156–63. doi: 10.1016/j.cub.2005.05.050. [DOI] [PubMed] [Google Scholar]
- Repicky SE, Broadie K. Metabotropic Glutamate Receptor Mediated Use-Dependent Down-regulation of Synaptic Excitability Involves the Fragile X Mental Retardation Protein. J Neurophysiol. 2008;101:672–87. doi: 10.1152/jn.90953.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siller SS, Broadie K. Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis Model Mech. 2011;4:673–85. doi: 10.1242/dmm.008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinakevitch I, Grau Y, Strausfeld NJ, Birman S. Dynamics of glutamatergic signaling in the mushroom body of young adult Drosophila. Neural Dev. 2010;5:10–0. doi: 10.1186/1749-8104-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, O'Dowd DK. Fast synaptic currents in Drosophila mushroom body Kenyon cells are mediated by alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors and picrotoxin-sensitive GABA receptors. J Neurosci. 2003;23:9246–53. doi: 10.1523/JNEUROSCI.23-27-09246.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka NK, Tanimoto H, Ito K. Neuronal assemblies of the Drosophila mushroom body. J Comp Neurol. 2008;508(5):711–55. doi: 10.1002/cne.21692. [DOI] [PubMed] [Google Scholar]
- Tessier CR, Broadie K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development. 2008;135:1547–57. doi: 10.1242/dev.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier CR, Broadie K. The fragile X mental retardation protein developmentally regulates the strength and fidelity of calcium signaling in Drosophila mushroom body neurons. Neurobiol Dis. 2011;41:147–159. doi: 10.1016/j.nbd.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol. 1985;157:263–77. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- Urbach R, Technau G. Molecular markers for identified neuroblasts in the developing brain of Drosophila. Development. 2003;130:3621–37. doi: 10.1242/dev.00533. [DOI] [PubMed] [Google Scholar]
- Waddell S. Dopamine reveals neural circuit mechanisms of fly memory. Trends Neurosci. 2010;33(10):457–64. doi: 10.1016/j.tins.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo H, Pologruto T, Hannan F, Hakker I, Svoboda K, Zhong Y. Stereotyped odor-evoked activity in the mushroom body of Drosophila revealed by green fluorescent protein-based Ca2+ imaging. J Neurosci. 2004;24:6507–14. doi: 10.1523/JNEUROSCI.3727-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mamiya A, Chiang AS, Zhong Y. Imaging of an early memory trace in the Drosophila mushroom body. J Neurosci. 2008;28:4368–76. doi: 10.1523/JNEUROSCI.2958-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JS, Luo L. A protocol for mosaic analysis with a repressible cell marker (MARCM) in Drosophila. Nat Protoc. 2006;1:2583–9. doi: 10.1038/nprot.2006.320. [DOI] [PubMed] [Google Scholar]
- Wu Y, Ren Q, Li H, Guo A. The GABAergic anterior paired lateral neurons facilitate olfactory reversal learning in Drosophila. Learning & memory (Cold Spring Harbor, N.Y.) 2012;19:478–86. doi: 10.1101/lm.025726.112. [DOI] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. The J Physiol. 2004;557:829–41. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Akalal DB, Davis RL. Drosophila alpha/beta mushroom body neurons form a branch-specific, long-term cellular memory trace after spaced olfactory conditioning. Neuron. 2006;52:845–55. doi: 10.1016/j.neuron.2006.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Awasaki T, Schroeder M, Long F, Yang J, He Y, Ding P, Kao J, Wu G, Peng H, Myers G, Lee T. Clonal development and organization of the adult Drosophila central brain. Curr Biol. 2013;23:633–43. doi: 10.1016/j.cub.2013.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, de Rubeis S, Di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, Bagni C. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10:578–87. doi: 10.1038/nn1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]