

Abstract
RNA-binding proteins (RBPs) are key players in the post-transcriptional regulation of gene expression. Precise knowledge about their binding sites is therefore critical to unravel their molecular function and to understand their role in development and disease. Individual-nucleotide resolution UV crosslinking and immunoprecipitation (iCLIP) identifies protein–RNA crosslink sites on a genome-wide scale. The high resolution and specificity of this method are achieved by an intramolecular cDNA circularization step that enables analysis of cDNAs that truncated at the protein–RNA crosslink sites. Here, we describe the improved iCLIP protocol and discuss critical optimization and control experiments that are required when applying the method to new RBPs.
Keywords: iCLIP, UV crosslinking and immunoprecipitation (CLIP), Protein–RNA interaction, High-throughput sequencing, RNA-binding protein, RNA, Post-transcriptional regulation
1. Introduction
Post-transcriptional regulation critically contributes to the ability of cells to adjust gene expression in the face of a changing external or internal environment [1]. RNA-binding proteins (RBPs) are the primary regulatory factors of the various post-translational stages, including alternative splicing, polyadenylation, mRNA localization, translation and degradation [2]. Their RNA binding is mediated by modular RNA-binding domains (RBDs), such as the RNA recognition motif (RRM), hnRNP K-homology domain or zinc fingers (Znf) [3]. Although many RBDs recognize RNA in a sequence-specific manner, sequence information is not sufficient to reliably predict RBP binding sites throughout the transcriptome. In particular, RBPs cooperate and compete when binding to RNA; therefore it is crucial to study protein–RNA interactions in the cellular environment.
The first approaches to investigate protein–RNA complexes in vivo employed affinity purification or immunoprecipitation combined with microarray analysis (RIP-CHIP). However, these approaches were prone to identifying non-physiologic or indirect interactions and their low resolution made it difficult to narrow down actual binding sites [4,5]. Development of in vivo UV-crosslinking and immunoprecipitation (CLIP) enabled the study of protein–RNA interactions with high positional resolution and specificity. Combined with high-throughput sequencing, CLIP became the standard tool for the genome-wide analysis of protein–RNA interactions [6].
In the original CLIP approach, reverse transcription needs to proceed from a universal 3′ ligated adapter to a universal 5′ ligated adapter, since both adapters are required for PCR amplification. However, in over 80% of cases, the reverse transcriptase stalls at the short polypeptide left at the UV-induced crosslink site, resulting in truncated cDNAs that lack the 5′ adapter, and are therefore not amplified in CLIP [7]. We previously developed individual-nucleotide resolution CLIP (iCLIP), which enables PCR amplification of truncated cDNAs, and thereby identifies protein–RNA crosslink sites with nucleotide resolution (Fig. 1) [8]. iCLIP has been successfully used to study the function of RBPs in alternative splicing [8–13], alternative polyadenylation [14], RNA methylation [15] and mRNA stability [16]. It determined high-resolution RNA splicing maps of different RBPs, which enabled to assess how the position of RBP binding around alternative exons determines their splicing function [8–13,17]. Importantly, in addition to identifying the RBP binding sites of an RBP, iCLIP can also quantitate genome-wide changes in protein–RNA interactions; for instance, it was demonstrated that the splicing factor U2AF65 gains access to hundreds of Alu elements after knockdown of hnRNP C, which prevents their erroneous recognition under normal conditions [18].
Fig. 1.

Schematic representation of the iCLIP procedure identifying RNA–protein interactions in intact cells. Cells are irradiated with UV-C light on ice, leading to formation of a covalent bond between protein and RNA. This is followed by partial RNase digestion and an immunoprecipitation with protein-specific antibodies. For the library preparation and visualization, the RNA is dephosphorylated, a 3′ end adapter is ligated and the 5′ end is radioactively labeled. The complexes are separated by SDS–PAGE and isolated from a nitrocellulose membrane according to the expected size. The protein is then digested by proteinase K, and reverse transcription (RT) is performed truncating at the remaining polypeptide. The RT primer introduces two cleavable adapter regions and barcode sequences. The free RT primers are removed by size selection and circularization of the cDNA is carried out. Linearization generates suitable templates for PCR amplification. In the last step, high-throughput sequencing generates reads in which the barcode sequences are immediately followed by the last nucleotide of the cDNA.
The iCLIP protocol starts with UV irradiation, which forms covalent bonds at sites of protein–RNA interactions and thereby preserves the in vivo binding pattern (Fig. 1, step 1). Next, the cells are lysed and the RNAs are partially digested to obtain RNA fragments in an optimal size range (Fig. 1, steps 2 and 3). After immunoprecipitation of the protein–RNA complexes, the RNA is dephosphorylated, an adapter is ligated to the 3′ end of the RNA and the 5′ end is radioactively labeled (Fig. 1, steps 4–7). SDS–PAGE and transfer to nitrocellulose membrane enable to remove free RNA and to stringently purify the crosslinked protein–RNA complexes (Fig. 1, step 8). The RNA is recovered from the nitrocellulose membrane by digesting the protein with proteinase K, which leaves a polypeptide at the crosslink site (Fig. 1, step 9). The RNA is then reverse transcribed into cDNA, which most often truncates at the polypeptide remaining at the crosslink site (Fig. 1, step 10). Size selection of the cDNA removes free reverse transcription (RT) primers, and is followed by cDNA circularization, which attaches the second adapter to the 3′ end of cDNA (Fig. 1, steps 11–13). Restriction enzyme digestion linearizes the cDNA before PCR amplification (Fig. 1, steps 14 and 15). To increase the quantitative power of the method, the reverse transcription primers contain a randomized sequence (random barcode), which enables computational filtering of artifacts caused by variable PCR amplification of the cDNAs [19]. After high-throughput sequencing, the barcode sequence precedes the cDNA sequence. Truncated cDNAs represent over 80% of the cDNA library, and after mapping their sequence to the genome, the position of the preceding nucleotide corresponds to the crosslinking site [7].
In the following section, we provide a detailed description of required materials and experimental procedures of the iCLIP protocol. In particular, we introduce important controls and optimization steps including some example experiments. Throughout the protocol, we point out critical steps and give advice on how to optimize them.
2. Materials
2.1. Buffers for standard iCLIP protocol
| Buffer | Ingredients |
|---|---|
| Lysis buffer | 50 mM Tris–HCl, pH 7.4 |
| 100 mM NaCl | |
| 1% Igepal CA-630 | |
| 0.1% SDS | |
| 0.5% sodium deoxycholate | |
| On the day: 1/100 volume of Protease Inhibitor Cocktail Set III, for tissues: 1/1000 volume of ANTI-RNase | |
| High-salt Wash | 50 mM Tris–HCl, pH 7.4 |
| 1 M NaCl | |
| 1 mM EDTA | |
| 1% Igepal CA-630 | |
| 0.1% SDS | |
| 0.5% sodium deoxycholate | |
| PNK buffer | 20 mM Tris–HCl, pH 7.4 |
| 10 mM MgCl2 | |
| 0.2% Tween-20 | |
| 5x PNK pH 6.5 buffer | 350 mM Tris–HCl, pH 6.5 |
| 50 mM MgCl2 | |
| 5 mM dithiothreitol | |
| Freeze aliquots of the buffer, do not thaw and freeze again | |
| 4x Ligation buffer | 200 mM Tris–HCl, pH 7.8 |
| 40 mM MgCl2 | |
| 4 mM dithiothreitol | |
| Freeze aliquots of the buffer, do not thaw and freeze again | |
| PK buffer | 100 mM Tris–HCl, pH 7.4 |
| 50 mM NaCl | |
| 10 mM EDTA | |
| PK buffer + 7 M urea | 100 mM Tris–HCl, pH 7.4 |
| 50 mM NaCl | |
| 10 mM EDTA | |
| 7 M urea |
2.2. Materials for UV crosslinking and immunoprecipitation
| Material | Manufacturer |
|---|---|
| Stratalinker UV crosslinker 2400 | Stratagene |
| 4-Thiouridine | Sigma (T4509) |
| Protein G Dynabeads | Life Technologies (10004D) |
| Protein A Dynabeads | Life Technologies (10002D) |
| Protease Inhibitor Cocktail Set III | Calbiochem/Merck (539134-1SET) |
| ANTI-RNase | Life Technologies (AM2692) |
| RNase I | Life Technologies (AM2295) |
| Turbo DNase | Life Technologies (AM2238) |
| T4 PNK plus 10x PNK buffer | NEB (M0201L) |
| RNasin | Promega (N2515) |
| Proteus Clarification Mini Spin Columns | Generon (GEN-MSF500) |
| T4 RNA Ligase I | NEB (M0204L) |
| Pre-adenylated adapter L3-App | IDT (rAppAGATCGGAAGAGCGGTTCAG/ddC/) |
| ATP [γ-32P] | Perkin Elmer (NEG502A250UC) |
2.3. SDS PAGE and nitrocellulose transfer
| Material | Manufacturer |
|---|---|
| 4–12% NuPAGE gels | Life Technologies (NP0322BOX) |
| Electrophoresis chamber | Life Technologies (EI0002) |
| Transfer apparatus | Life Technologies (EI0002) |
| LDS-4x sample buffer | Life Technologies (NP0007) |
| Pre-stained protein marker | Fermentas (SM1811) |
| Nitrocellulose membrane protran BA85 | VWR (732-4174) |
| Sponge pads for XCell II blotting | Life Technologies (EI9052) |
| 20 × transfer buffer | Life Technologies (NP0006-1) |
| 20 × MOPS-SDS running buffer | Life Technologies (NP0001) |
| Whatman filter paper | GE Healthcare (3030917) |
| Film | Fuji (4741019236) |
2.4. RNA isolation
| Material | Manufacturer |
|---|---|
| Proteinase K | Fisher Scientific (YSJ-762-Q) |
| 19G syringe needles | BD Microlance (304000) |
| Phenol/chloroform | Sigma (P3803) |
| Phase lock gel heavy tube | VWR (713-2536) |
| Glycoblue | Ambion (9510) |
| 3 M sodium acetate pH 5.5 | Life Technologies (AM9740) |
2.5. Reverse transcription
| Material | Manufacturer |
|---|---|
| PCR tubes | VWR (732-0545) |
| dNTPs | Promega (1009310) |
| Superscript III | Life Technologies (18080085) |
| 5X First-strand buffer | Life Technologies (18080085) |
| DTT | Life Technologies (18080085) |
| 1 M HEPES pH 7.3 | Thermo Scientific (BP299-500) |
| TE buffer | VWR (A2575) |
| Name | Sequence (IDT) |
|---|---|
| Rt1clip | /5Phos/NNAACCNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt2clip | /5Phos/NNACAANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt3clip | /5Phos/NNATTGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt4clip | /5Phos/NNAGGTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt6clip | /5Phos/NNCCGGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt7clip | /5Phos/NNCTAANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt8clip | /5Phos/NNCATTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt9clip | /5Phos/NNGCCANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt11clip | /5Phos/NNGGTTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt12clip | /5Phos/NNGTGGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt13clip | /5Phos/NNTCCGNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt14clip | /5Phos/NNTGCCNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt15clip | /5Phos/NNTATTNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
| Rt16clip | /5Phos/NNTTAANNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC |
2.6. cDNA isolation
| Material | Manufacturer |
|---|---|
| 2x TBE – urea loading buffer | Life Technologies (LC6876) |
| 6% TBE – urea pre-cast gels | Life Technologies (EC68652B) |
| Low molecular weight marker | NEB (N3233L) |
| TBE running buffer | Life Technologies (LC6675) |
| SYBR green II | Life Technologies (S-7564) |
| 19G syringe needle | BD Microlance (300637) |
| Glass pre-filters | Whatman (1823010) |
| Phase lock gel heavy | VWR (713-2536) |
| Costar SpinX column | Corning Incorporated (8161) |
2.7. Ligation of the primer to the 5′ end of the cDNA
| Material | Manufacturer |
|---|---|
| 10x CircLigase buffer | Cambio (CL9025K) |
| CircLigase II | Cambio (CL9025K) |
| MnCl2 | Cambio (CL9025K) |
| Cut_oligo | GTTCAGGATCCACGACGCTCTTCaaaa |
| BamHI | Fermentas, FD0055 |
| Fast digest buffer | Fermentas, FD0055 |
2.8. PCR amplifcation
| Material | Manufacturer |
|---|---|
| Accuprime Supermix I | Life Technologies (12342028) |
| SYBR green I | Life Technologies (S-7563) |
| P5 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| P3 | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT |
2.9. qPCR quantitation
| Material | Manufacturer |
|---|---|
| Accuprime Supermix I | Life Technologies (12342028) |
| PhiX control library (RNAseq/ChIPseq) | Illumina (PC-110-3001) |
| Platinium Taq mastermix | Life Technologies (11743100) |
| DLP | [6FAM] CCCTACACGACGCTCTTCCGATCT [TAMRA] |
| Primer 1 | AATGATACGGCGACCACCGAGATC |
| Primer 2 | CAAGCAGAAGACGGCATACGAGATC |
2.10. Buffers for stringent urea iCLIP protocol
| Buffer | Ingredients |
|---|---|
| Urea cracking buffer | 50 mM Tris–HCl, pH 7.4 |
| 6 M urea | |
| 1% SDS | |
| 25% PBS | |
| T-20 IP buffer | 50 mM Tris–HCl, pH 7.4 |
| 150 mM NaCl | |
| 0.5% Tween 20 | |
| 0.1 mM EDTA |
2.11. Stringent urea iCLIP
| Material | Manufacturer |
|---|---|
| Dynabeads Antibody Coupling Kit | Life Technologies (14311D) |
| SUPERase• in™ RNase inhibitor | Life Technologies (AM2694) |
| NuPAGE sample reducing agent | Life Technologies (NP0004) |
| NuPAGE antioxidant | Life Technologies (NP0005) |
| Linear acrylamide | Life Technologies (AM9520) |
3. iCLIP protocol
In this manuscript, we provide a detailed iCLIP protocol with comments, tips and explanations provided in the footnotes. For the most critical steps, we describe the results of example experiments at the end of the subchapters. We also provide a table summarizing the recent improvements and quality assessments, which facilitate an optimal library preparation for your protein of interest (Table 1). We present tests for various critical steps including dephosphorylation, sonication and alkaline hydrolysis. We also describe how to optimize the concentration of RNase I, and test the efficiency of reverse transcription primers. This optimized protocol enables the user to overcome the technical challenges of iCLIP, and ensures the comprehensive identification of protein–RNA interactions.
Table 1.
Improvements compared to the previous protocol, which was published in the Journal of Visualized Experiments [19].
| Original protocol | Improved protocol | Reason for the change | Benefit of the change |
|---|---|---|---|
| 25 and 40 mM dithiothreitol in the 5x PNK pH 6.5 and the 4x ligation buffer, respectively | 5 and 4 mM dithiothreitol in the 5x PNK pH 6.5 and the 4x ligation buffer, respectively | High concentration of DTT can decrease the immunoprecipitation efficiency | Increased recovery of protein–RNA complexes, as visualized on the radiograph |
| No 4-thiouridine pre-incubation | Optional 4-thiouridine pre-incubation and UV-A crosslinking | Special modifications are required for certain proteins | 4-thiouridine enhances crosslinking of certain proteins |
| Optional sonication | Use of bioruptor enables uniform sonication of multiple samples | Sonication shears the DNA, which releases proteins from the chromatin | Increased recovery of protein–RNA complexes, as visualized on the radiograph (Fig. 2D) |
| Use phenol/chloroform pH 4.5 | Use phenol/chloroform pH 6.7 | L3 is a DNA adapter ligated to the RNA creating an DNA–RNA hybrid | Reduction of DNA-RNA hybrids in phenol phase |
| Radioactive RNA carried over into PCR | Alkaline hydrolysis degrades RNA after reverse transcription | The carryover RNA is radioactive and might inhibit cDNA circularization | Reduction of radioactive signal after reverse transcription (Fig. 4B) |
| No phenol/chloroform extraction after cDNA recovery | Additional phenol/ chloroform extraction after cDNA recovery from TBE-urea gel | Carryover of urea and polyacrylamide after gel extraction can decrease CircLigase II efficiency | Increased efficiency of cDNA circularization due to removal of potential contaminants |
| Preparative PCR in the same volume as the PCR optimization | Increased the volume of the preparative PCR | High cDNA concentration in the preparative PCR can introduce salts that may inhibit PCR amplification | Consistent efficiency of PCR amplification |
| No stringent urea treatment | Optional stringent purification strategy with urea | Special modifications are required for certain proteins | Removal of co-purification of RBPs that strongly interact with the protein of interest (Fig. 5) |
Moreover, we provide protocols for two alternative procedures. In Section 3.1.2, we describe pre-incubation of cells with photo-reactive ribonucleosides, such as 4-thiouridine, which enables crosslinking with UV-A rather than UV-C light [20]. Since pre-incubation of cells with 4-thiouridine enhances crosslinking of some proteins [21], we tested and integrated it as an alternative entry point into the iCLIP protocol (PAR-iCLIP). In Section 3.10, we describe a stringent purification strategy that employs denaturation with urea, which can be used to avoid co-purification of RBPs that strongly interact with the protein of interest.
3.1. Protein–RNA crosslinking
This chapter describes the experimental steps for UV-C or UV-A crosslinking. All further steps of the iCLIP protocol (3.2–3.10) are the same, regardless of the crosslinking procedure. We estimate that for a standard experiment (one 10 cm diameter dish), we use ∼8–10 million HeLa cells or ∼10–12 million HEK293 cells. The input for other cell lines needs to be optimized.
3.1.1. UV-C crosslinking
-
•
After culturing adherent cells in a 10 cm diameter plate, remove the medium, add 6 ml of ice-cold PBS and place on ice.
-
•
Irradiate cells with 150 mJ/cm2 at 254 nm in a Stratalinker 2400 or an equivalent instrument.1
-
•
Scrape off the cells with cell lifters.
-
•
Transfer the cell suspension into three 2 ml microtubes. Centrifuge at 514g at 4 °C for 1 min to pellet cells, and then remove supernatant.
-
•
Snap-freeze the cell pellets on dry ice and store at −80 °C until further use. Continue with step 3.2 or 3.10.
3.1.2. 4-Thiouridine labeling and UV-A crosslinking (alternative to step 3.1.1)
-
•
Add 50 μl of 4SU (stock concentration: 100 mM 4SU) to a 10 cm plate of cells cultured in 10 ml DMEM supplemented with FBS and Pen/Strep to get a final concentration of 500 μM 4SU [22].2 Alternatively, add 10 μl of 4-thiouridine (stock concentration: 100 mM) to get a final concentration of 100 μM 4SU [23].
-
•
Incubate the cells with 4-thiouridine for 60 min at 500 μM or for 8 h at 100 μM; afterward check the viability of your cells under the microscope.
-
•
Aspirate the medium and add 6 ml of ice-cold PBS to cells growing in a 10 cm plate (enough for three immunoprecipitations). Remove the lid and place on ice.
-
•
Irradiate once with 2 × 400 mJ/cm2 in a Stratalinker 2400 with 365 nm bulbs, or an equivalent instrument.
-
•
Scrape off the cells with cell lifters.
-
•
Transfer the cell suspension into three 2 ml microtubes. Centrifuge at 514g at 4 °C for 1 min to pellet cells, and then remove supernatant.
-
•
Snap-freeze the cell pellets on dry ice and store at −80 °C until further use. Continue with step 3.2.
Test: crosslinking technique
Pre-incubation of cells with photo-reactive ribonucleosides, such as 4-thiouridine or 6-thioguanosine, enables crosslinking with UV-A light as commonly used in photoactivatable-ribonucleoside-enhanced CLIP (PAR-CLIP) [20], which can enhance the crosslinking efficiency for certain RBPs [21]. However, excess incorporation of these nucleotide analogs can impair cellular function.
In order to test if photo-activatable nucleosides affect cell viability, we measured the proliferation of HeLa and HEK293 cells upon 6-thioguanosine or 4-thiouridine incubation (Fig. 2A and B). We seeded the same number of cells for all conditions, let them settle for a day and then added either 4-thiouridine or 6-thioguanosine. The viable and dead cells were counted after a period of 0–96 h using trypan blue staining, and the number of viable cells was normalized by the initial number of seeded cells.
Fig. 2.

Tests using photo-reactive nucleosides and sonication. (A) Increasing concentrations of 4-thiouridine (4SU) or 6-thioguanosine (6GU) lead to cell death in HeLa cells. HeLa cells were treated with 4SU (4SU; 25 μM, 100 μM and 500 μM) or 6SG (6SG; 25 μM, 100 μM) over a time course of 96 h. Viable cells were counted using trypan blue staining at different time points, and normalized to the initial cell numbers (depicted as log2). Dead cells were counted at each time point using trypan blue and displayed as percentage of total cell numbers at this time point. (B) Increasing concentrations of 4SU or 6SG inhibit growth of HEK293 cells. HEK293 cells were treated with 4SU (25 μM, 100 μM and 500 μM) or 6SG (5 μM, 25 μM, 100 μM) over a time course of 96 or 48 h, respectively. Display as in (A). (C) hnRNP C can be crosslinked with either UV-C or UV-A light in combination with 4SU. Autoradiograph showing radioactively labeled hnRNP C protein–RNA complexes. 4SU incubation for 30 and 60 min was followed by crosslinking by UV-A light and compared to UV-C crosslinking. Samples without antibody serve as controls. The size of uncrosslinked hnRNP C protein is labeled on the right. (D) Autoradiograph of radioactively labeled protein–RNA complexes immunoprecipitated with antibodies against endogenous TIA1 or hnRNP C. Since sonication can free chromatin-associated proteins, samples were either sonicated or left untreated before immunoprecipitation, and RNA was digested with a 1/50 dilution of RNase I. Samples without antibody serve as controls. Sizes of uncrosslinked proteins are labeled on the right. The asterisk indicates the hnRNP C dimer.
These experiments indicated that the incubation with photo-reactive nucleosides is toxic for the cells. Proliferation of HEK293 cells was stalled when incubated with 100 μM 4-thiouridine or 25 μM of 6-thioguanosine, and the number of HeLa cells decreased at these concentrations, suggesting that photo-reactive nucleosides lead to decreased proliferation in HEK293, and cell death in HeLa cells (Fig. 2A and B). Only the lowest concentration (25 μM of 4-thiouridine or 5 μM of 6-thioguanosine) allowed a limited proliferation of HEK293 cells (Fig. 2A).
In order to minimize the toxic effects of photo-reactive nucleosides, the optimal duration of incubation with the photo-activatable nucleoside should be determined. Combining 4-thiouridine-enhanced UV-A crosslinking with the iCLIP protocol (PAR-iCLIP), we used immunoprecipitation of the RNA-binding protein hnRNP C as a reference to estimate the optimal 4-thiouridine incubation period (Fig. 2C). HEK293 cells were incubated for 30 or 60 min with 4-thiouridine, followed by UV-A crosslinking. For the labeling of nascent RNA, short incubations were chosen with a higher concentration of 4-thiouridine [22]. For comparison, untreated cells were crosslinked using standard UV-C irradiation. All experiments were performed under low RNase conditions (see below). 30 and 60 min of incubation with 4-thiouridine recovered equal amounts of radioactive protein–RNA complexes, which was similar to the amount recovered from UV-C treated cells (Fig. 2C). Independent of the crosslinking technique, a radioactive smear was seen extending upwards from about 40 kDa (close to the molecular weight of hnRNP C), which represents proteins crosslinked to RNA fragments of varying lengths. Control experiments omitting the antibody confirmed that non-specific background signal is absent when using the iCLIP purification conditions with either crosslinking method. We conclude that the iCLIP protocol is appropriate for studies of either UV-A or UV-C crosslinked protein–RNA complexes. While UV-A does not increase the efficiency of hnRNP C crosslinking, use of photo-activatable nucleoside and UV-A crosslinking may increase crosslinking efficiency for other RBPs [20]. It is important to keep in mind that hnRNP C is a predominantly nuclear RBP, which mainly binds to pre-mRNA. When studying cytoplasmic RBPs, prolonged incubation with 4-thiouridine might be required in order to enable accumulation of labeled RNAs in the cytoplasm. For the analysis of PAR-iCLIP data, it is important to keep in mind that this protocol may require adjustment of the mapping procedure, allowing an additional mismatch to accommodate the frequent T-to-C transition in the reads (for UV-C crosslinking experiments, we usually allow max. two mismatches within a read). A thorough analysis of the relative occurrence of transitions versus truncations in PAR-iCLIP remains to be done.
3.2. Immunoprecipitation
3.2.1. Bead preparation
-
•
Add 100 μl of protein G Dynabeads per experiment to a fresh microtube (use protein A Dynabeads for rabbit antibodies).
-
•
Wash beads twice with lysis buffer (without protease inhibitor). This and all following washing steps of the beads are performed with 900 μl of the respective buffer.
-
•
Resuspend the beads in 100 μl lysis buffer with 2–10 μg antibody per experiment.3
-
•
Rotate tubes for 30–60 min at room temperature.
-
•
Wash 3 times with 900 μl lysis buffer and leave in the last wash until ready to proceed to step 3.2.3.
3.2.2. Cell lysis and partial RNA digestion
-
•
Resuspend the cell pellet in 1 ml lysis buffer (with 1:100 Protease Inhibitor Cocktail Set III) and transfer to a 1.5 ml microtube.4 Prepare enough lysate for two experimental samples (low and high RNase) and appropriate controls.5
-
•
Sonication of samples (Optional).
EITHER
-
•
Sonicate the sample on ice using a probe sonicator. The probe should be approximately 0.5 cm from the bottom of the tube and not touching the tube sides in order to avoid foaming. Sonicate twice with 10 s bursts at five decibels. Clean the probe by sonicating H2O before and after sample treatment.
OR
-
•
Use Bioruptor plus for five cycles with alternating 30 s on/30 s off at low intensity.
-
•
Prepare two dilutions of RNase I in PBS6; we recommend to initially use a 1:50 dilution for high RNase and a 1:250 dilution for low RNase, but this needs to be carefully optimized (see Fig. 3A and comments at the end of this subchapter).
-
•
Add 10 μl of low or high RNase I dilution and 2 μl Turbo DNase to the cell lysate and immediately place the samples at 37 °C for 3 min, shaking at 1100 rpm. Immediately afterward transfer to ice for >3 min.
-
•
Centrifuge for 10 min at 22,000g and at 4 °C to clear the lysate. Carefully collect the supernatant (leave about 50 μl lysate on the pellet to prevent carryover).
-
•
Optional: load 500 μl of the lysate onto a Proteus Clarification Mini Spin Column. Centrifuge for 1 min at 22,000g at 4 °C. Transfer flow-through to a new tube and place on ice. Repeat for the rest of the lysate and combine the fractions.7
Fig. 3.

Tests for dephosphorylation and partial RNase digestion. (A) Optimization of RNase I concentration is crucial to obtain optimal fragment sizes. Autoradiograph showing hnRNP C protein–RNA complexes that were treated with decreasing concentrations of RNase I before immunoprecipitation. Markings on the right (A–E) indicate five size ranges that were cut from the nitrocellulose membrane. A sample without antibody serves as control. The sizes of uncrosslinked hnRNP C and hnRNP C dimers are marked by an arrowhead and an asterisk, respectively. (B) Partial RNase digestion with a 1/250 dilution yields an optimal distribution of recovered RNA fragment sizes. Size separation of RNA fractions isolated from hnRNP C protein–RNA complexes through proteinase K digestion. RNA was isolated from five regions of the nitrocellulose membrane (A–E, see labeling in (A)) for three differently treated samples (1/50, 1/250 and 1/500 dilutions of RNase I). (C) PNK treatment for 20 min is the most efficient for 3′ end dephosphorylation. PTB protein–RNA complexes were treated either with Shrimp Alkaline Phosphatase (SAP) for 20 min or with Polynucleotide Kinase (PNK) for 20 min or 2 h, and the cDNA yield was compared after library preparation. A sample without antibody serves as control.
3.2.3. Immunoprecipitation
-
•
Add the lysate to the beads and rotate for 1 h or overnight at 4 °C.
-
•
Place on magnet and discard the supernatant8, and wash twice with high-salt wash (rotate the second wash for at least 1 min at 4 °C).
-
•
Wash twice with PNK buffer and then leave in 1 ml PNK buffer and proceed to step 3.3.
Test: sonication
Genomic DNA can impair the purity and efficiency of immunoprecipitation, in particular if the protein of interest directly interacts with chromatin. In addition to the Turbo DNase treatment, an optional sonication step can be included to shear the DNA. We tested the impact of this treatment on two proteins commonly studied in our lab. Sonication led to a minor improvement in the efficiency of TIA1-RNA immunoprecipitation, but had no visible effect on hnRNP C-RNA immunoprecipitation (Fig. 2D). However, it is important to keep in mind that a specific subset of protein–RNA complexes could be lost without sonication, even if no effect is visible on the gel. The sonication step is optional, however for nuclear RBPs either syringe treatment or sonication are crucial for efficient immunoprecipitation.
Test: partial RNase digestion
The conditions of the partial RNase digestion need to be optimized for successful library preparation. The target size of purified RNAs is between 50 and 300 nucleotides. Keep in mind that the crosslink site of the protein is at a random position within these RNA molecules. Most often the reverse transcriptase stalls at the short polypeptide attached to the RNAs. The percentage of truncated cDNAs can vary depending on the protein, but we previously found that it is generally >80% [7].
Therefore, on average, the resulting cDNA molecules will be half the size of the RNA. To determine the optimal RNase I concentration, we tested dilutions ranging from 1:50 to 1:2000. In our test experiment, the optimal digestion was observed with the 1:250 dilution, which generated a diffuse signal of protein–RNA complexes starting from 40 kDa, which is close to the molecular weight of hnRNP C (Fig. 3A). In preliminary experiments, we recommend to run a Western blot for your protein of interest to determine its exact position on the membrane (see Supplementary Figure 1A in [8] as an example). This can be used to guide the later cutting process.
At the optimal RNase concentration (1:250), the recovered RNAs are in the range between 50 and 300 nucleotides (Fig. 3B). Higher RNase concentration (1:50) leads to a sharp band of protein–RNA complexes (Fig. 3A), which corresponds to RNA molecules smaller than 60 nt (Fig. 3B). RNase concentration of 1:500 or lower resulted mainly in RNA molecules longer than 200 nt, and reduced the overall signal (Fig. 3B), indicating that the large size of protein–RNA complexes obstructed gel migration and/or transfer to the nitrocellulose membrane. These results demonstrate the importance of optimizing the RNase concentration. In agreement with a previous study [24], the distinct size ranges acquired from different regions of the nitrocellulose membrane show that 70 nt of attached RNA shift the migration of protein–RNA complexes by approximately 20 kDa (compare size fragment C in Fig. 3A and B). In order to collect a broad spectrum of RNAs, we recommend cutting a wide region corresponding to fragments B–D in Fig. 3A for the iCLIP cDNA library preparation.9
For RBPs that bind relatively low amounts of RNA, autoradiographs usually result in faint bands or smear (depending on the extent of partial digestion), due to the small amount of crosslinked RNA. The protocol can be used for the library preparation with small amounts of input RNA. However, it is important to compare the PCR gel and sequencing results of a weak RBP with results from a no-antibody control, in order to confirm that the cDNAs do not originate from non-specifically bound or contaminating RNAs.
Beside RNase I, we have tried other nucleases such as micrococcal nuclease, RNase T2 Benzonase and Cyanase (data not shown). Most of these had limited RNase digestion activity compared to RNase I, probably due to buffer incompatibility. RNase T2 showed good RNase activity in the iCLIP lysis buffer, but was not used further because of a preference for cleaving after adenosines.
3.3. RNA 3′ end dephosphorylation10
-
•
Discard the supernatant and resuspend the beads in 20 μl of the following mixture:
| 4 μl | 5 × PNK pH 6.5 buffer |
| 0.5 μl | PNK |
| 0.5 μl | RNasin |
| 15 μl | H2O |
-
•
Incubate for 20 min at 37 °C in a thermomixer at 1100 rpm.
-
•
Wash with PNK buffer.
-
•
Wash with high-salt wash (rotate wash in the cold room for at least 1 min).
-
•
Wash twice with PNK buffer.
Test: dephosphorylation. The dephosphorylation step is critical to remove the cyclic phosphate that remains at the RNA 3’ end after partial RNase I digestion since it prevents adapter ligation (see below). Current CLIP protocols use either Polynucleotide Kinase (PNK; later also used in the 5′ end labeling reaction) or Shrimp Alkaline Phosphatase (SAP) for this purpose. We compared the efficiency of both enzymes in the iCLIP protocol by measuring the amount of amplified cDNA after library preparation (Fig. 3C). PTB–RNA complexes treated with PNK for 20 min showed the highest library concentration, indicating efficient removal of the inhibitory cyclic phosphate from 3′ ends. Prolonged treatment with PNK up to 2 h decreased rather than improved the cDNA yield, probably due to loss of immunopurified protein–RNA complexes (not shown). In contrast to PNK, the SAP treatment showed almost no effect when compared to no treatment (Fig. 3C). Consistently, enzymatic activity to remove cyclic phosphate has been described for PNK but not SAP [25]. We therefore recommend using PNK for 3′ end dephosphorylation.
3.4. L3 adapter ligation
-
•
Carefully remove the supernatant and resuspend the beads in 20 μl of the following mix11:
| 8 μl | H2O |
| 5 μl | 4X ligation buffer |
| 1 μl | RNA ligase11 |
| 0.5 μl | RNasin |
| 1.5 μl | pre-adenylated adapter L3-App (20 μM) |
| 4 μl | PEG400 |
-
•
Incubate overnight at 16 °C in a thermomixer at 1100 rpm.
-
•
Add 500 μl PNK buffer.
-
•
Wash twice with high-salt buffer, rotating each wash in the cold room for 5 min, and twice with PNK buffer.
3.5. 5′ end labeling
-
•
Collect 200 μl (20%) of beads from step 3.5 and remove the supernatant.
-
•
Add 4 μl of hot PNK mix:
| 0.2 μl | PNK |
| 0.4 μl | γ-32P-ATP |
| 0.4 μl | 10x PNK buffer |
| 3 μl | H2O |
-
•
Incubate 5 min at 37 °C in a thermomixer at 1100 rpm.
-
•
Discard the supernatant as radioactive waste and add 20 μl 1x NuPAGE loading buffer.
-
•
Remove the supernatant from the remaining cold beads. Then add the radioactively labeled beads to the cold beads.
-
•
Incubate 5 min at 70 °C.
-
•
Place on the magnet, collect the supernatant and load it on the gel.
3.6. SDS–PAGE and nitrocellulose transfer
-
•
Load the samples on a 4–12% NuPAGE Bis-Tris gel12. Also load 5 μl of a pre-stained protein size marker. Use 0.5 l 1x MOPS running buffer.
-
•
Run the gel at 180 V for 50 min.
-
•
Cut off the dye front and discard it as solid radioactive waste (it contains the unincorporated radioactive ATP).
-
•
Transfer the protein–RNA complexes from the gel to a Protran Nitrocellulose Membrane using the Novex wet transfer apparatus according to the manufacturer’s instructions.
-
•
Transfer at 30 V for 1 h.
-
•
After the transfer, rinse the membrane in PBS buffer, then wrap it in saran wrap and expose it to a Fuji film at −80 °C. Perform exposures for 30 min, 1 h and overnight.13
3.7. Library preparation
3.7.1. RNA isolation
-
•
Isolate the protein–RNA complexes from the low RNase condition using the autoradiograph as a mask for cutting the respective region out of the nitrocellulose membrane. Place the membrane fragments into 1.5 ml tubes.14
-
•
Add 10 μl proteinase K in 200 μl PK buffer to the nitrocellulose pieces (all should be submerged). Incubate for 20 min shaking at 1100 rpm at 37 °C.
-
•
Add 200 μl PK buffer + 7 M urea and incubate for further 20 min at 37 °C and 1100 rpm.
-
•
Collect the solution and add it together with 400 μl phenol/chloroform to a 2 ml Phase Lock Gel Heavy tube.
-
•
Incubate for 5 min at 30 °C shaking at 1100 rpm. Separate the phases by spinning for 5 min at full speed and room temperature.
-
•
Transfer the aqueous layer into a new tube (be careful not to touch the gel matrix with the pipette). Optional: spin again for 1 min and transfer into a new tube.
-
•
Precipitate by adding 0.75 μl glycoblue and 40 μl 3 M sodium acetate pH 5.5. Then mix and add 1 ml 100% ethanol, mix again and place at −20 °C overnight.
-
•
Centrifuge at 15,000 rpm at 4 °C for 20 min. Remove the supernatant and wash the pellet with 0.9 ml 80% ethanol and spin again for 5 min.15 Resuspend the pellet in 5 μl H2O and transfer to a PCR tube.
3.7.2. Reverse transcription
-
•
Add the following reagents to the resuspended pellet16:
| 1 μl primer Rt#clip (0.5 pmol/μl) |
| 1 μl dNTP mix (10 mM) |
-
•
RT thermal program:
-
•
70 °C 5 min
-
•
25 °C hold until the RT mix is added, mix by pipetting
-
•
RT mix
| 7 μl | H2O |
| 4 μl | 5 × First Strand Buffer |
| 1 μl | 0.1 M DTT |
| 0.5 μl | RNasin |
| 0.5 μl | Superscript III |
-
•
25 °C 5 min, 42 °C 20 min, 50 °C 40 min, 80 °C 5 min, 4 °C hold
-
•
Add 1.65 μl 1 M NaOH and incubate at 98 °C for 20 min. Then add 20 μl 1 M Hepes–NaOH pH 7.3 to eliminate radioactivity from strongly labeled samples and to prevent RNA from interfering with subsequent reactions.17
-
•
Add 350 μl TE buffer, 0.75 μl glycoblue and 40 μl 3 M sodium acetate, pH 5.5, mix, then add 1 ml 100% ethanol. Mix again and precipitate at −20 °C overnight.
Test: RT primers
All reverse transcription primers are equipped with 5-nt random barcodes to enable removal of PCR artifacts, plus a 4-nt barcode sequences that allow multiplexing. The 4-nt barcodes differ by at least two nucleotides, insuring that a single point mutation is not sufficient to convert one barcode into another. Since the quality of the library depends on the efficiency of the individual primer, it is necessary to test each new primer (Fig. 4A). In order to do so, a full library preparation is needed (a well-established antibody can be used for immunoprecipitation of protein–RNA complexes, which are then used for reverse transcription with different RT primers). It is advisable to include an already optimized primer as a reference for the newly ordered primers. The intensity of the PCR-amplified library on a TBE gel acquired from the preparative PCR can be used to estimate the quality of a specific RT primer. From Fig. 4A it can be concluded that the Rt#clip primers 1, 2, 6, 9, 10 and 13–16 were the most reliable in our hands. However, this depends on the individual batch and can vary between different orders. We advice to keep negative controls separate to estimate the unspecific background signal before the sequencing reaction.
Fig. 4.

Quality improvements of library preparation and urea treatment. (A) Reverse transcription (RT) primers can differ in their efficiency. Different reverse transcription primers were tested by analyzing hnRNP C iCLIP libraries using gel electrophoresis. RT primers Rt#clip 1, 2, 6, 9, 10 and 13–16 were identified as the most useful for library preparation. Note that in this experiment also the control omitting the antibody shows product on the PCR gel, indicating some impurities in the purification. For the evaluation of the primer quality this does not have an impact. Furthermore, the signal of the product is much weaker when compared to the sample that was reverse transcribed with the same RT primer (Rt9clip). (B) Alkaline hydrolysis efficiently removes radioactively labeled RNA and does not impair library preparation. Gel image of amplified cDNA libraries using RT primers Rt1clip and Rt2clip and extraction of low and medium size ranges (L, 70–80 nt; M, 80–100 nt). Samples were treated with or without alkaline hydrolysis. Radioactive counts per second before RT and after gel extraction are given above. (C) Increased numbers of PCR amplification cycles lead to the formation of secondary products. hnRNP C iCLIP libraries were amplified using between 21 and 39 cycles. The appearance of secondary products beyond 31 cycles indicates overamplification. 1/10 and 1/100 dilutions of cycle number 35 and 39 are shown for comparison.
Test: alkaline hydrolysis
After the RNA is released with proteinase K, the samples can still be radioactive. Alkaline hydrolysis can be used to remove the carry-over of radioactively labeled RNA after reverse transcription. In this test, the counts/s of each sample were measured before and after the alkaline treatment. This confirmed that alkaline hydrolysis degrades the RNA and removes all remaining radioactive signal (Fig. 4B). Use of two different RT primers confirmed that the same amount of PCR product is recovered independent of the alkaline hydrolysis, indicating that this treatment did not influence the quality of the library preparation.
3.7.3. Gel purification
-
•
Centrifuge for 15 min at 15,000 rpm at 4 °C. Remove the supernatant and wash the pellet with 0.5 ml 80% ethanol. Centrifuge again, remove supernatant and resuspend the pellet in 6 μl H2O.
-
•
Add 6 μl 2 × TBE-urea loading buffer to the cDNA. It is recommended, at least in initial experiments, to add the loading buffer also to 6 μl DNA size marker. Heat samples to 80 °C for 5 min immediately before loading. Leave one lane free between each sample to avoid cross-contamination between samples.
-
•
Prepare 0.8 l 1x TBE running buffer and fill the upper chamber with 0.2 l and the lower chamber with 0.6 l. Use a p1000 pipette to flush remaining urea out of the wells before loading 12 μl of each sample. Load the marker into the last lane.
-
•
Run 6% TBE-urea gel for 40 min at 180 V until the lower (dark blue) dye is close to the bottom.
-
•
Cut off the last lane containing the size marker and incubate it gently shaking for 10 min in 20 ml TBE buffer with 2 μl SYBR green II. Wash once with TBE and visualize by UV transillumination. Print the result with 100% scale, and use it as a mask to guide the excision of cDNA bands from the rest of the gel.
-
•
Together with the full L3-App sequence, the primer sequence accounts for 52 nt of the cDNA. Cut three bands, at 70–80 nt, 80–100 nt and 100–150 nt.18
-
•
Add 400 μl TE to each gel piece and crush the gel piece into small pieces with a 1 ml syringe plunger.
OR
-
•
Prepare 0.5 ml tubes by piercing a hole in the bottom using a 19G needle. Place a gel fragment inside and then place the tubes into a 2 ml collection tube. Centrifuge 2 min at full speed. Remove 0.5 ml tube and add 400 μl TE.
-
•
Incubate shaking for 1 h at 1100 rpm at 37 °C for, then place on dry ice for 2 min, and place back for 1 h at 1100 rpm at 37 °C. Transfer the liquid portion of the supernatant into a Costar SpinX column, into which you placed two 1 cm glass pre-filters.
-
•
Centrifuge at full speed for 1 min. Collect the solution and add it together with 400 μl RNA phenol/chloroform to a 2 ml Phase Lock Gel Heavy tube.
-
•
Incubate for 5 min shaking at 1100 rpm at 30 °C. Separate the phases by spinning 5 min at full speed at room temperature.
-
•
Transfer the aqueous layer into a new tube. Centrifuge again for 1 min and transfer into a new tube.
-
•
Add 1 μl glycoblue and 40 μl 3 M sodium acetate, pH 5.5. Mix, then add 1 ml 100% ethanol. Mix again and precipitate at −20 °C overnight.
3.7.4. Ligation of the primer to the 5′ end of the cDNA (circularization)
-
•
Pellet the precipitated cDNA by centrifugation, wash as described above and resuspend in 8 μl ligation mix:
| 6.5 μl H2O |
| 0.8 μl 10x CircLigase buffer II |
| 0.4 μl 50 mM MnCl2 |
| 0.3 μl CircLigase II |
-
•
Transfer to PCR tubes and incubate at 60 °C for 1 h.
-
•
Add 30 μl oligo annealing mix:
| 26 μl | H2O |
| 3 μl | Fast digest buffer |
| 1 μl | 10 μM Cut_oligo |
-
•
Anneal the oligonucleotide with the following program:
| 95 °C 2 min. |
| Successive cycles of 20 s, starting from 95 °C and decreasing the temperature by 1 °C each cycle down to 25 °C. |
| 25 °C hold. |
-
•
Add 2 μl BamHI and incubate for 30 min at 37 °C, then incubate for 5 min at 80 °C.
-
•
Add 350 μl TE, 0.75 μl glycoblue and 40 μl 3 M sodium acetate, pH 5.5, and mix. Then add 1 ml 100% ethanol. Mix again and precipitate at −20 °C overnight.
3.8. PCR amplification
3.8.1. PCR optimization
-
•
Centrifuge and wash the cDNA, then resuspend it in 21 μl H2O.
-
•
Prepare the following PCR mix:
| 1 μl cDNA |
| 0.25 μl primer mix P5Solexa/P3Solexa, 10 μM each |
| 5 μl Accuprime Supermix 1 |
| 3.75 μl H2O |
-
•
Run the following PCR:
| 94 °C 2 min |
| 25–35 cycles of: 94 °C 15 s, 65 °C 30 s, 68 °C 30 s |
| 68 °C 3 min |
| 25 °C hold |
-
•
Mix 8 μl PCR product with 2 μl 5x TBE loading buffer, load on a 6% TBE gel and stain with SYBR green I.19
3.8.1.1. Test: PCR amplification
The size of the cDNA insert to be mapped to the genome will be the size of product minus the combined length of the P3/P5Solexa primers and the barcode (128 nt). Therefore, a band cut at 70–80 nt on the cDNA gel (with 20–30 nt cDNA + 52 nt primer) is expected to generate 145–155 nt PCR products. A common problem is overamplification during the PCR, which results in secondary products that migrate as a diffuse band of higher size (Fig. 4C). To avoid overamplification of your library, it is necessary to determine the optimal PCR cycle number for your cDNA. To demonstrate this, an hnRNP C library was amplified with cycles ranging from 21 to 39, increasing by two cycles between the lanes (Fig. 4C). Amplification with less than 27 cycles resulted in a weak signal, indicating that the DNA concentration is not high enough for high-throughput sequencing. On the other hand, amplification with more than 31 cycles led to the appearance of the secondary band, and with more than 33 cycles reduced the intensity of the expected band (80–100 nt).
The secondary band could correspond to PCR artifacts, or it could be an artifact of gel migration, caused by the high concentration of DNA. To test this, we loaded the gel with 1/10 or 1/100 diluted PCR products obtained after 35 and 39 cycles. The dilution of the amplified cDNA did not remove the secondary products, but just weakened the overall signal (Fig. 4C). Since the secondary products could not be removed by dilution, we conclude that they are PCR artifacts, which need to be avoided before submitting the cDNA library for sequencing. For the cDNA used here, cycle numbers between 27 and 29 would be appropriate for sequencing, since they produce the correct PCR product of ±150 nt, while the secondary products are hardly detectable.
3.8.2. Preparative PCR
Estimate the minimum number of PCR cycles based on the results in Section 3.8.1, and use it to amplify ½ of the library. Consider that a 2.5-fold more concentrated cDNA is used, therefore we recommend to use one cycle less than in the test PCR.
-
•
Prepare the following PCR mix:
| 10 μl | cDNA |
| 9 μl | H2O |
| 1 μl | primer mix P5Solexa/P3Solexa, 10 μM each |
| 20 μl | Accuprime Supermix 1 |
-
•
Run the same PCR program as in step 3.8.1.
-
•
Mix 8 μl PCR product with 2 μl 5x TBE loading buffer, load on a 6% TBE gel and run at 180 V and 120 mA for 30 min. Stain and visualize with SYBR green I.
-
•
If under-amplified (i.e., the band is not clearly visible on the gel), immediately place back into the PCR machine for two or more cycles and re-run the gel.
-
•
Now amplify the remaining ½ of the cDNA with the appropriate number of cycles, and combine it with the other ½ PCR reaction.
-
•
If the resulting product sizes are correct, mix the PCR reactions from cDNAs that were excised from the different portions of the gel (3.7.3) in the following ratios: lower:middle:high = 1:5:5.20
3.9. qPCR quantitation
-
•
Set up serial dilutions of all libraries to be tested in a 96-well plate. Prepare a 1:10 dilution to start with. Use 1:100, 1:1000, 1:10,000 and 1:10,000, ideally with a multichannel pipette for maximum consistency. Also set up dilutions of an old iCLIP library of known concentration at 1000,100, 10, 1 and 0.1 pM.
-
•
Prepare a master mix with the following reagents (18 μl for each qPCR reaction):
| 10 μl | Invitrogen Platinum qPCR Supermix w/ROX |
| 0.5 μl | DLP oligo (10 μM) |
| 0.6 μl | primer 1 (10 μM) |
| 0.6 μl | primer 2 (10 μM) |
| 6.3 μl | H2O |
-
•
Add 18 μl per well in a qPCR plate (MicroAmp Fast Optical plates).
-
•
Add 2 μl template from the dilution plate using a multichannel pipette.
-
•
Seal the plate well with the MicroAmp optical adhesive film.
-
•
Run the plate with the following program in the qPCR machine (normal setting, detector = FAM-TAMRA, passive reference = ROX): 54 °C 2 min, 94 °C 10 min, 40 cycles of: 94 °C 15 s, 62 °C 1 min, 72 °C 30 s.
-
•
To calculate the concentrations, plot concentration vs. Ct value for the standard on a log scale and fit a line to the graph (the R2 value of the line should be >0.99). Use this line to obtain concentrations of the unknown samples using their Ct values.
-
•
Dilute the iCLIP library to 10 nM, submit 10 μl for sequencing and store the rest.
iCLIP libraries can be sequenced using standard Illumina protocols. For cost effectiveness, we recommend 50-nucleotide single-end runs. Bioinformatic analysis requires de-multiplexing if multiple samples were run within one sequencing lane, mapping reads to the genome, collapsing of random barcodes and definition of binding sites [7]. For further details, readers are referred to a recent review [6].
3.10. Immunoprecipitation under denaturing urea conditions (alternative procedure to 3.2)
In the case that standard iCLIP conditions are not stringent enough to specifically purify the protein of interest, a dual immunoprecipitation with urea denaturation can be used instead of the protocol described in 3.2. This urea denaturation protocol is adapted from Kiel et al. [26] and presented in the following steps for FLAG-tagged proteins of interest.
3.10.1. Antibody coupling
-
•
Covalently couple 30 μg of M2 FLAG antibody to 1 mg of Dynabeads M-270 Epoxy by following manufacturer’s protocol (Dynabeads Antibody Coupling Kit, Life Technologies), and resuspend the beads in 100 μl SB (contained in Dynabeads Antibody Coupling Kit) per 1 mg beads.
3.10.2. Preparation of antibody-coupled beads for immunoprecipitation
-
•
Wash 40 μl of the antibody-coupled beads prepared in 3.10.1 with high-salt wash.
-
•
Wash the beads with lysis buffer and resuspend in 40 μl of lysis buffer.
-
•
Use 40 μl of the beads for each immunoprecipitation experiment.
3.10.3. Partial RNA digestion and centrifugation
-
•
Resuspend the cell pellet (from step 3.1) in 1 ml lysis buffer with 1 μl ANTI-RNase and 10 μl Protease Inhibitor Cocktail Set III, EDTA-free.
-
•
Sonicate with Bioruptor plus for five cycles with alternating 30 s on/30 s off at low intensity.
-
•
Add 2 μl Turbo DNase and 10 μl RNase I (1:50 or 1:250 dilution for high and low RNase condition) to the cell lysate for immunoprecipitation of the FLAG-tagged protein.
-
•
Incubate at 37 °C shaking at 1100 rpm for 3 min. After incubation transfer to ice for >3 min.
-
•
Centrifuge at 22,000g at 4 °C for 10 min and transfer the supernatant to a new 1.5 ml microtube. Repeat this step.
3.10.4. 1st round of immunoprecipitation
-
•
Add the cell extract to the beads prepared in 3.10.2.
-
•
Rotate the beads/lysate mix at 4 °C for 2 h.
-
•
Discard the supernatant and wash twice with high-salt wash (rotate the second wash for at least 1 min in the cold room).
-
•
Wash the beads with PNK buffer, leave the beads in PNK buffer until antibody-attached beads are prepared.
3.10.5. Preparation of antibody-attached beads
Antibody-attached beads were prepared as described in 3.2.1 with minor modifications.
-
•
Add 20 μl of protein G Dynabeads per experiment to a fresh microtube.
-
•
Wash beads twice with lysis buffer (without protease inhibitor). This and all following washing steps of the beads are performed with 900 μl of the respective buffer.
-
•
Resuspend the beads in 20 μl lysis buffer with 2 μg M2 FLAG antibody per experiment.
-
•
Rotate tubes for 1 h at room temperature.
-
•
Wash with lysis buffer, and wash twice with T-20 IP buffer and resuspend in 20 μl of T-20 IP buffer. Leave in T-20 IP buffer until ready to proceed to step 3.10.6.
3.10.6. 2nd round of immunoprecipitation
-
•
Add 100 μl urea cracking buffer to the beads obtained from 3.10.4.
-
•
Incubate at 65 °C, shaking at 1100 rpm for 3 min to elute the immunoprecipitated protein.
-
•
Transfer the supernatant to a new 1.5 ml microtube using a magnetic stand, and add 900 μl T-20 IP buffer with 1 μl ANTI-RNase, 10 μl Protease Inhibitor Cocktail Set III, EDTA-free, and 1 μl SUPERaseIn.
-
•
Place on magnetic stand.
-
•
Transfer the supernatant to new 1.5 ml microtube in order to remove any residual beads and add 20 μl of freshly prepared antibody-attached beads (from step 3.10.5).
-
•
Rotate the beads/supernatant mix at 4 °C for 2 h.
-
•
Discard the supernatant and wash the beads twice with high-salt wash (rotate the second wash for at least 1 min in the cold room).
-
•
Wash the beads twice with PNK buffer.
Test: iCLIP with denaturing conditions
To assess the efficiency of removing co-purifying proteins with denaturing conditions, FLAG-tagged Staufen 1 (STAU1) was immunoprecipitated with a M2-FLAG antibody using either the protocol 3.2 or 3.10, followed by RNA labeling as described in 3.4 and SDS–PAGE analysis. An RBP of around 40 kDa co-purifies with STAU1 when using the standard iCLIP protocol (see no urea treatment in Fig. 5). The denaturing protocol as described in 3.3 removes the signal of the 40 kDa protein without significant loss in the signal of STAU1-RNA complexes (Fig. 5). The efficiency of the protocol depends on the protein of interest. In case of additional bands in the high RNase control, the urea protocol would be preferred. However, the input, cost and duration of the protocol are higher than for the standard protocol.
Fig. 5.
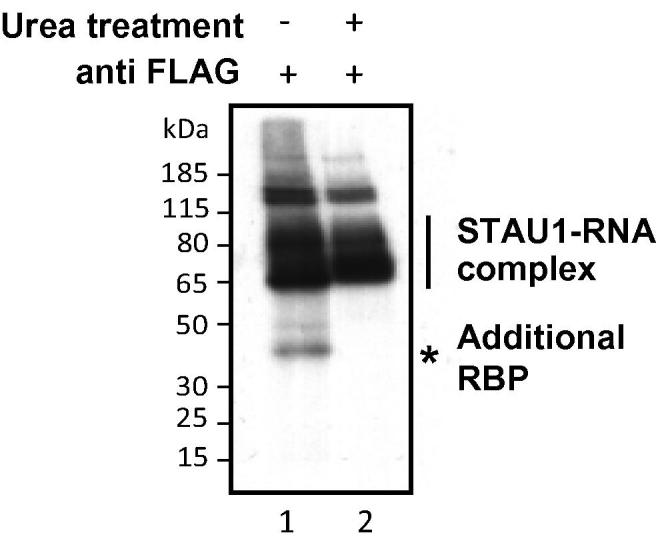
Alternative IP using urea treatment. Denaturing urea treatment prevents co-purification of a second RBP of about 40 kDa. FLAG-tagged Staufen 1 (STAU1) was purified using the standard iCLIP protocol (no urea treatment) and compared to an immunoprecipitation procedure using denaturing urea treatment.
4. Conclusion
Here, we provide a detailed iCLIP protocol, with explanations and experiments that demonstrate how to prepare a high-quality cDNA library. Moreover, the protocol can be used with UV-A crosslinking or with denaturing urea purification, which broaden the range of RBPs that can be investigated with iCLIP.
Acknowledgments
We would like to thank the large number of iCLIP users for many valuable comments that helped to improve this protocol. We thank Dr. Kathi Zarnack for critical reading of the manuscript. This work was supported by the Marie Curie Initial Training Network RNPnet 289007, the European Research Council Grant 206726-CLIP and the Medical Research Council Grant number U105185858. Y.S. is supported by the Nakajima Foundation and J.A. is supported by the Boehringer Ingelheim Fonds.
Footnotes
Crosslinking conditions can be optimized when working with a new protein, however we do not see large increases in the efficiency of UV-C crosslinking with energies higher than recommended in the protocol. Special care should be taken when further increasing the dose, since this could distort library preparation due to damaged RNA and could trigger DNA-damage response pathways in the cells.
4-Thiouridine is light-sensitive, therefore return the cells to the incubator quickly after its addition.
The amount of required antibody depends on its quality and purity. This should be optimized in preliminary experiments.
We suggest that the RNA or protein concentration is determined with Nanodrop or Bradford assay, respectively, and normalized to the concentration of the sample with the lowest amount. We recommend 2 mg/ml as the optimal protein concentration.
Other recommended controls include a control where the RBP is absent from the original material (such as knockout or knockdown cells), a control where no crosslinking is done and a control where no antibody is used during IP.
We find that RNase I is deactivated after prolonged incubation with 0.1% SDS in the lysis buffer.
To test a new antibody, collect 15 μl at this step for Western blot comparison of lysate before and after IP and visualize depletion of protein.
If monitoring depletion efficiency, save 15 μl supernatant for Western blot analysis (see Footnote 6).
If contaminating bands of other protein–RNA complexes are present above your protein–RNA complex, only cut up to, but not including these bands. If the contaminating bands run below your RNA–protein complex, cut an additional band between the contaminating band and your protein–RNA complex. The RNA sequences cloned from this band can later be used to compare with those purified with your protein–RNA complexes to control for the specificity of your experiment.
Sections 3.3 and 3.4 do not need to be carried out on no-UV and high-RNase controls. However, they should be performed on the no-antibody control to use it as a background estimate for the complete library preparation.
Alternatively, truncated RNA ligase T4 R55K, K227Q can be used. Truncated T4 does not use ATP and can only ligate a 5′ pre-adenylated linker to the 3′-OH of the RNA, thereby precluding RNA self-circularization. In our hands, it did not improve the yield of iCLIP libraries, presumably because loss of RNA molecules due to self-circularization is minor.
The Novex NuPAGE gels are critical. A pour-your-own SDS-PAGE gel (Laemmli) changes its pH during the run which can get to pH ∼9.5 leading to alkaline hydrolysis of the RNA. Instead, the Novex NuPAGE buffer system keeps the pH around 7 throughout the whole run. We use MOPS NuPAGE running buffer.
Unlike other RNases, RNase I has no base preference, and therefore cleaves after all four nucleotides. Under high-RNase conditions, the size of the radioactive band has to change in comparison to low RNase conditions, confirming that the band corresponds to a protein–RNA complex. Furthermore, the high-RNase condition helps to determine the size of the immunoprecipated RBP, as the RBP will be bound to short RNAs and thus will migrate as a less diffuse band ∼5 kDa above the expected molecular weight of the protein.
In order to determine the specificity of the protein–RNA complex, it is necessary to check the high RNase condition. The radioactive band of the high RNase experiment should be ∼5 kDa above the molecular weight, there should be no band appearing in any of the controls and the band should shift up and become more diffuse in the low-RNase condition. On this basis, proceed to RNA isolation using the following guidelines: the average molecular weight of 70 nt RNA is ∼20 kDa. To isolate a broad range of RNAs between 40 and 300 nt in size (including the adapter), we recommend cutting a wide band of ∼15–80 kDa above the expected molecular weight of the protein.
Remove the wash first with a p1000 and then with a p20 or p10. Try not to disturb the pellet, but if you do, spin it down again. Leave on the bench for exactly 3 min with the cap open to dry. When resuspending, make sure to pipette along the back area of the tube.
Use distinct primers (Rt1clip–Rt16clip) for the control and the different experimental replicates. The different primers contain individual 4-nt barcode sequences that allow multiplexing of samples and control for cross-contamination between the samples.
It is possible to mix up to three samples that shall be multiplexed at this point. Alternatively, cDNA libraries of each sample can be amplified separately and mixed after the PCR.
The 70–80 nt band is prone to producing primer artifacts in the PCR, and even if containing specific cDNAs, sequences are often too short to map to the genome. Therefore, if binding to miRNAs or other short RNAs is not of interest, it is not necessary to isolate this band.
All work done post PCR must be carried out on a specially designated bench. This cDNA must never be taken to an area where work with iCLIP RNA is done.
The ratio of the bands can be adjusted according to the following criteria. Adjust the ratio by the size of cDNAs of interest; for instance, if miRNAs are important, use more of the lower bands. Take the quality of each PCR sample into account; if a sample contains the primer artifact band at 128 nt, then use less of this sample, or exclude it. Also take the quality of the amplification into account; if one of the ½ PCR reactions was overamplified, use more of the reaction that is in the right range of amplification.
Contributor Information
Julian König, Email: j.koenig@imb-mainz.de.
Jernej Ule, Email: j.ule@ucl.ac.uk.
References
- 1.Glisovic T., Bachorik J.L., Yong J., Dreyfuss G. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lunde B.M., Moore C., Varani G. Nat. Rev. Mol. Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auweter S.D., Oberstrass F.C., Allain F.H. Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mili S., Steitz J.A. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trifillis P., Day N., Kiledjian M. RNA. 1999;5:1071–1082. doi: 10.1017/s1355838299981803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.König J., Zarnack K., Luscombe N.M., Ule J. Nat. Rev. Genet. 2011;13:77–83. doi: 10.1038/nrg3141. [DOI] [PubMed] [Google Scholar]
- 7.Sugimoto Y., Konig J., Hussain S., Zupan B., Curk T., Frye M., Ule J. Genome Biol. 2012;13:R67. doi: 10.1186/gb-2012-13-8-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anko M.L., Muller-McNicoll M., Brandl H., Curk T., Gorup C., Henry I., Ule J., Neugebauer K.M. Genome Biol. 2012;13:R17. doi: 10.1186/gb-2012-13-3-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogelj B., Easton L.E., Bogu G.K., Stanton L.W., Rot G., Curk T., Zupan B., Sugimoto Y., Modic M., Haberman N., Tollervey J., Fujii R., Takumi T., Shaw C.E., Ule J. Sci. Rep. 2012;2:603. doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schor I.E., Lleres D., Risso G.J., Pawellek A., Ule J., Lamond A.I., Kornblihtt A.R. PLoS One. 2012;7:e48084. doi: 10.1371/journal.pone.0048084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tollervey J.R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., Konig J., Hortobagyi T., Nishimura A.L., Zupunski V., Patani R., Chandran S., Rot G., Zupan B., Shaw C.E., Ule J. Nat. Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z., Kayikci M., Briese M., Zarnack K., Luscombe N.M., Rot G., Zupan B., Curk T., Ule J. PLoS Biol. 2010;8:e1000530. doi: 10.1371/journal.pbio.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao C., Biesinger J., Wan J., Weng L., Xing Y., Xie X., Shi Y. Proc. Nat. Acad. Sci. USA. 2012;109:18773–18778. doi: 10.1073/pnas.1211101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussain S., Sajini A.A., Blanco S., Dietmann S., Lombard P., Sugimoto Y., Paramor M., Gleeson J.G., Odom D.T., Ule J., Frye M. Cell Rep. 2013;4:255–261. doi: 10.1016/j.celrep.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagnon J.L., Briese M., Sun W., Mahaffey C.L., Curk T., Rot G., Ule J., Frankel W.N. PLoS Genet. 2012;8:e1003067. doi: 10.1371/journal.pgen.1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witten J.T., Ule J. Trends Genet. 2011;27:89–97. doi: 10.1016/j.tig.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zarnack K., König J., Tajnik M., Martincorena I., Eustermann S., Stevant I., Reyes A., Anders S., Luscombe N.M., Ule J. Cell. 2013;152:453–466. doi: 10.1016/j.cell.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. J. Vis. Exp. 2011 doi: 10.3791/2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jr., Jungkamp A.C., Munschauer M., Ulrich A., Wardle G.S., Dewell S., Zavolan M., Tuschl T. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M., Krijgsveld J., Hentze M.W. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 22.Windhager L., Bonfert T., Burger K., Ruzsics Z., Krebs S., Kaufmann S., Malterer G., L’Hernault A., Schilhabel M., Schreiber S., Rosenstiel P., Zimmer R., Eick D., Friedel C.C., Dolken L. Genome Res. 2012;22:2031–2042. doi: 10.1101/gr.131847.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jungkamp A.C., Munschauer M., Ulrich A., Wardle G.S., Dewell S., Zavolan M., Tuschl T. J. Vis. Exp. 2010 doi: 10.3791/2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ule J., Jensen K., Mele A., Darnell R.B. Methods. 2005;37:376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 25.Galburt E.A., Pelletier J., Wilson G., Stoddard B.L. Structure. 2002;10:1249–1260. doi: 10.1016/s0969-2126(02)00835-3. [DOI] [PubMed] [Google Scholar]
- 26.Kiel J.A., Emmrich K., Meyer H.E., Kunau W.H. J. Biol. Chem. 2005;280:1921–1930. doi: 10.1074/jbc.M403632200. [DOI] [PubMed] [Google Scholar]