

Abstract
Apolipoprotein D (ApoD) is an ancient member of the lipocalin family with a high degree of sequence conservation from insects to mammals. It is not structurally related to other major apolipoproteins and has been known as a small, soluble carrier protein of lipophilic molecules that is mostly expressed in neurons and glial cells within the central and peripheral nervous system. Recent data indicate that ApoD not only supplies cells with lipophilic molecules, but also controls the fate of these ligands by modulating their stability and oxidation status. Of particular interest is the binding of ApoD to arachidonic acid and its derivatives, which play a central role in healthy brain function. ApoD has been shown to act as a catalyst in the reduction of peroxidized eicosanoids and to attenuate lipid peroxidation in the brain. Manipulating its expression level in fruit flies and mice has demonstrated that ApoD has a favorable effect on both stress resistance and life span. The APOD gene is the gene that is upregulated the most in the aging human brain. Furthermore, ApoD levels in the nervous system are elevated in a large number of neurologic disorders including Alzheimer's disease, schizophrenia, and stroke. There is increasing evidence for a prominent neuroprotective role of ApoD because of its antioxidant and anti-inflammatory activity. ApoD emerges as an evolutionarily conserved anti-stress protein that is induced by oxidative stress and inflammation and may prove to be an effective therapeutic agent against a variety of neuropathologies, and even against aging.
Keywords: Aging, Alzheimer's disease, Apolipoprotein, Inflammation, Lipid peroxidation, Lipocalin, Neurodegeneration, Oxidative stress, Reactive oxygen species, Schizophrenia, Stress response, Stroke
1. Introduction
A major focus of today's research in neuroscience is on the molecular mechanisms leading to age-related neurologic decline. Given the continuous increase in life expectancy in human populations worldwide, finding strategies to maintain health in old age is becoming a matter of increasing socio-economic relevance (Oeppen and Vaupel, 2002; Wilmoth, 2000). Age-related disorders are predicted to have dramatic repercussions for health and social systems over the next few decades. Aging is the most significant risk factor for cognitive decline and neurodegenerative disorders, a set of pathologies that has collectively been referred to as the silent epidemic (Larson, 2001). After the age of 65 years, the incidence and prevalence of Alzheimer's disease (AD) double every 5 years (Kukull et al., 2002). After the age of 45 years, the incidence of stroke doubles with each decade (Kelly-Hayes, 2010). Two of the main drivers of age-related and disease-related neurologic decline seem to be oxidative stress and chronic inflammation (Bishop et al., 2010; Salminen et al., 2012). Both forms of cellular stress increase with age because of the organism's declining capacity for maintenance and repair. Reactive oxygen species (ROS) are inevitably generated by the aerobic metabolism, but are controlled in early life by a variety of antioxidant mechanisms. With increasing age, however, ROS production gradually overwhelms these lines of defense, resulting not only in structural damage to molecules but also in the destabilization of the redox-sensitive signaling pathways and inducement of inflammation. Levels of oxidative stress and inflammation are also elevated in the brain in a number of neurologic and psychiatric disorders, and aggravate pathogenesis by progressively disrupting tissue homeostasis (Amor et al., 2010; Federico et al., 2012; Glass et al., 2010; Reynolds et al., 2007). There is growing evidence that apolipoprotein D (ApoD) is dispatched as part of the organism's natural defense against oxidative and inflammatory stress in the aging and degenerative brain. Although small in size it appears to have a major impact on brain health. In this paper we review the evidence for the neuroprotective role of ApoD during aging, and also in acute and chronic neurologic and psychiatric disorders, and we explore its potential as a therapeutic agent against a number of neuropathologies, and even against aging.
2. ApoD
2.1. ApoD: an atypical apolipoprotein
ApoD is a secreted 29-kDa glycoprotein and a member of the lipocalin family (Rassart et al., 2000). Lipocalins are phylogenetically old proteins found in prokaryotes and eukaryotes, and their functional hallmark is the binding and transport of small hydrophobic molecules (Grzyb et al., 2006). At a structural level, lipocalins are characterized by a ligand-binding pocket typically formed by an 8-strand antiparallel beta-barrel and the presence of up to 3 highly conserved short sequence motifs, known as short conserved regions (SCRs), within this fold. Lipocalins are subdivided on the basis of the number of these SCRs present into core (or kernel) lipocalins harboring 3 SCRs, and outlier lipocalins with only 1 or 2 SCRs. ApoD is a kernel lipocalin and has historically been classified as an apolipoprotein, the heterogenous group of lipid-binding proteins present in lipoprotein particles. Although originally isolated from human plasma high-density lipoproteins (HDLs) (McConathy and Alaupovic, 1973) ApoD is expressed in a variety of organs, with high levels of expression being found in the testes, peripheral nerves, the brain, and the spinal cord. ApoD is also readily detected in a variety of body fluids. It is most abundant in plasma, with the concentration in human plasma being approximately 100 times higher than in human cerebrospinal fluid (CSF) (Rassart et al., 2000; Terrisse et al., 1998). It is interesting that the expression of ApoD in rodents seems to be largely restricted to the central nervous system (CNS) tissue (Rassart et al., 2000). It is also of note that there are distinct differences between the composition of lipoprotein particles present in the CSF and that of lipoprotein particles in the serum, and that ApoD is found in all CSF lipoprotein classes (Borghini et al., 1995; Koch et al., 2001). Overall, the expression pattern of ApoD is in striking contrast to that of the other apolipoproteins, which are mainly produced in the liver and the intestinal epithelium. Moreover, the ApoD amino acid sequence does not show any similarity to that of any other apolipoproteins (except for ApoM, which is a member of the outlier lipocalin subfamily), but within mammals, it has the highest degree of homology to the retinol binding protein, RBP4. In view of these peculiarities, ApoD has been termed an atypical apolipoprotein (Weech et al., 1991).
Determination of the 3-dimensional structure of ApoD has confirmed the lipocalin-like beta-barrel fold that forms a cup, or calyx, with a central cavity serving as a ligand-binding pocket (Eichinger et al., 2007). Unlike other lipocalins, however, ApoD has a number of extended loops at the entrance to the ligand pocket that contain hydrophobic residues. It is through these exposed hydrophobic side chains that ApoD is believed to be inserted into the lipid envelope of the HDL particle or cell membranes. In fact, ApoD is not a classical “integral” apolipoprotein embedded in lipoprotein particles, but rather associates peripherally with lipoprotein particles. The physiological orientation of ApoD might thus appear to resemble a cup with its opening facing toward the lipid surface and its base forming a spike that could interact with a transmembrane receptor (Eichinger et al., 2007). Exactly which receptor might be involved has not yet been established; many apolipoproteins have been shown to bind to 1 or more members of the low-density lipoprotein receptor family (Elliott et al., 2010), but the receptor(s) for ApoD, if any, remain(s) elusive.
The widespread pattern of ApoD expression is reflected in a multitude of possible ligands. While the binding of ApoD to retinol and retinoic acid, sphingomyelin, progesterone, arachidonic acid (AA), and anandamide has been confirmed by independent studies and found to lie in the low micromolar or sub-micromolar range, there are discrepancies with regard to the interactions between ApoD and palmitic acid, cholesterol, ß-estradiol, pregnenolone, bilirubin, and the axillary odorant E-3M2H (Morais Cabral et al., 1995; Rassart et al., 2000; Ruiz et al., 2013; Vogt and Skerra, 2001). These controversial data could possibly be explained by technical issues, such as whether ApoD was prepared as a bacterial recombinant protein or purified from native sources. Alternatively, some interactions may be weak because of low affinity binding of ligands to the exposed hydrophobic residues at the surface of the binding pocket, or they may simply be indirect interactions, such as has been suggested for cholesterol which can be associated with sphingomyelin and hence cotransported with ApoD (Ruiz et al., 2013). Molecular dynamics analyses suggest that binding of AA and its derivatives to ApoD occurs in a rather flexible manner at the opening of the ligand binding pocket of ApoD and relies on some exposed hydrophobic residues (Oakley et al., 2012). The principal binding site of AA in ApoD thus seems to differ from the binding site of progesterone, which was shown to lie deeply buried in the binding pocket, as revealed by crystallography (Eichinger et al., 2007).
There is increasing evidence that the function of ApoD extends well beyond the simple transport of lipophilic molecules, and that it has direct relevance to healthy brain function. ApoD has recently been demonstrated to catalyze the reduction of hydroperoxides of AA (i.e., eicosatetraenoic acid) to lipid hydroxides via a highly conserved methionine residue (Bhatia et al., 2012). Specifically, human ApoD has been shown to reduce peroxidized, potentially radical-generating hydroperoxy eicosatetraenoic acids to the non-reactive hydroxide forms, thereby preventing lipid peroxidation chain reactions. This finding could readily explain the observed increase in lipid peroxidation in the ApoD knockout mouse brain, which occurs together with a decreased resistance to oxidative stress (Ganfornina et al., 2008). Overexpression of human ApoD in the mouse has, in contrast, been shown to result in reduced levels of lipid peroxidation and increased resistance to oxidative stress (Ganfornina et al., 2008). It is of note that ApoD was recently identified as a major cardioprotective protein in a mouse model of myocardial infarction. Overexpression of ApoD reduced the rate of myocardial infarction and protected cardiomyocytes from hypoxia and/or reperfusion-related stress, a protective effect that was shown to depend on ApoD's antioxidant activity (Tsukamoto et al., 2013).
ApoD may not only act as an antioxidant, with a focus on the eicosanoid metabolism, but it may also influence inflammatory pathways. The ratio of peroxidized to reduced eicosanoids is known to regulate the synthesis of downstream inflammatory leukotrienes (Phillis et al., 2006). Moreover, in a mouse model of viral infection, overexpression of human ApoD has been shown to reduce T-cell infiltration into the CNS, to decrease production of pro-inflammatory cytokines including IL-1ß and TNFα, and to downregulate the activity of phospholipase A2 (PLA2) (Do Carmo et al., 2008). Because PLA2 is the enzyme that releases AA from membrane phospholipids, ApoD may thereby restrain the availability of free AA, which is the major precursor of leukotrienes and prostaglandins, 2 groups of potent inflammatory modulators (Calder, 2005). ApoD also appears to stabilize membrane-associated AA and attenuate its release from phospholipids. Once released, ApoD might trap free AA by sequestration and prevent its subsequent conversion into pro-inflammatory eicosanoids, or its peroxidation to highly toxic compounds (Thomas et al., 2003b).
ApoD, thus, seems not only to bind and transport lipophilic molecules but also to actively interfere with their metabolism and signaling in an antioxidant and anti-inflammatory manner.
2.2. Phylogeny of ApoD
Lipocalins are ancient proteins and ApoD belongs to the most ancient group of lipocalins. Genome mining and sequence comparisons support the view that lipocalins arose in gram-negative bacteria and were passed on to primordial eukaryotes by horizontal gene transfer (Bishop, 2000). From there they spread into present-day plant, metazoan, and “protist” phyla. Lipocalins have undergone a remarkable radiation in chordates and in particular in mammals, with 21 lipocalin genes having been described in humans (http://www.uniprot.org/). In contrast, only a small number of lipocalins have been identified in arthropods (e.g., Drosophila melanogaster), “protists” (e.g., Dictyostelium discoideum), and plants (e.g., Arabidopsis thaliana), and they have apparently been lost completely from taxa such as nematodes (Caenorhabditis elegans) and fungi (Saccharomyzes cerevisiae) (http://www.uniprot.org/). Phylogenetic analyses based on gene structure and protein sequence have invariably revealed that ApoD is at the base of the chordate lipocalin tree. ApoD is thus the earliest lipocalin within the chordate lineage. The other lipocalins were derived from early ApoD in a monophyletic manner during the course of chordate diversification through multiple gene and genome duplication events, with RBP4 being the closest paralog of ApoD (Sanchez et al., 2003; Wang et al., 2007). Matching its status as the primordial chordate lipocalin, ApoD seems to be a genuine ortholog of the arthropod lipocalins Lazarillo from the grasshopper Schistocerca americana (Ganfornina et al., 1995), and the 2 Lazarillo proteins in D. melanogaster, Glial-Lazarillo (GLaz), and Neural-Lazarillo (NLaz) (Sanchez et al., 2000) (Fig. 1). The protein sequence of human ApoD is 25.2% identical to GLaz, 31.3% identical to NLaz, and 26.0% identical to Lazarillo. When chemically similar amino acids are included in the alignment, the sequence identity between human ApoD and its insect orthologs is well above 50%. In contrast, the sequence identity between human ApoD and its closest paralog, human RBP4, is only 23.0% (http://www.uniprot.org/).
Fig. 1.
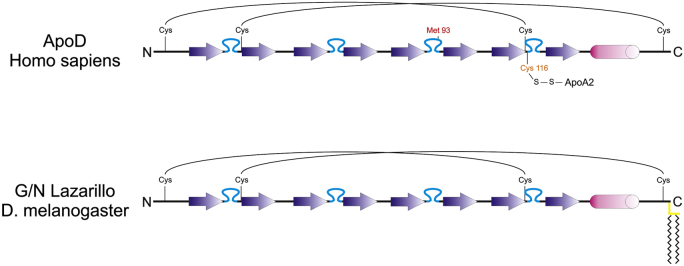
ApoD is a lipocalin that has been evolutionarily conserved from insects to mammals. The secondary structure and sequence elements of human ApoD are compared with those of the Drosophila Lazarillo proteins, GLaz, and NLaz. The backbone of all 3 proteins is comprised of 8 beta sheets (arrows) that give rise to a barrel-like shape, which constitutes the ligand-binding pocket and is characteristic for lipocalins. This fold is stabilized by 4 conserved cysteine residues forming 2 disulfide bridges. All 3 orthologs have a signal peptide at their N-terminus (not shown) and a short alpha helix at the C-terminus (cylinder). While the highest sequence conservation is found in the beta sheets, the sequence stretches in between often contain hydrophobic residues which in human ApoD were shown to be organized as protruding loops framing the entrance to the binding pocket (Eichinger et al., 2007). These hydrophobic loops (blue) are assumed to be involved in defining the ligand specificity and also to facilitate the interaction of ApoD and Lazarillo proteins with hydrophobic surfaces. Lazarillo proteins in fact associate with membranes through a GPI-anchor (yellow bars), whereas human ApoD, but not the ApoD orthologs of other species, was found to be covalently linked to ApoA2 through a disulfide bridge. Met93 in human ApoD has been shown to have reducing activity and to prevent peroxidation of lipids. This methionine residue seems to be restricted to ApoD orthologs in higher vertebrates (mammals and birds). Abbreviation: ApoD, apolipoprotein D.
This high degree of sequence conservation between human ApoD and its insect orthologs is reflected in an evolutionarily conserved functionality. As is the case with ApoD, the insect Lazarillo proteins are predominantly expressed in the nervous system. As with ApoD in the mouse (Ganfornina et al., 2008), GLaz has been implicated in resistance against oxidative stress in Drosophila (Sanchez et al., 2006; Walker et al., 2006). Intriguingly, overexpression of human ApoD in Drosophila both increases stress resistance and extends life span (Muffat et al., 2008), reproducing the phenotype obtained by overexpression of GLaz in Drosophila (Walker et al., 2006). ApoD and GLaz are thus functionally interchangeable across phyla. Although the overall structure and function of ApoD has been well preserved over large evolutionary distances, the protein sequence of ApoD continues to be varied. For example, unlike all other ApoD orthologs, human ApoD has a cysteine residue in the hydrophobic region at the entrance to the ligand-binding pocket, which forms an intermolecular disulfide bond with the HDL-associated apolipoprotein A2 (Yang et al., 1994) (Fig. 1). Although the functional consequences of this covalent interaction are not yet fully understood, this observation suggests that the association of ApoD with HDL is more stable in humans than any other species.
3. ApoD in the aging brain
3.1. Oxidative stress and inflammaging are major drivers of aging in the brain
3.1.1. Evolutionary biology of aging
Biological aging has been defined as the persistent decline in Darwinian fitness over an organism's typical lifetime because of internal physiological deterioration (Rose, 1991). Because individual organismal fitness is determined by survival and fertility, this definition implies that both the chances of survival and the degree of fecundity of a species shrink with increasing age until they reach zero at the end of the species' maximum life span. At the first glance this might seem counter-intuitive to the concept of natural selection since aging, with death occurring at the end of this process, is clearly bad for the individual but does not seem to be a necessary, inevitable phenomenon of simple wear-and-tear, as illustrated by the immortality of the germ line. Why then do somatic cells age, but not germ cells? An explanation is provided by the evolutionary theory of aging, and in particular the disposable soma theory (Kirkwood, 2005; Kirkwood and Austad, 2000; Kirkwood and Rose, 1991). The essence of this theory is that during an individual's lifetime there is a trade-off between short-term benefits at a young age and long-term survivorship. The theory is based on the realization that organisms have a finite metabolic energy budget that must be allocated among somatic maintenance, that is, survival and reproduction. Investment in somatic maintenance and repair requires metabolic resources that are then not longer available for reproduction, and vice versa. An optimal resource allocation therefore evolved in the form of hard-wired genetic programs that maximize the fitness of an individual. An important aspect of this solution is the clear-cut separation of an organism's cell mass into germ cells and somatic cells. Because the germ cells are far fewer in number than the somatic cells, an increased investment in maintenance and repair of germ cells is affordable to the organism resulting in their indefinite physiological homeostasis. The soma, on the other hand, invests more metabolic energy in competitive reproduction, at the expense of maintenance and repair, therefore accumulating damage and finally succumbing to wear-and-tear. Two major questions that arise from this theoretical framework are (1) what is the nature of the damage inflicted on an organism's soma over its lifetime; and (2) what are the defense mechanisms that have evolved to contain this progressive damage, but which apparently are inadequate in the soma because of metabolic energy limitations? Answering these questions could pave the way for some genetic engineering aimed at abolishing aging or, at least, delaying the appearance of age-related and damage-induced pathologies.
At the molecular level, a number of mechanisms have been identified that are believed to underlie the damage-induced aging process including somatic mutations, telomere shortening, protein denaturation and aggregation, and oxidative stress (Zglinicki, 2003). Each of these stressors can, in theory, be counterbalanced by the cell: an array of DNA repair enzymes removes molecular lesions from nucleotides, the telomerase restores the ends of chromosomes, proteins are stabilized by chaperones and if irreversibly denatured, removed and recycled by the proteasome or autophagosome, and ROS are neutralized by numerous antioxidants. The relative contribution of each of these stressors to the aging process remains unclear, as does the effectiveness and metabolic cost of the cellular stress response and repair pathways (Haigis and Yankner, 2010). The extent and type of damage that the soma suffers over a lifetime is presumably not homogenous for all parts of the body, but depends significantly on the type of tissue and its cellular and molecular specifics.
3.1.2. Oxidative stress and inflammaging in the aging brain
Because most of the cells in a brain are postmitotic, telomeric shortening is not considered likely to be a major cause of aging, but oxidative stress is likely to play a major role. Firstly, the (human) brain is the organ with the highest energy demand and the highest adenosine triphosphate (ATP) production in the body (Kety, 1957; Sokoloff, 1960). ATP production in mitochondria by oxidative phosphorylation is, in turn, the main source of ROS. Secondly, the brain is the most lipid-rich organ in the body (McIlwain and Bachelard, 1985) and because lipids are a prime target of ROS, the brain is prone to lipid peroxidation, resulting in membrane disruption and eventually in the generation of toxic and mutagenic compounds such as malondialdehyde and 4-hydroxynonenal. Moreover, in contrast to oxidative protein and DNA damage, lipid peroxidation proceeds through a self-propagating chain reaction mechanism that spreads the damage and requires antioxidants for its termination (Esterbauer, 1993; Gueraud et al., 2010; Marnett, 1999; Yin et al., 2011).
The idea of ROS being essential drivers of aging was first formulated by Denham Harman and became known as the “Free Radical Theory of Aging” (Harman, 1956). The theory considers ROS to be inevitable by-products of aerobic metabolism, aggressively reacting with and thereby damaging cellular macromolecules. Because of inadequate antioxidant defense mechanisms, macromolecular damage accumulates and eventually leads to age-dependent physiological decline. Although this hypothesis of ROS-induced aging based on structural damage has received a great deal of experimental support over the last few decades, more recent data, in particular from transgenic mouse models, have cast some doubt over its relevance (Lapointe and Hekimi, 2010; Muller et al., 2007; Speakman and Selman, 2011).
A new hypothesis on ROS-induced aging, termed the “redox stress hypothesis”, has recently been put forward by Sohal and Orr (2012). This new hypothesis shifts the focus from ROS-induced structural damage to ROS-triggered disruption of the redox-regulated signaling pathways. In essence, it is based on the realization that the catalytic or regulatory activity of a variety of proteins involved in basic signaling pathways, such as, for instance, Ras or MEKK1, is modulated by the oxidation and/or reduction reactions of specific cysteinyl thiol groups (Brandes et al., 2009; Klomsiri et al., 2011; Winterbourn and Hampton, 2008). With increasing age, a progressive ROS-induced pro-oxidative shift is assumed to occur in the cellular redox state, leading to the over-oxidation of redox-sensitive proteins and to the subsequent disruption of redox-regulated signaling pathways.
Another deleterious effect of ROS is the induction of cellular senescence and inflammation (Lu and Finkel, 2008; Naik and Dixit, 2011; Ramsey and Sharpless, 2006). Cellular senescence is a cell fate similar to apoptosis, differentiation, or proliferation that is characterized by essentially irreversible growth arrest and occurs when cells experience potentially oncogenic insults including DNA damage, telomeric shortening, protein aggregation, and increased levels of ROS (Campisi, 2007; Tchkonia et al., 2013). In contrast to apoptosis, however, cells survive initially but remain locked in a non-proliferating state, thereby reducing the risk of tumorigenesis. Senescence has presumably evolved as an alternative to apoptosis for handling insulted cells. It is evidently a less harsh measure than apoptosis and the decision between apoptosis and senescence may depend on the severity of the insult. Because senescent cells are still metabolically active, tissue homeostasis is preserved for some time before inflicted cells are apparently removed by the immune system. For a characteristic of senescent cells is the so-called senescence-associated secretory phenotype (SASP), which refers to the continued secretion of pro-inflammatory cytokines, chemokines, and proteases (Kuilman and Peeper, 2009; Tchkonia et al., 2013) and leads to infiltration of immune cells for the removal of damaged cells. Although aimed at resolving local and time-limited tissue damage, SASP becomes chronic in aged tissue, which because of continuous exposure to a variety of stressors, is enriched in senescent cells. This age-dependent chronic, low-level inflammatory response has been termed sterile inflammation (indicating an absence of pathogens) or “inflammaging”, because of its intimate relationship with the damage-induced aging process (Franceschi et al., 2000). Interestingly, inflammaging seems to be self-propagating through a positive feedback loop or vicious cycle. As indicated previously, oxidative phosphorylation in mitochondria is the main source of ROS in a cell. ROS-induced damage to mitochondria including the mitochondrial DNA is believed to result in progressively defective oxidative phosphorylation, which in turn increases ROS production. The rate of mitochondrial ROS production tends to increase during aging, particularly in long-lived postmitotic cell types such as neurons (Sohal and Sohal, 1991; Wang et al., 2013). This mechanism of self-inflicted mitochondrial damage is aggravated by an age-dependent decline in autophagic clearing capacity (Cuervo, 2008; Rubinsztein et al., 2011). Autophagy is an ancient housekeeping mechanism that controls cellular homeostasis by sequestering aggregated proteins and dysfunctional organelles including mitochondria (in this case referred to as mitophagy) in vesicles and fusing them with lysosomes for degradation and recycling (Ravikumar et al., 2010). Damaged mitochondria consequently accumulate within senescent cells in aged tissue and produce increasing levels of ROS, thereby triggering an inflammatory response through the SASP. There is, moreover, evidence that damaged mitochondria activate the NRLP3-dependent inflammasome, a protein platform that acts as a sensor of intracellular damage including elevated levels of ROS and elicits an inflammatory response (Gross et al., 2011; Zhou et al., 2011). Recent data suggest that the SASP and the inflammasome are tightly linked in the sense that activation of the inflammasome precedes the implementation of the SASP (Acosta et al., 2013).
Taken together, aging occurs because of a cell's progressive inability to cope with various forms of stress. Elevated levels of ROS are a major stressor in the aging brain, but not the only one. The main cellular sources of ROS are mitochondria; in a vicious cycle ROS damage mitochondrial enzymes and mitochondrial DNA such that even more ROS are generated because of defective oxidative phosphorylation reactions. ROS have detrimental effects on cells through at least 3 distinct mechanisms. First, they inflict direct chemical damage to cellular macromolecules because of their high reactivity. Second, they disrupt basic redox-sensitive signaling pathways by shifting a cell's redox balance to a pro-oxidative state. Third, elevated ROS induce a senescent state and activate the endogenous inflammasome, with both processes being mechanistically linked and triggering a chronic inflammatory response and subsequent disruption of tissue homeostasis.
3.2. ApoD: a natural defense against age-related stress
In the CNS, ApoD is mainly expressed in white matter, within neurons, oligodendrocytes, and astrocytes, whereas in the peripheral nervous system (PNS) ApoD is mostly detected in Schwann cells and fibroblasts. Within the brain there are marked regional differences in ApoD expression. Areas of high ApoD expression include the prefrontal cortex, the substantia nigra, and the hippocampal formation (Elliott et al., 2010; Rassart et al., 2000).
A number of studies have investigated the dynamics of ApoD expression in the aging brain. A marked increase in the number of ApoD positive cells, mostly glial cells, has been documented through immunohistochemistry in the cortex of the aging human brain (Kalman et al., 2000). ApoD in the human prefrontal cortex has been shown to be upregulated 6- to 8-fold at the messenger RNA and protein level between the early postnatal period of life and an age of 49 years (Kim et al., 2009). In a cross-species comparative analysis of transcription changes during brain aging, ApoD was identified as the most upregulated gene in the aged brain. Specifically, up-regulation of ApoD mRNA in the cortex during aging was not only conserved between human, macaque, and mouse, but it was also the strongest upregulation of any gene in each of these 3 species (Loerch et al., 2008). This study confirms previous results obtained with the mouse, which indicate a global upregulation of oxidative stress and inflammation induced genes in the aging brain including ApoD (Lee et al., 2000). In a meta-analysis of age-related gene expression profiles that involved 27 datasets from mouse, rat, and human, ApoD was found to be the most significant of the age-dependent up-regulated genes (de Magalhaes et al., 2009).
As with all correlational studies, the question of cause or consequence arises. In this case the question is whether the observed upregulation of ApoD during aging causes, or is a consequence of, the aging process. The answer has been obtained through a series of experiments on Drosophila and the mouse that clearly indicated a highly conserved neuroprotective, antioxidant function for ApoD during aging. First, overexpression of GLaz, an ApoD ortholog, in Drosophila resulted in an increased resistance to oxidative stress and a marked extension in life span by 30% (Walker et al., 2006). Second, an increase in both life span and stress resistance has also been observed as a result of overexpression of Lazarillo, the grasshopper ortholog of ApoD, in Drosophila. Moreover, Lazarillo has been demonstrated to functionally restore loss of Drosophila NLaz by rescuing stress resistance in a neuron-specific manner (Ruiz et al., 2012). Intriguingly, overexpression of human ApoD in Drosophila led to a reduction in age-associated lipid peroxidation, concomitant with an increase in stress resistance and longevity (Muffat et al., 2008). Overexpression of human ApoD in Drosophila thus reproduces the phenotype of overexpression of GLaz, indicating a physiological role for ApoD that has been highly conserved over long evolutionary distances. Similar results have been obtained from a mammalian model organism. When exposed to acute oxidative stress, the mouse brain responds by up-regulating ApoD (Ganfornina et al., 2008). Upon oxidative stress transgenic mice overexpressing human ApoD in the brain exhibit a reduced rate of lipid peroxidation and an improved rate of survival (Ganfornina et al., 2008). Conversely, ApoD loss-of-function resulted in elevated levels of peroxidized lipids in the mouse brain, together with increased sensitivity to oxidative stress (Ganfornina et al., 2008). Loss of GLaz function in Drosophila has been reported to increase lipid peroxidation and sensitivity to oxidative stress, and to decrease the life span (Sanchez et al., 2006). In line with these observations is the finding that, upon oxidative stress, ApoD stabilizes dopaminergic circuits in the substantia nigra, presumably by lowering the load of lipid peroxidation in, and decreasing the inflammatory reactivity of, astrocytes (Bajo-Graneras et al., 2011).
Taken together, these results indicate that upregulation of ApoD is part of an evolutionarily conserved stress response pathway in the aging brain. By virtue of its antioxidant and anti-inflammatory activity, ApoD protects against the age-associated oxidative stress that builds up because of increasing ROS levels. ApoD is likely to achieve these neuroprotective effects by restraining lipid peroxidation and by modulating AA metabolism in an anti-inflammatory manner.
4. ApoD in neurologic disorders
The main determinant of ApoD expression is cellular stress. Specifically, ApoD expression has been shown to be induced through oxidative stress (e.g., H2O2), UV light, and pro-inflammatory lipopolysaccharides and IL-1α (Blais et al., 1994; Do Carmo et al., 2007). At the cellular level, this regulatory pattern is reflected by potent up-regulation of ApoD upon growth arrest or senescence, a cellular fate that is brought about by various forms of stress and inflammation (Do Carmo et al., 2002, 2008; Provost et al., 1991).
Cellular stress and inflammation increase gradually during aging, and consistent upregulation of ApoD in the aging brain is therefore not surprising. Aside from aging, ApoD is also upregulated in a range of neurologic disorders including AD and other forms of dementia, Parkinson's disease, motor neuron disease, Niemann–Pick disease, meningoencephalitis, multiple sclerosis, major depression disease, bipolar disorder, and schizophrenia, as well as stroke and traumatic brain injury (Rassart et al., 2000; Sutcliffe and Thomas, 2002). Although many of these pathologies appear to have genetic risk factors, their manifestation is often linked to cellular stress and inflammation (Andersen, 2004; Di Carlo et al., 2012; Glass et al., 2010; Pizza et al., 2011). In fact aging, which is characterized by elevated levels of stress and chronic inflammation, is the primary risk factor for a number of neurodegenerative diseases including AD and other forms of dementia, Parkinson's disease, and major depression disease. Normal brain aging, thus, often entails pathologic forms of neurodegeneration and identifying the underlying mechanisms of age-associated neurodegeneration is of utmost societal importance (Yankner et al., 2008). Upregulation of ApoD appears to be a shared response occurring in both the aging brain and in the pathologic, degenerative brain. Given the neuroprotective role of ApoD in normal brain aging, it is tempting to speculate that it may also have a beneficial effect under pathologic conditions. We present in the following sections the relevant evidence for 3 major brain pathologies: AD, stroke, and schizophrenia.
4.1. ApoD in chronic neurodegenerative disorders: AD
AD is the most common form of dementia (Kandel, 2012). It is a progressive disease with symptoms that range from mild memory lapses to loss of speech and self-awareness, apathy, and immobility. AD is a typical age-related neurodegenerative disease with its incidence and prevalence doubling every 5 years after the age of 65 years (Kukull et al., 2002). The ultimate cause of AD is a progressive loss of neurons, mostly in the hippocampus and the cerebral cortex. While the disease mechanism remains essentially unknown, most current research on AD centers on 2 basic findings. First, in an AD-affected brain, amyloid plaques that consist mainly of insoluble aggregates of beta amyloid peptides (Aß), a collection of proteolytic cleavage products of the amyloid precursor protein, are found around and between neurons. Second, neurofibrillary tangles, comprised of aggregates of the microtubule-associated tau protein, accumulate within neurons themselves. What is the cause and what is the consequence remains a matter of debate, as does exactly how these protein aggregates are initiated, and how they ultimately lead to neuronal cell death (Kandel, 2012).
Elevated levels of ApoD mRNA and/or protein have been found in the CSF, hippocampus, and entorhinal cortex of AD subjects (Terrisse et al., 1998) and the number of ApoD immunopositive neurons has been shown to be markedly increased in AD-affected brains (Belloir et al., 2001; Kalman et al., 2000). In contrast to other apolipoproteins, ApoD is co-distributed with compact (dense fibrillar) but not diffuse (amorphous), Aß plaques. ApoD immunopositive cells within these plaques appear to be reactive microglia, suggesting an inflammation-induced upregulation of ApoD in AD (Desai et al., 2005). Alternatively, Aß itself might induce upregulation of ApoD, as has recently been shown with a hippocampal cell line (Martinez et al., 2012). It remains unclear whether this induction would be direct or indirect, but it is known that Aß triggers production of ROS, which may in turn lead to ApoD upregulation (Butterfield, 2002). A causal link between Aß and expression of ApoD is also indicated by a mouse model of AD. Specifically, ApoD has been demonstrated to be significantly upregulated in the brain of a transgenic mouse with an overexpressed highly amyloidogenic, that is, Aβ producing, form of human amyloid precursor protein. As is typical for AD, this effect was observed only in aged (26-month-old), but not young (6-month-old) animals (Thomas et al., 2001c). While these results do not necessarily imply a neuroprotective role for ApoD in AD, recent biochemical data do in fact suggest precisely such a role. As mentioned previously, ApoD has been demonstrated to reduce lipid hydroperoxides to lipid hydroxides through a highly conserved methionine residue (Met93 in human ApoD) (Bhatia et al., 2012). This reaction results in oxidation of the methionine sulfhydryl to form methionine sulfoxide (MetSO), and subsequently to dimerization of ApoD because of structural destabilization by Met93-SO, as suggested by molecular dynamics simulation (Oakley et al., 2012). While MetSO is usually reduced back to methionine sulfhydryl by MetSO reductases, a substantial decline in this enzymatic activity has been found in AD brains (Gabbita et al., 1999). If ApoD is to act as an antioxidant in AD one would expect to find elevated levels of the dimeric form, given that the reductive regeneration is diminished, and this is in fact exactly the case. In the brains of AD patients ApoD has predominantly been detected as a dimerized aggregate (Bhatia et al., 2012, 2013). Elevated levels of lipid peroxidation were measured in the same AD samples, confirming previous results that showed increased lipid peroxidation in the brain of AD patients (Butterfield and Lauderback, 2002; Pratico and Sung, 2004). Detailed studies have even revealed breakdown products of lipid peroxidation in amyloid plaques of AD brains (Ando et al., 1998), co-localizing with plaque-associated ApoD (Desai et al., 2005).
Taken together, these data are consistent with a neuroprotective role for ApoD in AD. The picture that emerges is that increased oxidative stress in AD promotes lipid peroxidation and elicits upregulation of ApoD as part of the brain's stress response. As the disease progresses, ApoD is used up by ever increasing levels of peroxidized molecules, a situation that is aggravated by the failure to regenerate ApoD because of reduced MetSO reductase activity. Oxidized ApoD accumulates as dimeric aggregates and is eventually deposited in amyloid plaques, together with peroxidized lipids.
It is of note that a series of studies have detected some polymorphisms of the human APOD gene that appear to predispose carriers to AD (Chen et al., 2008; Desai et al., 2003; Helisalmi et al., 2004; Shibata et al., 2013). However, evidence that certain APOD variants are a risk factor for AD is much less well established than for the ε4 allele of APOE (Mahley et al., 2006; Strittmatter et al., 1993).
4.2. ApoD in acute neurodegenerative disorders: stroke
A stroke is the disruption of brain function because of a lack of blood supply (Donnan et al., 2008). There are 2 types of stroke: ischemic stroke, which is caused by obstruction of flow in a blood vessel; and hemorrhagic stroke, which is caused by the rupture of a blood vessel in the brain. The large majority of strokes are ischemic and hemorrhagic strokes are often associated with ischemia in that part of the brain that was fed by the ruptured artery. Unlike any other organ, the brain is exceptionally sensitive to reduced levels of oxygen because it relies entirely on aerobic metabolism. Depriving the brain of blood and/or oxygen leads to what is called the ischemic cascade. Falling levels of ATP quickly result in the failure of ion pumps and a breakdown of ion gradients, which in turn triggers a series of interrelated events including a release of the excitotoxic neurotransmitter glutamate, an influx of calcium, and the generation of ROS. The damage spreads beyond the immediately affected area and neurons die by apoptosis (Brouns and De Deyn, 2009).
ApoD has been shown to be upregulated following a stroke. Specifically, in a stroke model of the rat, ApoD expression was found to be increased within the first 24 hours in post-ischemic peri-infarct regions, with a further increase 48 hours post-ischemia (Rickhag et al., 2006). Immunohistochemical studies revealed that ApoD within the infarct core was markedly upregulated by degenerating pyramidal neurons, whereas in the surviving and regenerating peri-infarct region ApoD was localized in mature oligodendrocytes where it remained upregulated for more than 1 week (Rickhag et al., 2008). These findings are in agreement with those of Franz et al. (1999), who detected an increase in ApoD expression at the mRNA and protein level in the cortex and hippocampus of the rat 2–14 days after experimental traumatic brain injury. ApoD expression was even further enhanced relative to control levels in ischemic rats housed in an enriched environment, compared with those housed in a standard environment. This extra upregulation of ApoD under enriched housing conditions correlated with an improved rate of functional recovery (Rickhag et al., 2008) This observation is of particular relevance to post-stroke recovery. Recovery from stroke is based on structural and functional remodeling activities, including the recruitment of alternative neuronal circuits and axonal sprouting (Dancause et al., 2005; Wieloch and Nikolich, 2006). Post-ischemic recovery has been shown to be promoted by an enriched environment, which has been attributed to enhanced dendritic arborization, increased spine density, and upregulation of genes coding for synaptic proteins and growth factors (Johansson and Ohlsson, 1996; Nygren and Wieloch, 2005). Neurologic recovery also depends on a substantial level of plasma antioxidant capacity, as shown in Leinonen et al. (2000). Studies that report on increased levels of lipid peroxidation in stroke patients (Polidori et al., 1998) and that show a direct correlation between the extent of lipid peroxidation and severity of neurologic outcome after stroke (Ferretti et al., 2008) underscore the relevance of oxidative damage in stroke pathogenesis. ApoD is likely to reduce the load of oxidative stress in this scenario by virtue of its antioxidant activity. Aside from its antioxidant role during the acute stroke phase, ApoD is believed to play a beneficial role in the regenerating phase by providing lipids such as cholesterol or free fatty acids, thereby facilitating remodeling processes, including remyelination. This notion is supported by data obtained from a lesion model of the sciatic nerve (Ganfornina et al., 2010).
In conclusion, ApoD may exert a neuroprotective influence in the acute stroke phase and may also support neuronal regeneration and remyelination in the extended post-stroke recovery phase.
4.3. ApoD in psychiatric disorders: schizophrenia
Schizophrenia is a chronic psychiatric disorder characterized by disruption of mental processing and inappropriate emotional behavior (Kandel, 2012). Common symptoms include delusions, illusions, hallucinations, disorganized speech, and flattened or absent emotional responsiveness. These symptoms are typically accompanied by social withdrawal or isolation. While the causes for this disease remain unknown, it appears to result from an interplay between genetic and environmental factors. There is a large body of evidence that an imbalance in neurotransmitter activities, with a special focus on dopamine, can give rise to the symptoms produced by this disorder, but deregulation of serotonergic and glutamatergic transmission also seems to be of relevance (Jones and Pilowsky, 2002; Konradi and Heckers, 2003; van Os and Kapur, 2009).
ApoD has been found to be significantly upregulated in the brain of schizophrenic and bipolar subjects. Specifically, ApoD expression was selectively increased in the dorsolateral prefrontal cortex and the caudate, which are the brain regions that are implicated in the pathogenesis of these neuropsychiatric disorders (Thomas et al., 2001b). In a more detailed follow-up study, specific differences in neuroanatomic sites of ApoD elevation were found between patients with schizophrenic disorder and those with bipolar disorder (Thomas et al., 2003a). ApoD levels have also been found to be significantly increased in the plasma of schizophrenic patients, including first-episode patients, with the increase in ApoD presumably predating the first clinical symptoms (Mahadik et al., 2002).
Apart from its upregulation in well-defined brain areas in schizophrenia, there is an interesting link between ApoD and clozapine, as well as other atypical neuroleptics. Clozapine is an atypical antipsychotic drug that has been shown to be effective in acute and long-term treatment of patients with bipolar disorder and schizophrenia, in particular of patients who had proved refractory to other antipsychotic treatments (Ciapparelli et al., 2000). In contrast to typical antipsychotic drugs, which have high affinities for dopamine receptors, clozapine binds to multiple neurotransmitter receptors. In a study that searched for clozapine-induced changes in gene expression within the mouse brain, ApoD was found to increase both at the mRNA and protein level over a period of 2 weeks of continued clozapine treatment (Thomas et al., 2001a). Upregulation of ApoD seems to be a distinguishing feature between typical and atypical neuroleptics because the atypical drugs risperidone and olanzapine increase the expression of ApoD in the brain as well, whereas haloperidol, a typical antipsychotic drug, does not (Khan et al., 2003). Because atypical neuroleptics have a better treatment outcome in terms of improved cognitive performance and ameliorated negative symptoms (Bilder et al., 2002), these observations raise the intriguing possibility that upregulation of ApoD is a mechanism underlying the beneficial effect of atypical antipsychotics in the treatment of schizophrenia. Exactly how ApoD is able to have a positive modulating effect on schizophrenic symptoms remains uncertain. However, the well-described role that ApoD plays in the transport and modification of lipids may provide an explanation, because altered membrane phospholipids are known to be involved in the pathophysiology of schizophrenia (Berger et al., 2006; Horrobin and Bennett, 1999). AA is of particular interest, because this fatty acid is a prominent ligand of ApoD (Morais Cabral et al., 1995), but also because reduced concentrations of AA and AA-enriched phospholipids have been measured in the brains of schizophrenic subjects (Arvindakshan et al., 2003; Skosnik and Yao, 2003). This reduction in AA concentration is consistent with the increased levels of PLA2 activity that have been detected in the serum and prefrontal cortex of schizophrenic patients (Gattaz et al., 1987; Ross et al., 1999). Moreover, ApoD has been found to stabilize plasma membrane-associated as well as free AA and to attenuate its peroxidation to highly toxic compounds or its metabolic conversion into pro-inflammatory eicosanoids (Thomas et al., 2003b). AA is a polyunsaturated omega-6 fatty acid (20:4 n-6). It is abundant in neuronal membranes and, together with docosahexaenoic acid (DHA; 22:6 n-3), makes up more than 90% of the polyunsaturated fatty acids in the CNS (O'Brien and Sampson, 1965; Youdim et al., 2000). Polyunsaturated fatty acids are key determinants of membrane order and fluidity and affect the binding and signaling characteristics of membrane proteins, including ion channels and neurotransmitter receptors (Luchtman and Song, 2013). They have been implicated in the pathogenesis of schizophrenia and their neuroprotective potential as medication is currently being investigated (Horrobin and Bennett, 1999; Mossaheb et al., 2012; Perica and Delas, 2011). ApoD may therefore have a protective effect in schizophrenia by stabilizing AA for use as a membrane building block and preventing its peroxidation or metabolic conversion with associated deleterious consequences. The notion of an AA-specific role for ApoD in schizophrenia is supported by the observation that atypical antipsychotics, but not typical ones, prevent the decrease of membrane phospholipid–AA levels in schizophrenic patients (van der Kemp et al., 2012). Like in AD and stroke, oxidative stress and lipid peroxidation seem to be part of the pathophysiology of schizophrenia (Boskovic et al., 2011) and ApoD might exert a positive influence because of its overall antioxidant activity.
Of relevance to the possible beneficial role of ApoD in schizophrenia may also be the impact that ApoD has on the endocannabinoid system. Anandamide has recently been identified as a novel, high affinity, ligand of ApoD (Ruiz et al., 2013). Because the endocannabinoid system is dysregulated in schizophrenic patients (Zamberletti et al., 2012), it would be very interesting to explore any functional relationship between ApoD and endocannabinoids, under both physiological and pathophysiological conditions.
In conclusion, ApoD appears to play a neuroprotective role in schizophrenia, which is further augmented by treatment with atypical neuroleptics, including clozapine. Elevating the expression level of ApoD may in fact reflect a novel molecular mechanism underlying the favorable effects of atypical antipsychotics, as opposed to typical antipsychotics, in schizophrenic patients.
5. Outlook
Characterizing the complete ligand repertoire of ApoD will be the key to appreciating its full physiological relevance. Anandamide has only recently been identified as a novel ApoD ligand, through a candidate screening approach (Ruiz et al., 2013). Although the known ligands of ApoD belong to quite different groups of lipophilic molecules, there is a remarkable specificity in the binding pattern. For instance, ApoD binds free AA and anandamide (N-arachidonoylethanolamine) with high affinity, but not the chemically similar 2-arachidonyl glycerol (Ruiz et al., 2013). This makes it difficult to predict novel ligands. With regard to the length of the hydrocarbon chain, ApoD has been shown to accommodate C16 (palmitic acid), C18 (sphingosine), and C20 (AA) ligands, each in all-cis configuration (Morais Cabral et al., 1995; Ruiz et al., 2013). To our knowledge, there have been no investigations to date into whether ApoD binds omega-3 fatty acids, in particular α-linolenic acid (18:3 n-3), which is an essential fatty acid, eicosapentaenoic acid (EPA; 20:5 n-3), and DHA (22:6 n-3), although such investigations would be of considerable interest from a neurologic point of view. Omega-3 fatty acids, also known as n-3 fatty acids, are vital building blocks for neuronal membranes and indispensable for the maintenance of healthy brain function and for regeneration of the injured or stressed brain. Omega-3 fatty acids become deficient in the aging and degenerative brain, but dietary supplements have been shown to restore their membrane levels and to attenuate cognitive decline (Luchtman and Song, 2013; Yehuda et al., 2002). In this context, it would also be interesting to investigate any relationship between ApoD and resolvins, which are derivatives of EPA and DHA, and neuroprotectins, which are derived from DHA. Both classes of eicosanoids and docosanoids have been demonstrated to be potent anti-inflammatories and antioxidants and to exert an overall pro-survival, anti-apoptotic influence on neurons (Bazan, 2006; Farooqui, 2012; Ji et al., 2011).
While in chronic disease or aging ApoD is upregulated approximately 2 to 3-fold at the mRNA and protein level, its dynamic range of expression appears to be far greater. In a peripheral nerve crush model of the rat, ApoD protein was dramatically induced in the regenerating nerve, by a factor of 500 (Boyles et al., 1990). The question that arises is what would be the beneficial effect in AD or aging if ApoD were upregulated to a similar extent and expression maintained at that high level for an extended period of time? Would the progress of the disease or aging be attenuated and symptoms ameliorated in a clinically relevant manner? After all, in AD the increase in endogenous ApoD levels may be ineffective as the dimerized form of the protein that is predicted to lack antioxidant activity, is elevated because of high levels of lipid peroxidation and reduced MetSO reductase activity (Bhatia et al., 2012). Moreover, atypical neuroleptics induce endogenous ApoD and thereby appear to produce favorable effects in the treatment of psychiatric diseases. Exogenously added ApoD may therefore attenuate the progress of a number of neurologic diseases without the deleterious side effects that have, for instance, been described for antipsychotic drugs. Although ApoD can be efficiently produced as a functional recombinant protein in microbial host cells, its therapeutic value for long-term systemic application is restricted by the blood-brain barrier (Vogt and Skerra, 2001). It might therefore be more promising to try to induce upregulation of endogenous ApoD. Its promoter has been characterized at some length (Do Carmo et al., 2002; Levros et al., 2010) but it still remains unclear by which signaling pathways the dramatic induction as described in (Boyles et al., 1990) is attained, or how atypical antipsychotic drugs such as clozapine elevate ApoD expression levels. Switching ApoD expression into acute injury mode over an extended period of time may have visible clinical benefits in a series of neurologic and psychiatric disorders and could possibly even delay the aging process in the brain.
6. Conclusion
ApoD has initially been characterized as a carrier of a range of small lipophilic molecules. It is prominently expressed in the central and peripheral nervous system, by both glial cells and neurons. In the past few years it has become apparent that the function of ApoD is not merely limited to supplying cells with membrane building blocks, hormones, and messengers. One of its best characterized ligand interactions is with AA, which is enriched in the CNS in the same way as ApoD. It has been demonstrated in a series of studies that ApoD modulates AA metabolism in an anti-inflammatory and antioxidant manner. This may in part explain why ApoD confers potent resistance to oxidative stress when overexpressed in Drosophila or the mouse. Overexpression of ApoD results in an extension of the mean life span in Drosophila and increases survival under stress in the mouse; the ApoD gene therefore belongs to the small selection of genes whose manipulation is life extending. Conversely, endogenous ApoD is upregulated during aging and under a large variety of neurologic and psychiatric disorders, suggesting that induction of ApoD is part of a basic evolutionarily conserved anti-stress response. ApoD, thus, appears to be an ancient anti-stress protein that counteracts cellular stress, and in particular oxidative stress and inflammation. Both forms of stress are widespread throughout the body and increase with advancing age. Oxidative stress and inflammation are also generated at some point in time, either directly or indirectly, in most neurologic and psychiatric disorders. Even if ApoD does not target the primary cause of a disorder, it may well ameliorate the clinical symptoms and delay the progress by reducing the stress load that accompanies a disorder. Augmenting its neuroprotective properties could be particularly relevant for chronic diseases and aging, which are characterized by a continuous rate of oxidative and inflammatory stress, but exhibit only moderate upregulation of ApoD compared with the dramatic but temporary elevation of ApoD levels observed in acute injuries. ApoD may therefore be a promising therapeutic target for a wide range of neurologic diseases, as well as for aging, because of its anti-stress activity; designing strategies for its delivery to the CNS for long-term application, or for manipulating its endogenous expression levels, will provide a challenge for future research.
Disclosure statement
The authors have no conflicts of interest to disclose.
Acknowledgements
This work was supported by the Austrian Science Fund (FWF; grant number #P 19908-B05 to R.S.).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
References
- Acosta J.C., Banito A., Wuestefeld T., Georgilis A., Janich P., Morton J.P., Athineos D., Kang T.W., Lasitschka F., Andrulis M., Pascual G., Morris K.J., Khan S., Jin H., Dharmalingam G., Snijders A.P., Carroll T., Capper D., Pritchard C., Inman G.J., Longerich T., Sansom O.J., Benitah S.A., Zender L., Gil J. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biology. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor S., Puentes F., Baker D., van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J.K. Oxidative stress in neurodegeneration: cause or consequence? Nat. Med. 2004;10(Suppl.):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Ando Y., Brannstrom T., Uchida K., Nyhlin N., Nasman B., Suhr O., Yamashita T., Olsson T., El Salhy M., Uchino M., Ando M. Histochemical detection of 4-hydroxynonenal protein in Alzheimer amyloid. J. Neurol. Sci. 1998;156:172–176. doi: 10.1016/s0022-510x(98)00042-2. [DOI] [PubMed] [Google Scholar]
- Arvindakshan M., Sitasawad S., Debsikdar V., Ghate M., Evans D., Horrobin D.F., Bennett C., Ranjekar P.K., Mahadik S.P. Essential polyunsaturated fatty acid and lipid peroxide levels in never-medicated and medicated schizophrenia patients. Biol. Psychiatry. 2003;53:56–64. doi: 10.1016/s0006-3223(02)01443-9. [DOI] [PubMed] [Google Scholar]
- Bajo-Graneras R., Ganfornina M.D., Martin-Tejedor E., Sanchez D. Apolipoprotein D mediates autocrine protection of astrocytes and controls their reactivity level, contributing to the functional maintenance of paraquat-challenged dopaminergic systems. Glia. 2011;59:1551–1566. doi: 10.1002/glia.21200. [DOI] [PubMed] [Google Scholar]
- Bazan N.G. The onset of brain injury and neurodegeneration triggers the synthesis of docosanoid neuroprotective signaling. Cell. Mol. Neurobiol. 2006;26:901–913. doi: 10.1007/s10571-006-9064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloir B., Kovari E., Surini-Demiri M., Savioz A. Altered apolipoprotein D expression in the brain of patients with Alzheimer disease. J. Neurosci. Res. 2001;64:61–69. doi: 10.1002/jnr.1054. [DOI] [PubMed] [Google Scholar]
- Berger G.E., Smesny S., Amminger G.P. Bioactive lipids in schizophrenia. Int. Rev. Psychiatry. 2006;18:85–98. doi: 10.1080/09540260600583072. [DOI] [PubMed] [Google Scholar]
- Bhatia S., Jenner A.M., Li H., Ruberu K., Spiro A.S., Shepherd C.E., Kril J.J., Kain N., Don A., Garner B. Increased apolipoprotein D dimer formation in Alzheimer's disease hippocampus is associated with lipid conjugated diene levels. J. Alzheimers Dis. 2013;35:475–486. doi: 10.3233/JAD-122278. [DOI] [PubMed] [Google Scholar]
- Bhatia S., Knoch B., Wong J., Kim W.S., Else P.L., Oakley A.J., Garner B. Selective reduction of hydroperoxyeicosatetraenoic acids to their hydroxy derivatives by apolipoprotein D: implications for lipid antioxidant activity and Alzheimer's disease. Biochem. J. 2012;442:713–721. doi: 10.1042/BJ20111166. [DOI] [PubMed] [Google Scholar]
- Bilder R.M., Goldman R.S., Volavka J., Czobor P., Hoptman M., Sheitman B., Lindenmayer J.P., Citrome L., McEvoy J., Kunz M., Chakos M., Cooper T.B., Horowitz T.L., Lieberman J.A. Neurocognitive effects of clozapine, olanzapine, risperidone, and haloperidol in patients with chronic schizophrenia or schizoaffective disorder. Am. J. Psychiatry. 2002;159:1018–1028. doi: 10.1176/appi.ajp.159.6.1018. [DOI] [PubMed] [Google Scholar]
- Bishop N.A., Lu T., Yankner B.A. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop R.E. The bacterial lipocalins. Biochim. Biophys. Acta. 2000;1482:73–83. doi: 10.1016/s0167-4838(00)00138-2. [DOI] [PubMed] [Google Scholar]
- Blais Y., Sugimoto K., Carriere M.C., Haagensen D.E., Labrie F., Simard J. Potent stimulatory effect of interleukin-1 alpha on apolipoprotein D and gross cystic disease fluid protein-15 expression in human breast-cancer cells. Int. J. Cancer. 1994;59:400–407. doi: 10.1002/ijc.2910590319. [DOI] [PubMed] [Google Scholar]
- Borghini I., Barja F., Pometta D., James R.W. Characterization of subpopulations of lipoprotein particles isolated from human cerebrospinal fluid. Biochim. Biophys. Acta. 1995;1255:192–200. doi: 10.1016/0005-2760(94)00232-n. [DOI] [PubMed] [Google Scholar]
- Boskovic M., Vovk T., Kores Plesnicar B., Grabnar I. Oxidative stress in schizophrenia. Curr. Neuropharmacol. 2011;9:301–312. doi: 10.2174/157015911795596595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles J.K., Notterpek L.M., Anderson L.J. Accumulation of apolipoproteins in the regenerating and remyelinating mammalian peripheral nerve. Identification of apolipoprotein D, apolipoprotein A-IV, apolipoprotein E, and apolipoprotein A-I. J. Biol. Chem. 1990;265:17805–17815. [PubMed] [Google Scholar]
- Brandes N., Schmitt S., Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns R., De Deyn P.P. The complexity of neurobiological processes in acute ischemic stroke. Clin. Neurol. Neurosurg. 2009;111:483–495. doi: 10.1016/j.clineuro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Butterfield D.A. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic. Res. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- Butterfield D.A., Lauderback C.M. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Calder P.C. Polyunsaturated fatty acids and inflammation. Biochem. Soc. Trans. 2005;33(Pt 2):423–427. doi: 10.1042/BST0330423. [DOI] [PubMed] [Google Scholar]
- Campisi J. Aging and cancer cell biology, 2007. Aging Cell. 2007;6:261–263. doi: 10.1111/j.1474-9726.2007.00292.x. [DOI] [PubMed] [Google Scholar]
- Chen Y., Jia L., Wei C., Wang F., Lv H., Jia J. Association between polymorphisms in the apolipoprotein D gene and sporadic Alzheimer's disease. Brain Res. 2008;1233:196–202. doi: 10.1016/j.brainres.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Ciapparelli A., Dell'Osso L., Pini S., Chiavacci M.C., Fenzi M., Cassano G.B. Clozapine for treatment-refractory schizophrenia, schizoaffective disorder, and psychotic bipolar disorder: a 24-month naturalistic study. J. Clin. Psychiatry. 2000;61:329–334. doi: 10.4088/jcp.v61n0502. [DOI] [PubMed] [Google Scholar]
- Cuervo A.M. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancause N., Barbay S., Frost S.B., Plautz E.J., Chen D., Zoubina E.V., Stowe A.M., Nudo R.J. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhaes J.P., Curado J., Church G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P.P., Hendrie H.C., Evans R.M., Murrell J.R., DeKosky S.T., Kamboh M.I. Genetic variation in apolipoprotein D affects the risk of Alzheimer disease in African-Americans. Am. J. Med. Geneti. B Neuropsychiatr. Genet. 2003;116B:98–101. doi: 10.1002/ajmg.b.10798. [DOI] [PubMed] [Google Scholar]
- Desai P.P., Ikonomovic M.D., Abrahamson E.E., Hamilton R.L., Isanski B.A., Hope C.E., Klunk W.E., DeKosky S.T., Kamboh M.I. Apolipoprotein D is a component of compact but not diffuse amyloid-beta plaques in Alzheimer's disease temporal cortex. Neurobiol. Dis. 2005;20:574–582. doi: 10.1016/j.nbd.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Di Carlo M., Giacomazza D., Picone P., Nuzzo D., San Biagio P.L. Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Radic. Res. 2012;46:1327–1338. doi: 10.3109/10715762.2012.714466. [DOI] [PubMed] [Google Scholar]
- Do Carmo S., Jacomy H., Talbot P.J., Rassart E. Neuroprotective effect of apolipoprotein D against human coronavirus OC43-induced encephalitis in mice. J. Neurosci. 2008;28:10330–10338. doi: 10.1523/JNEUROSCI.2644-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Carmo S., Levros L.C., Jr., Rassart E. Modulation of apolipoprotein D expression and translocation under specific stress conditions. Biochim. Biophys. Acta. 2007;1773:954–969. doi: 10.1016/j.bbamcr.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Do Carmo S., Seguin D., Milne R., Rassart E. Modulation of apolipoprotein D and apolipoprotein E mRNA expression by growth arrest and identification of key elements in the promoter. J. Biol. Chem. 2002;277:5514–5523. doi: 10.1074/jbc.M105057200. [DOI] [PubMed] [Google Scholar]
- Donnan G.A., Fisher M., Macleod M., Davis S.M. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- Eichinger A., Nasreen A., Kim H.J., Skerra A. Structural insight into the dual ligand specificity and mode of high density lipoprotein association of apolipoprotein D. J. Biol. Chem. 2007;282:31068–31075. doi: 10.1074/jbc.M703552200. [DOI] [PubMed] [Google Scholar]
- Elliott D.A., Weickert C.S., Garner B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin. Lipidol. 2010;51:555–573. doi: 10.2217/CLP.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am. J. Clin. Nutr. 1993;57(5 Suppl.):779S–785S. doi: 10.1093/ajcn/57.5.779S. discussion 85S-86S. [DOI] [PubMed] [Google Scholar]
- Farooqui A.A. n-3 fatty acid-derived lipid mediators in the brain: new weapons against oxidative stress and inflammation. Curr. Med. Chem. 2012;19:532–543. doi: 10.2174/092986712798918851. [DOI] [PubMed] [Google Scholar]
- Federico A., Cardaioli E., Da Pozzo P., Formichi P., Gallus G.N., Radi E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012;322:254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Ferretti G., Bacchetti T., Masciangelo S., Nanetti L., Mazzanti L., Silvestrini M., Bartolini M., Provinciali L. Lipid peroxidation in stroke patients. Clin. Chem. Lab. Med. 2008;46:113–117. doi: 10.1515/CCLM.2008.011. [DOI] [PubMed] [Google Scholar]
- Franceschi C., Bonafe M., Valensin S., Olivieri F., De Luca M., Ottaviani E., De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Franz G., Reindl M., Patel S.C., Beer R., Unterrichter I., Berger T., Schmutzhard E., Poewe W., Kampfl A. Increased expression of apolipoprotein D following experimental traumatic brain injury. J. Neurochem. 1999;73:1615–1625. doi: 10.1046/j.1471-4159.1999.0731615.x. [DOI] [PubMed] [Google Scholar]
- Gabbita S.P., Aksenov M.Y., Lovell M.A., Markesbery W.R. Decrease in peptide methionine sulfoxide reductase in Alzheimer's disease brain. J. Neurochem. 1999;73:1660–1666. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- Ganfornina M.D., Do Carmo S., Lora J.M., Torres-Schumann S., Vogel M., Allhorn M., Gonzalez C., Bastiani M.J., Rassart E., Sanchez D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell. 2008;7:506–515. doi: 10.1111/j.1474-9726.2008.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina M.D., Do Carmo S., Martinez E., Tolivia J., Navarro A., Rassart E., Sanchez D. ApoD, a glia-derived apolipoprotein, is required for peripheral nerve functional integrity and a timely response to injury. Glia. 2010;58:1320–1334. doi: 10.1002/glia.21010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina M.D., Sanchez D., Bastiani M.J. Lazarillo, a new GPI-linked surface lipocalin, is restricted to a subset of neurons in the grasshopper embryo. Development. 1995;121:123–134. doi: 10.1242/dev.121.1.123. [DOI] [PubMed] [Google Scholar]
- Gattaz W.F., Kollisch M., Thuren T., Virtanen J.A., Kinnunen P.K. Increased plasma phospholipase-A2 activity in schizophrenic patients: reduction after neuroleptic therapy. Biol. Psychiatry. 1987;22:421–426. doi: 10.1016/0006-3223(87)90164-8. [DOI] [PubMed] [Google Scholar]
- Glass C.K., Saijo K., Winner B., Marchetto M.C., Gage F.H. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O., Thomas C.J., Guarda G., Tschopp J. The inflammasome: an integrated view. Immunol. Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- Grzyb J., Latowski D., Strzalka K. Lipocalins - a family portrait. J. Plant Physiol. 2006;163:895–915. doi: 10.1016/j.jplph.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Gueraud F., Atalay M., Bresgen N., Cipak A., Eckl P.M., Huc L., Jouanin I., Siems W., Uchida K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010;44:1098–1124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- Haigis M.C., Yankner B.A. The aging stress response. Mol. Cell. 2010;40:333–344. doi: 10.1016/j.molcel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Helisalmi S., Hiltunen M., Vepsalainen S., Iivonen S., Corder E.H., Lehtovirta M., Mannermaa A., Koivisto A.M., Soininen H. Genetic variation in apolipoprotein D and Alzheimer's disease. J. Neurol. 2004;251:951–957. doi: 10.1007/s00415-004-0470-8. [DOI] [PubMed] [Google Scholar]
- Horrobin D.F., Bennett C.N. New gene targets related to schizophrenia and other psychiatric disorders: enzymes, binding proteins and transport proteins involved in phospholipid and fatty acid metabolism. Prostaglandins Leukot. Essent. Fatty Acids. 1999;60:141–167. doi: 10.1054/plef.1999.0027. [DOI] [PubMed] [Google Scholar]
- Ji R.R., Xu Z.Z., Strichartz G., Serhan C.N. Emerging roles of resolvins in the resolution of inflammation and pain. Trends. Neurosci. 2011;34:599–609. doi: 10.1016/j.tins.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B.B., Ohlsson A.L. Environment, social interaction, and physical activity as determinants of functional outcome after cerebral infarction in the rat. Exp. Neurol. 1996;139:322–327. doi: 10.1006/exnr.1996.0106. [DOI] [PubMed] [Google Scholar]
- Jones H.M., Pilowsky L.S. Dopamine and antipsychotic drug action revisited. Br. J. Psychiatry. 2002;181:271–275. doi: 10.1192/bjp.181.4.271. [DOI] [PubMed] [Google Scholar]
- Kalman J., McConathy W., Araoz C., Kasa P., Lacko A.G. Apolipoprotein D in the aging brain and in Alzheimer's dementia. Neurol. Res. 2000;22:330–336. doi: 10.1080/01616412.2000.11740678. [DOI] [PubMed] [Google Scholar]
- Kandel E.R. fifth ed. McGraw-Hill; New York: 2012. Principles of Neural Science. [Google Scholar]
- Kelly-Hayes M. Influence of age and health behaviors on stroke risk: lessons from longitudinal studies. J. Am. Geriatr. Soc. 2010;58(Suppl. 2):S325–S328. doi: 10.1111/j.1532-5415.2010.02915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kety S.S. The cerebral circulation in man. Triangle. 1957;3:47–52. [PubMed] [Google Scholar]
- Khan M.M., Parikh V.V., Mahadik S.P. Antipsychotic drugs differentially modulate apolipoprotein D in rat brain. J. Neurochem. 2003;86:1089–1100. doi: 10.1046/j.1471-4159.2003.01866.x. [DOI] [PubMed] [Google Scholar]
- Kim W.S., Wong J., Weickert C.S., Webster M.J., Bahn S., Garner B. Apolipoprotein-D expression is increased during development and maturation of the human prefrontal cortex. J. Neurochem. 2009;109:1053–1066. doi: 10.1111/j.1471-4159.2009.06031.x. [DOI] [PubMed] [Google Scholar]
- Kirkwood T.B. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Kirkwood T.B., Austad S.N. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- Kirkwood T.B., Rose M.R. Evolution of senescence: late survival sacrificed for reproduction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1991;332:15–24. doi: 10.1098/rstb.1991.0028. [DOI] [PubMed] [Google Scholar]
- Klomsiri C., Karplus P.A., Poole L.B. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S., Donarski N., Goetze K., Kreckel M., Stuerenburg H.J., Buhmann C., Beisiegel U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 2001;42:1143–1151. [PubMed] [Google Scholar]
- Konradi C., Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol. Ther. 2003;97:153–179. doi: 10.1016/s0163-7258(02)00328-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T., Peeper D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- Kukull W.A., Higdon R., Bowen J.D., McCormick W.C., Teri L., Schellenberg G.D., van Belle G., Jolley L., Larson E.B. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch. Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- Lapointe J., Hekimi S. When a theory of aging ages badly. Cell Mol. Life Sci. 2010;67:1–8. doi: 10.1007/s00018-009-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson E.B. Dementia in the elderly: the “silent epidemic” no more. Trans. Am. Clin. Climatol. Assoc. 2001;112:136–146. discussion 46-8. [PMC free article] [PubMed] [Google Scholar]
- Lee C.K., Weindruch R., Prolla T.A. Gene-expression profile of the ageing brain in mice. Nat. Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- Leinonen J.S., Ahonen J.P., Lonnrot K., Jehkonen M., Dastidar P., Molnar G., Alho H. Low plasma antioxidant activity is associated with high lesion volume and neurological impairment in stroke. Stroke. 2000;31:33–39. doi: 10.1161/01.str.31.1.33. [DOI] [PubMed] [Google Scholar]
- Levros L.C., Jr., Do Carmo S., Edouard E., Legault P., Charfi C., Rassart E. Characterization of nuclear factors modulating the apolipoprotein D promoter during growth arrest: implication of PARP-1, APEX-1 and ERK1/2 catalytic activities. Biochimi Biophys. Acta. 2010;1803:1062–1071. doi: 10.1016/j.bbamcr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerch P.M., Lu T., Dakin K.A., Vann J.M., Isaacs A., Geula C., Wang J., Pan Y., Gabuzda D.H., Li C., Prolla T.A., Yankner B.A. Evolution of the aging brain transcriptome and synaptic regulation. PloS One. 2008;3:e3329. doi: 10.1371/journal.pone.0003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T., Finkel T. Free radicals and senescence. Exp. Cell Res. 2008;314:1918–1922. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchtman D.W., Song C. Cognitive enhancement by omega-3 fatty acids from child-hood to old age: findings from animal and clinical studies. Neuropharmacology. 2013;64:550–565. doi: 10.1016/j.neuropharm.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Mahadik S.P., Khan M.M., Evans D.R., Parikh V.V. Elevated plasma level of apolipoprotein D in schizophrenia and its treatment and outcome. Schizophr. Res. 2002;58:55–62. doi: 10.1016/s0920-9964(01)00378-4. [DOI] [PubMed] [Google Scholar]
- Mahley R.W., Weisgraber K.H., Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc. Nat. Acad. Sci. U.S.A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnett L.J. Lipid peroxidation-DNA damage by malondialdehyde. Mutat. Res. 1999;424:83–95. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- Martinez E., Navarro A., Ordonez C., Del Valle E., Tolivia J. Amyloid-beta25-35 induces apolipoprotein D synthesis and growth arrest in HT22 hippocampal cells. J. Alzheimers Dis. 2012;30:233–244. doi: 10.3233/JAD-2012-112102. [DOI] [PubMed] [Google Scholar]
- McConathy W.J., Alaupovic P. Isolation and partial characterization of apolipoprotein D: a new protein moiety of the human plasma lipoprotein system. FEBS Lett. 1973;37:178–182. doi: 10.1016/0014-5793(73)80453-3. [DOI] [PubMed] [Google Scholar]
- McIlwain H., Bachelard H.S. fifth ed. Churchill Livingstone; Edinburgh; New York: 1985. Biochemistry and the Central Nervous System. [Google Scholar]
- Morais Cabral J.H., Atkins G.L., Sanchez L.M., Lopez-Boado Y.S., Lopez-Otin C., Sawyer L. Arachidonic acid binds to apolipoprotein D: implications for the protein's function. FEBS Lett. 1995;366:53–56. doi: 10.1016/0014-5793(95)00484-q. [DOI] [PubMed] [Google Scholar]
- Mossaheb N., Schloegelhofer M., Schaefer M.R., Fusar-Poli P., Smesny S., McGorry P., Berger G., Amminger G.P. Polyunsaturated fatty acids in emerging psychosis. Curr. Pharm. Des. 2012;18:576–591. doi: 10.2174/138161212799316055. [DOI] [PubMed] [Google Scholar]
- Muffat J., Walker D.W., Benzer S. Human ApoD, an apolipoprotein up-regulated in neurodegenerative diseases, extends lifespan and increases stress resistance in Drosophila. Proc. Nat. Acad. Sci. U.S.A. 2008;105:7088–7093. doi: 10.1073/pnas.0800896105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F.L., Lustgarten M.S., Jang Y., Richardson A., Van Remmen H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Naik E., Dixit V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011;208:417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren J., Wieloch T. Enriched environment enhances recovery of motor function after focal ischemia in mice, and downregulates the transcription factor NGFI-A. J. Cereb. Blood Flow Metab. 2005;25:1625–1633. doi: 10.1038/sj.jcbfm.9600157. [DOI] [PubMed] [Google Scholar]
- O'Brien J.S., Sampson E.L. Fatty acid and fatty aldehyde composition of the major brain lipids in normal human gray matter, white matter, and myelin. J. Lipid Res. 1965;6:545–551. [PubMed] [Google Scholar]
- Oakley A.J., Bhatia S., Ecroyd H., Garner B. Molecular dynamics analysis of apolipoprotein-D-lipid hydroperoxide interactions: mechanism for selective oxidation of Met-93. PloS One. 2012;7:e34057. doi: 10.1371/journal.pone.0034057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeppen J., Vaupel J.W. Demography. Broken limits to life expectancy. Science. 2002;296:1029–1031. doi: 10.1126/science.1069675. [DOI] [PubMed] [Google Scholar]
- Perica M.M., Delas I. Essential fatty acids and psychiatric disorders. Nutr. Clinic. Pract. 2011;26:409–425. doi: 10.1177/0884533611411306. [DOI] [PubMed] [Google Scholar]
- Phillis J.W., Horrocks L.A., Farooqui A.A. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res. Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Pizza V., Agresta A., D'Acunto C.W., Festa M., Capasso A. Neuroinflamm-aging and neurodegenerative diseases: an overview. CNS Neurol. Disord. Drug Targets. 2011;10:621–634. doi: 10.2174/187152711796235014. [DOI] [PubMed] [Google Scholar]
- Polidori M.C., Frei B., Cherubini A., Nelles G., Rordorf G., Keaney J.F., Jr., Schwamm L., Mecocci P., Koroshetz W.J., Beal M.F. Increased plasma levels of lipid hydroperoxides in patients with ischemic stroke. Free Radic. Biol. Med. 1998;25:561–567. doi: 10.1016/s0891-5849(98)00085-9. [DOI] [PubMed] [Google Scholar]
- Pratico D., Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer's disease. J. Alzheimers Dis. 2004;6:171–175. doi: 10.3233/jad-2004-6209. [DOI] [PubMed] [Google Scholar]
- Provost P.R., Marcel Y.L., Milne R.W., Weech P.K., Rassart E. Apolipoprotein D transcription occurs specifically in nonproliferating quiescent and senescent fibroblast cultures. FEBS Lett. 1991;290:139–141. doi: 10.1016/0014-5793(91)81244-3. [DOI] [PubMed] [Google Scholar]
- Ramsey M.R., Sharpless N.E. ROS as a tumour suppressor? Nat. Cell Biol. 2006;8:1213–1215. doi: 10.1038/ncb1106-1213. [DOI] [PubMed] [Google Scholar]
- Rassart E., Bedirian A., Do Carmo S., Guinard O., Sirois J., Terrisse L., Milne R. Apolipoprotein D. Biochim. Biophys. Acta. 2000;1482:185–198. doi: 10.1016/s0167-4838(00)00162-x. [DOI] [PubMed] [Google Scholar]
- Ravikumar B., Sarkar S., Davies J.E., Futter M., Garcia-Arencibia M., Green-Thompson Z.W., Jimenez-Sanchez M., Korolchuk V.I., Lichtenberg M., Luo S., Massey D.C., Menzies F.M., Moreau K., Narayanan U., Renna M., Siddiqi F.H., Underwood B.R., Winslow A.R., Rubinsztein D.C. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Reynolds A., Laurie C., Mosley R.L., Gendelman H.E. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol. 2007;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- Rickhag M., Deierborg T., Patel S., Ruscher K., Wieloch T. Apolipoprotein D is elevated in oligodendrocytes in the peri-infarct region after experimental stroke: influence of enriched environment. J. Cereb. Blood Flow Metab. 2008;28:551–562. doi: 10.1038/sj.jcbfm.9600552. [DOI] [PubMed] [Google Scholar]
- Rickhag M., Wieloch T., Gido G., Elmer E., Krogh M., Murray J., Lohr S., Bitter H., Chin D.J., von Schack D., Shamloo M., Nikolich K. Comprehensive regional and temporal gene expression profiling of the rat brain during the first 24 h after experimental stroke identifies dynamic ischemia-induced gene expression patterns, and reveals a biphasic activation of genes in surviving tissue. J. Neurochem. 2006;96:14–29. doi: 10.1111/j.1471-4159.2005.03508.x. [DOI] [PubMed] [Google Scholar]
- Rose M.R. Oxford University Press; New York: 1991. Evolutionary Biology of Aging. [Google Scholar]
- Ross B.M., Turenne S., Moszczynska A., Warsh J.J., Kish S.J. Differential alteration of phospholipase A2 activities in brain of patients with schizophrenia. Brain Res. 1999;821:407–413. doi: 10.1016/s0006-8993(99)01123-3. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D.C., Marino G., Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Ruiz M., Sanchez D., Correnti C., Strong R.K., Ganfornina M.D. Lipid-binding properties of human ApoD and Lazarillo-related lipocalins: functional implications for cell differentiation. FEBS J. 2013;280:3928–3943. doi: 10.1111/febs.12394. [DOI] [PubMed] [Google Scholar]
- Ruiz M., Wicker-Thomas C., Sanchez D., Ganfornina M.D. Grasshopper Lazarillo, a GPI-anchored lipocalin, increases Drosophila longevity and stress resistance, and functionally replaces its secreted homolog NLaz. Insect Biochem. Mol. Biol. 2012;42:776–789. doi: 10.1016/j.ibmb.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Salminen A., Ojala J., Kaarniranta K., Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol. Life Sci. 2012;69:2999–3013. doi: 10.1007/s00018-012-0962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez D., Ganfornina M.D., Gutierrez G., Marin A. Exon-intron structure and evolution of the Lipocalin gene family. Mol. Biol. Evol. 2003;20:775–783. doi: 10.1093/molbev/msg079. [DOI] [PubMed] [Google Scholar]
- Sanchez D., Ganfornina M.D., Torres-Schumann S., Speese S.D., Lora J.M., Bastiani M.J. Characterization of two novel lipocalins expressed in the Drosophila embryonic nervous system. Int. J. Dev. Biol. 2000;44:349–359. [PubMed] [Google Scholar]
- Sanchez D., Lopez-Arias B., Torroja L., Canal I., Wang X., Bastiani M.J., Ganfornina M.D. Loss of glial lazarillo, a homolog of apolipoprotein D, reduces lifespan and stress resistance in Drosophila. Curr. Biol. 2006;16:680–686. doi: 10.1016/j.cub.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Shibata N., Nagata T., Shinagawa S., Ohnuma T., Shimazaki H., Komatsu M., Kuerban B., Tomson K., Nakayama K., Yamada H., Arai H. Genetic association between APOA1 and APOD polymorphisms and Alzheimer's disease in a Japanese population. J. Neural Transm. 2013 doi: 10.1007/s00702-013-1036-7. [DOI] [PubMed] [Google Scholar]
- Skosnik P.D., Yao J.K. From membrane phospholipid defects to altered neurotransmission: is arachidonic acid a nexus in the pathophysiology of schizophrenia? Prostaglandins Leukot. Essent. Fatty Acids. 2003;69:367–384. doi: 10.1016/j.plefa.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Sohal R.S., Orr W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012;52:539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal R.S., Sohal B.H. Hydrogen peroxide release by mitochondria increases during aging. Mech. Ageing Dev. 1991;57:187–202. doi: 10.1016/0047-6374(91)90034-w. [DOI] [PubMed] [Google Scholar]
- Sokoloff L. Quantitative measurements of cerebral blood flow in man. Methods Med. Res. 1960;8:253–261. [PubMed] [Google Scholar]
- Speakman J.R., Selman C. The free-radical damage theory: accumulating evidence against a simple link of oxidative stress to ageing and lifespan. Bioessays. 2011;33:255–259. doi: 10.1002/bies.201000132. [DOI] [PubMed] [Google Scholar]
- Strittmatter W.J., Saunders A.M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G.S., Roses A.D. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Nat. Acad. Sci. U.S.A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe J.G., Thomas E.A. The neurobiology of apolipoproteins in psychiatric disorders. Mol. Neurobiol. 2002;26:369–388. doi: 10.1385/mn:26:2-3:369. [DOI] [PubMed] [Google Scholar]
- Tchkonia T., Zhu Y., van Deursen J., Campisi J., Kirkland J.L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrisse L., Poirier J., Bertrand P., Merched A., Visvikis S., Siest G., Milne R., Rassart E. Increased levels of apolipoprotein D in cerebrospinal fluid and hippocampus of Alzheimer's patients. J. Neurochem. 1998;71:1643–1650. doi: 10.1046/j.1471-4159.1998.71041643.x. [DOI] [PubMed] [Google Scholar]
- Thomas E.A., Danielson P.E., Nelson P.A., Pribyl T.M., Hilbush B.S., Hasel K.W., Sutcliffe J.G. Clozapine increases apolipoprotein D expression in rodent brain: towards a mechanism for neuroleptic pharmacotherapy. J. Neurochem. 2001;76:789–796. doi: 10.1046/j.1471-4159.2001.00027.x. [DOI] [PubMed] [Google Scholar]
- Thomas E.A., Dean B., Pavey G., Sutcliffe J.G. Increased CNS levels of apolipoprotein D in schizophrenic and bipolar subjects: implications for the pathophysiology of psychiatric disorders. Proc. Nat. Acad. Sci. U.S.A. 2001;98:4066–4071. doi: 10.1073/pnas.071056198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas E.A., Dean B., Scarr E., Copolov D., Sutcliffe J.G. Differences in neuroanatomical sites of apoD elevation discriminate between schizophrenia and bipolar disorder. Mol. Psychiatry. 2003;8:167–175. doi: 10.1038/sj.mp.4001223. [DOI] [PubMed] [Google Scholar]
- Thomas E.A., George R.C., Sutcliffe J.G. Apolipoprotein D modulates arachidonic acid signaling in cultured cells: implications for psychiatric disorders. Prostaglandins leukot. Essent. Fatty Acids. 2003;69:421–427. doi: 10.1016/j.plefa.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Thomas E.A., Sautkulis L.N., Criado J.R., Games D., Sutcliffe J.G. Apolipoprotein D mRNA expression is elevated in PDAPP transgenic mice. J. Neurochem. 2001;79:1059–1064. doi: 10.1046/j.1471-4159.2001.00654.x. [DOI] [PubMed] [Google Scholar]
- Tsukamoto K., Mani D.R., Shi J., Zhang S., Haagensen D.E., Otsuka F., Guan J., Smith J.D., Weng W., Liao R., Kolodgie F.D., Virmani R., Krieger M. Identification of apolipoprotein D as a cardioprotective gene using a mouse model of lethal atherosclerotic coronary artery disease. Proc. Nat. Acad. Sci. 2013;110:17023–17028. doi: 10.1073/pnas.1315986110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kemp W.J., Klomp D.W., Kahn R.S., Luijten P.R., Hulshoff Pol H.E. A meta-analysis of the polyunsaturated fatty acid composition of erythrocyte membranes in schizophrenia. Schizophr. Res. 2012;141:153–161. doi: 10.1016/j.schres.2012.08.014. [DOI] [PubMed] [Google Scholar]
- van Os J., Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- Vogt M., Skerra A. Bacterially produced apolipoprotein D binds progesterone and arachidonic acid, but not bilirubin or E-3M2H. J. Mol. Recognit. 2001;14:79–86. doi: 10.1002/1099-1352(200101/02)14:1<79::AID-JMR521>3.0.CO;2-4. 1<79::AID-JMR521>3.0.CO;2–4. [DOI] [PubMed] [Google Scholar]
- Walker D.W., Muffat J., Rundel C., Benzer S. Overexpression of a Drosophila homolog of apolipoprotein D leads to increased stress resistance and extended lifespan. Curr. Biol. 2006;16:674–679. doi: 10.1016/j.cub.2006.01.057. [DOI] [PubMed] [Google Scholar]
- Wang C.H., Wu S.B., Wu Y.T., Wei Y.H. Oxidative stress response elicited by mitochondrial dysfunction: implication in the pathophysiology of aging. Exp. Biol. Med. (Maywood) 2013;238:450–460. doi: 10.1177/1535370213493069. [DOI] [PubMed] [Google Scholar]
- Wang L., Zhang S., Liu Z., Li H., Wang Y., Jiang S. Characterization and expression of amphioxus ApoD gene encoding an archetype of vertebrate ApoD proteins. Cell Biol. Int. 2007;31:74–81. doi: 10.1016/j.cellbi.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Weech P.K., Provost P., Tremblay N.M., Camato R.N., Milne R.W., Marcel Y.L., Rassart E. Apolipoprotein D–an atypical apolipoprotein. Prog. Lipid Res. 1991;30:259–266. doi: 10.1016/0163-7827(91)90023-x. [DOI] [PubMed] [Google Scholar]
- Wieloch T., Nikolich K. Mechanisms of neural plasticity following brain injury. Curr. Opin. Neurobiol. 2006;16:258–264. doi: 10.1016/j.conb.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Wilmoth J.R. Demography of longevity: past, present, and future trends. Exp. Gerontol. 2000;35:1111–1129. doi: 10.1016/s0531-5565(00)00194-7. [DOI] [PubMed] [Google Scholar]
- Winterbourn C.C., Hampton M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Yang C.Y., Gu Z.W., Blanco-Vaca F., Gaskell S.J., Yang M., Massey J.B., Gotto A.M., Jr., Pownall H.J. Structure of human apolipoprotein D: locations of the intermolecular and intramolecular disulfide links. Biochemistry. 1994;33:12451–12455. doi: 10.1021/bi00207a011. [DOI] [PubMed] [Google Scholar]
- Yankner B.A., Lu T., Loerch P. The aging brain. Annu Rev. Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- Yehuda S., Rabinovitz S., Carasso R.L., Mostofsky D.I. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiol. Aging. 2002;23:843–853. doi: 10.1016/s0197-4580(02)00074-x. [DOI] [PubMed] [Google Scholar]
- Yin H., Xu L., Porter N.A. Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev. 2011;111:5944–5972. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- Youdim K.A., Martin A., Joseph J.A. Essential fatty acids and the brain: possible health implications. Int. J. Dev. Neurosci. 2000;18:383–399. doi: 10.1016/s0736-5748(00)00013-7. [DOI] [PubMed] [Google Scholar]
- Zamberletti E., Rubino T., Parolaro D. The endocannabinoid system and schizophrenia: integration of evidence. Curr. Pharm. Des. 2012;18:4980–4990. doi: 10.2174/138161212802884744. [DOI] [PubMed] [Google Scholar]
- Zglinicki T.v. Kluwer Academic; Dordrecht; Boston: 2003. Aging at the Molecular Level. [Google Scholar]
- Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]