

Highlights
-
•
Development of an antigen array for high throughput monoclonal antibody selection.
-
•
Identification of cross-reactive antibodies at an early stage of screening.
-
•
Convenient screening method to identify functional recombinant antibodies.
Keywords: Protein microarray, Hybridoma, Monoclonal antibodies, High throughput
Abstract
Monoclonal antibodies are valuable laboratory reagents and are increasingly being exploited as therapeutics to treat a range of diseases. Selecting new monoclonal antibodies that are validated to work in particular applications, despite the availability of several different techniques, can be resource intensive with uncertain outcomes. To address this, we have developed an approach that enables early screening of hybridoma supernatants generated from an animal immunised with up to five different antigens followed by cloning of the antibody into a single expression plasmid. While this approach relieved the cellular cloning bottleneck and had the desirable ability to screen antibody function prior to cloning, the small volume of hybridoma supernatant available for screening limited the number of antigens for pooled immunisation. Here, we report the development of an antigen microarray that significantly reduces the volume of supernatant required for functional screening. This approach permits a significant increase in the number of antigens for parallel monoclonal antibody selection from a single animal. Finally, we show the successful use of a convenient small-scale transfection method to rapidly identify plasmids that encode functional cloned antibodies, addressing another bottleneck in this approach. In summary, we show that a hybrid approach of combining established hybridoma antibody technology with refined screening and antibody cloning methods can be used to select monoclonal antibodies of desired functional properties against many different antigens from a single immunised host.
1. Introduction
The high binding affinity and specificity of monoclonal antibodies for their targets have made them invaluable tools for biomedical research and an increasingly important class of drugs that have been exploited to treat a range of diseases [1,2]. To select new monoclonal antibodies to a defined antigen, host animals are immunized and the resulting antibody-secreting B-lymphocytes are fused to a myeloma cell line to create a hybridoma. Hybridomas that secrete monoclonal antibodies of the required properties are selected so that they can be cultured indefinitely to provide large amounts of antibody as necessary [2,3]. While well-established, selecting monoclonal antibodies using this approach has several limitations that have made selecting monoclonal antibodies to multiple different antigens in parallel difficult. The limitations for scaling this approach include the use of laboratory animals, with standard protocols typically recommending immunising several animals per target antigen. Furthermore, because of the additional chromosomes, hybridomas are genetically only metastable, often necessitating the repeated cellular cloning of the hybridoma cell line which can be lengthy and labour intensive. Finally, it takes up to 2 weeks after the cellular fusion procedure before single hybridomas have divided to form a colony that is large enough to secrete sufficient amounts of antibody to permit robust screening.
Because of the usefulness of monoclonal antibodies, a wide range of different techniques for selecting them have been developed that bypass some or all of these limitations. Approaches using libraries of antibody-based binding reagents and in vitro selection methods such as phage display [4] and similar methods [5] have been particularly successful and obviate the need for animals. The requirement to create and culture hybridomas can also be circumvented by sorting individual antigen-specific B-lymphocytes and amplifying the regions encoding the rearranged antibody light and heavy chain regions by single cell RT-PCR; once cloned, antibodies can be expressed recombinantly by transfecting mammalian cell lines [6]. Variations include B-cell panning [7], lithographic methods of single cell incubation [8] or spotting of single cells onto an antigen coated chip [9], each of which have their own advantages for certain applications.
While these alternative methods have specific advantages, animal immunisation and the generation of hybridomas have two important features. Firstly, the affinities of antibodies raised in vivo are often higher than those from in vitro selection methods due to the process of somatic hypermutation; and secondly, hybridoma colonies typically secrete sufficient amounts of antibody to permit some functional screening so that subsequent cloning efforts are focussed only on antibodies that have the required immunological or biochemical properties. With these points in mind, we developed a convenient method of selecting monoclonal antibodies against multiple antigens immunised as a pool into a single animal [10]. This hybrid approach ensured high-affinity antibodies were elicited, and that some hybridoma supernatant was available for screening to identify antibodies with desired functional properties prior to cloning. Selected antibodies were cloned by amplification of the rearranged antibody light and heavy chains by RT-PCR from the hybridomas, and ligated into a single expression plasmid that could be used to express the antibodies recombinantly [10]. Using this approach, we were able to immunise and screen up to five different antigens per mouse, a number that was restricted by the small volume (∼200 μl) of available antibody-containing supernatant per hybridoma and our use of a standard ELISA in our antibody selection screen. Because, in principle, antibodies to more antigens could be obtained from a single mouse, we sought to reduce the amount of hybridoma supernatant required for initial antibody screening and address an additional bottleneck in this method: the identification of functional antibody-encoding plasmids.
We now describe the development and use of a protein microarray that permits the screening of up to 100 different antigens with small volumes of undiluted hybridoma tissue culture supernatant which significantly increases the number of antibodies that can be cloned from a single mouse in parallel. In addition, we describe a refinement using the small scale transfection of HEK293 cells which facilitates the identification of functional antibody expression plasmids. Together, these refinements reduce the number of animals required for generating monoclonal antibodies and vastly increase the potential throughput of this method of monoclonal antibody generation.
2. Materials and methods
2.1. Recombinant protein production and purification
The extracellular domains of zebrafish proteins used in this study were expressed as monobiotinylated proteins using mammalian cells. Expression plasmids were made from published resources [11–13] by subcloning the NotI/AscI enzyme flanked ectodomains [14] into a plasmid containing a C-terminal rat Cd4 domains 3 and 4, an enzymatically biotinylatable peptide sequence, and a 6 His-tag [11,15]. The ectodomains of zebrafish proteins and recombinant antibodies were expressed by transient transfection of either HEK293E [16] or F (Invitrogen) cells. To monobiotinylate proteins during expression, cells were co-transfected with a plasmid encoding a secreted Escherichia coli BirA enzyme [11,15]. Supernatants were harvested after 6 days, filtered, and purified using Ni2+-NTA Sepharose (Invitrogen) [17]. Proteins were assessed by SDS–PAGE and protein biotinylation confirmed by ELISA [15]. Recombinant antibodies were purified using protein G columns (GE Healthcare) [10].
2.2. Immunizations and hybridoma generation
Six-week-old Balb/c mice were immunised intraperitoneally with pools of up to twenty antigens (5 μg each) mixed with Gold’s Adjuvant (Sigma) three times at 4 week intervals. Mice were given a final immunisation without adjuvant 3 days before the spleen was removed. Splenocytes (108) were fused to SP2/0 myeloma cells (107) in 50% PEG 1500 (Roche, Hertfordshire, UK) using standard procedures [10]. The hybridoma mixture was plated over twelve 96-well plates and supernatants were harvested after 10–14 days for screening.
2.3. Printing of protein microarrays
Purified biotinylated proteins were spotted at the base of streptavidin-coated 96-well microtitre plates (NUNC Immobilizer, Thermo Scientific, Denmark) using a Microgrid II arrayer (BioRobotics) by direct contact printing using 0.2 or 0.4 mm solid printing pins. Printed plates were left unwashed and plates were stored at 4 °C unless described otherwise. For screening, hybridoma supernatants were added directly to the wells and a specific blocking step was found not to be required. A biotinylated anti-rat Cd4 antibody was used as a positive control and orientation marker.
2.4. Screening of protein microarrays
Hybridoma supernatants were added to the antigen arrays and incubated overnight at 4 °C. After washing with PBT (PBS + 0.1% Tween), arrays were incubated for 2 hours with a goat anti-mouse Alexa 488 secondary antibody (Invitrogen), washed in PBT, followed by a rabbit anti-goat Alexa 488 antibody (Invitrogen). This second amplification step, although not absolutely required, increased signals to permit direct visual screening using an epifluorescence microscope. In some cases, antibodies were detected with an anti-mouse Alexa 568 secondary antibody (e.g. Fig. 1C) or an anti-mouse alkaline phosphatase secondary followed by NBT/BCIP (Roche) as a precipitating colourimetric substrate (e.g. Fig. 1B). Antigen arrays were analysed on a Leica MZ 16 FA microscope, images captured using an Axiocam HRC (Zeiss) and adjusted for brightness and contrast with Adobe Photoshop CS4.
Fig. 1.

Establishment of the optimal printing and storage conditions for biotinylated antigen arrays in streptavidin-coated 96-well plates. (A) Schematic representation of a 96 well plate with a 5 × 5 antigen array. (B–L) A purified biotinylated antibody (OX68-bio) was spotted in streptavidin-coated 96 well plates. (B and C) Detection with either a non-fluorescent alkaline phosphatase-conjugated secondary antibody followed by a precipitating colourimetric NBT/BCIP substrate (B); and, with a fluorescent Alexa 568-conjugated secondary antibody (C). (D) Different concentrations of OX68-bio, 1 = 0.5; 2 = 0.25; 3 = 0.1; 4 = 0.05 μg/μl were arranged vertically, and in quadruplicate horizontally. (E) Different amounts of Tween 20 added prior to printing, 1 = 10%; 2 = 1%; 3 = 0.1%; 4 = None. (F) Different amounts of glycerol added prior to printing, 1 = 10%; 2 = 1%; 3 = 0.1%; 4 = 1% Tween 20. (G) Incubation of different concentrations of spotted antibody (as in (D)) for 2 h at room temperature with a 4% formalin solution. Storage of a printed plate for 2 days at room temperature (H), and at −20 °C (I), prior to antigen detection. Examples of: a 25-spot array using 0.4 mm pins, 0.66 mm pitch (J); a 53-spot array using 0.2 mm pins, 0.45 mm pitch (K), and a 101-spot array using 0.2 mm pins, 0.32 mm pitch. In panels D–L printed antibody was detected with an Alexa-488 conjugated secondary antibody.
2.5. Identification of functional recombinant antibody plasmids by transient transfection
The rearranged variable heavy and light chain antibody regions were amplified from hybridoma cDNA by RT-PCR and recombined into one PCR product using a fusion PCR-based strategy that, after cloning into a suitable expression vector, enables the expression of a recombinant antibody from a single expression vector, as described [10]. In brief, the fused PCR products from each hybridoma were ligated into an expression vector and the mixture used to transform chemically competent bacteria. Plasmids were purified from 96 bacterial colonies for each hybridoma. Suspension cultures (1 ml) of HEK293 cells were transfected in 24-well plates with 2.5 μg plasmid DNA using Metafectin (Biontex Laboratories), supernatants harvested after 3 days and tested by ELISA. The plasmids encoding the antibodies described in the study are openly available from Addgene (http://www.addgene.org).
3. Results
3.1. The printing of protein arrays in 96-well plates permits the parallel screening of a large number of antigens
To increase the number of monoclonal antibodies that could be selected in parallel from a single mouse immunized with multiple antigens, we aimed to print small protein microarrays at the base of 96-well microtitre plates (Fig. 1A). We have developed an approach that enables the expression of enzymatically monobiotinylated proteins so that they can be captured on streptavidin-coated microtitre plates [11,15]. Therefore, to determine the optimal arraying parameters, and to investigate the effectiveness of different mouse antibody detection methods, we spotted a purified biotinylated mouse antibody as a control, and used different secondary antibodies on the resulting arrays (Fig. 1B–L). We found that both non-fluorescent (Fig. 1B) and fluorescent detection methods using either Alexa-568 (Fig. 1C) or Alexa-488 (Fig. 1D) conjugated secondary antibodies gave good signal to noise ratios. A dilution series of the biotinylated mouse antibody ranging from 50 to 500 ng/μl demonstrated that a printing concentration of 250 ng/μl produced good spot morphology, and was therefore used for future experiments (Fig. 1D). We also tested the influence of the non-ionic detergent, Tween-20, which has been previously reported to improve spot morphology [18]; we found that spot diameter increased with increasing Tween-20 concentration (Fig. 1E). Purified proteins can be stored unfrozen at −20 °C in 50% glycerol, and while desirable for long term antigen storage, we found that the addition of even small amounts (0.1%) of glycerol inhibited the efficient immobilisation of the biotinylated antibody to the streptavidin-coated surface (Fig. 1F). Because we are ultimately interested in selecting antibodies that can recognise antigen in fixed wholemount tissue, antibodies will be eventually screened for their ability to recognise fixed epitopes on the array. Therefore, we tested the impact of formalin fixation on the arrayed protein and found that this did not impair detection significantly (Fig. 3G); indeed, we noticed an overall stronger signal, possibly due to the cross-linking of proteins during fixation thereby increasing the amount of protein retained on the plate during washing steps. We also investigated the optimal storage conditions for the printed arrays. We found that robust signals were still apparent after 2 days, regardless of whether the arrays were stored either dry or in PBS (data not shown), or stored at room temperature (Fig. 1H) or at −20 °C (Fig. 1I). While we have not systematically tested longer-term storage, we can at least say that the arrays retain good signal to noise ratios when stored dry for up to 2 weeks at room temperature. Finally, we should add a concluding cautionary note that these printing and storage parameters were determined using a single monoclonal antibody rather than a diverse set of proteins, which could very well differ in their properties.
Fig. 3.
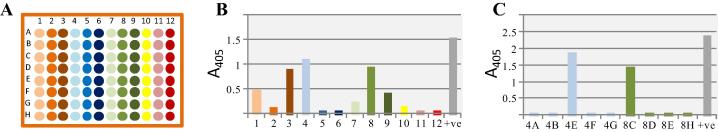
Small scale transfection of pooled plasmids facilitates the detection of those plasmids encoding functional recombinant antibodies. Plasmid DNA was purified from 96 bacterial colonies and pools of eight clones from each column were made. (B) All twelve pools were transfected into small HEK293 cell cultures and the resulting supernatants tested by ELISA, positive pools were from columns 1, 3, 4, 8 and 9. Positive (+ve) control is the anti-Cd4 antibody used to detect the Cd4-tag on the immobilised recombinant antigen. (C) Transfections and ELISAs from individual clones from columns 4 and 8 were repeated to identify plasmids 4E and 8C as encoding functionally-positive antibodies.
We also investigated the density of spots that could be arrayed in each well using solid printing pins with a diameter of either 0.2 or 0.4 mm. The larger pins were able to print a 5 × 5 array into each well (Fig. 1J) while the smaller pins enabled the printing of a 53-spot array (Fig. 1K), which could be increased to 101 spots by decreasing the distance between spots (Fig. 1L). Taken together, these experiments have established the parameters required for printing and storing antigen arrays that are suitable for monoclonal antibody screening against many antigens in parallel.
3.2. Antigen microarray screening enables the early and rapid detection of specific monoclonal antibodies
Previous work in the laboratory has identified a network of 188 extracellular protein interactions between 92 zebrafish cell surface and secreted proteins [11–13,19]. Elucidating the function of these interactions in early vertebrate development will be aided by selecting highly specific monoclonal antibodies that work on fixed wholemount tissue. Due to the paucity of high quality zebrafish antibody reagents, this has necessitated the development of methods to select antibodies to many different targets in parallel. We therefore wished to determine whether the protein antigen array could be used for the simultaneous screening of hybridoma supernatants from a single mouse immunised with many different zebrafish antigens. Hydridomas were generated from splenocytes taken from a mouse immunised with 20 different proteins and plated at a low density (on average, less than 1 hybridoma per well) over twelve 96-well plates. Arrays containing all 20 purified biotinylated proteins were printed (Fig. 2A), and the undiluted hybridoma supernatants were transferred to these plates to assess binding specificity; this approach therefore allows the convenient simultaneous screening of over 23,000 (20 × 12 × 96) antibody binding tests. Hybridomas secreting specific antibodies were easily identified as discrete positively-stained spots distinct from the controls on the array (Fig. 2B). An important feature of screening hybridoma supernatants against many antigens in parallel is that antibodies that are cross-reactive with multiple antigens can be identified at an early stage. Within our library of zebrafish cell surface receptor proteins, there are several examples of paralogous proteins that share a high amount of protein sequence identity [12,20]. Within the pool of 20 immunised proteins were two members of the same paralogous protein family: Pcam and Ncam, which share 66% amino acid identity [21]. During our antibody specificity screen, we were able to identify antibodies that were cross-reactive and could bind both Pcam and Ncam (Fig. 2C); these cross-reactive antibodies could therefore be excluded. Similarly, antibodies recognising the purification tags common to all arrayed proteins were easily and rapidly detected and also excluded (Fig. 2D). A smaller array of 11 antigens is shown as an example where specific antibodies against zebrafish Robo1, Brevican and Ncam2 were identified (Fig. 2E–H). Taken together, these data show that our array facilitates the early screening of antibody specificity against many antigens in parallel and is useful for identifying and excluding cross-reactive antibodies.
Fig. 2.

Antigen microarrays enable early screening of antibody specificity. (A) Twenty different recombinant biotinylated zebrafish cell surface and secreted proteins were purified and arrayed as shown, together with three control spots (No. 21). (B) An example of a positive, specific staining signal on antigen 19 (Fgfr1). (C) An example of a cross-reactive antibody that bound two paralogous antigens arrayed in position 10 (Pcam) and 18 (Ncam). (D) An antibody recognising the recombinant protein tags common to all arrayed proteins. (E) A smaller array with 11 different antigens and four control spots that was used to identify antibodies against: (F), antigen 4 (Robo1); (G), antigen 7 (Brevican), and (H), antigen 8 (Ncam2).
3.3. Transfecting plasmid pools of cloned antibodies rapidly identified those encoding functional antibodies
Once a well containing a hybridoma secreting an antibody of interest is identified, time-consuming cellular cloning is normally required to ensure a stable and clonal cell line. We have previously developed an approach to circumvent this by amplifying and cloning both the rearranged antibody heavy and light chain variable regions into a single plasmid so that the antibody can be expressed recombinantly [10]. Initially, we used sequence-based approaches to identify plasmids containing functional antibodies, but our experience from cloning more than a hundred hybridomas identified some limitations. Firstly, the need for two PCR steps to clone the antibodies increased the probability of PCR-based errors in the amplified sequence that would require complete sequencing of more than 1.5 kbp of plasmid to identify. Secondly, despite a low plating density, we sometimes observed more than one hybridoma colony growing per well, causing normally unpaired heavy and light chains to be amplified and paired by PCR, leading to a non-functional antibody. Finally, in some hybridomas, a large proportion of the amplified light chains originated from the aberrant SP2/0 myeloma light chain [10]. Together, these factors sometimes necessitated careful examination of a large number of bacterial clones to identify a specific functional recombinant antibody, slowing the cloning process considerably.
Because the antibodies are cloned into a single expression plasmid, we reasoned that pools and then individual plasmids could be transfected into cells to easily identify which plasmids contained a functional antibody. To test this approach, we purified plasmids from 96 bacterial clones that had been transformed with the amplified antibody regions from each positive hybridoma. Twelve pools of eight plasmids (Fig. 3A) were transfected into small-scale suspension cultures of HEK293 cells and supernatants tested for the presence of functional antibody by ELISA; plasmids within positive pools were individually transfected to identify those encoding a functional antibody. An example screen is shown in Fig. 3B; here, at least one plasmid encoding a functional antibody is present within the eight plasmids pooled from columns 1, 3, 4, 8 and 9. A further round of transfections using individual plasmids from the two columns with the highest signal by ELISA identified those containing the functional antibody (Fig. 3C). The use of this expression approach has considerably simplified the step of identifying which plasmids contain functional antibodies.
4. Discussion
Despite the importance of monoclonal antibodies in basic biomedical research, selecting new antibodies can be a very time- and labour-intensive process with uncertain chances of success. To streamline this process and reduce the number of animals used, we have developed a pooled immunisation strategy followed by recombinant cloning of the antibodies into a single expression plasmid [10]. Here, we have addressed two limitations that prevented further scaling of this approach. By developing an antigen array, we have reduced the volume of hybridoma supernatant required for screening so that the number of antigens per mouse can be increased from a maximum of five to over one hundred, an increase in scale that could not be achieved by miniaturising the screening ELISAs in either 384 or 1536-well plates. In addition, we have developed an expression-based approach using pooled plasmids to simplify the identification of those plasmids encoding functional antibodies.
In addition to increasing the throughput of the approach, screening hybridoma supernatants by antigen microarray had the advantage of identifying cross-reacting antibodies at an early stage during the screening process to exclude them. Target specificity can be a problem for monoclonal antibodies and lead to misleading results, or dangerous consequences if used therapeutically. The increasing availability of large recombinant protein libraries has highlighted this problem even for commercially-available antibodies [22,23].
There are many different methods developed to select affinity reagents which do not require the use of animals and/or bypass the need to generate hybridomas. While these methods streamline some aspects of monoclonal antibody selection, in vivo immunisation typically results in high affinity antibodies due to the processes of somatic hypermutation, and generating hybridomas results in enough antibody to permit functional screening prior to selecting them for cloning. Reducing the amount of supernatant required for specificity screening by microarray leaves enough hybridoma supernatant for further functional testing such as screening for the ability of the antibody to work in applications such as Western blotting, immunoprecipitation, immunohistochemistry, or other properties such as activating signalling pathways or blocking the binding of other proteins. Similar antigen-array methods have been used for antibody production procedures, but have been used either at a later stage after “shotgun” immunisation approaches for target deconvolution [24], or directly spotting the hybridoma supernatants making screening for cross-reactive antibodies more challenging [25]. A further advantage of cloning the antibodies recombinantly is that the amplified antibody light and heavy chains can be subcloned into a variety of expression plasmids that encode different antibody functionalities such as switching the isotype and adding recombinant protein tags for detection or purification of the antibodies.
In summary, by combining pooled animal immunisation, hybridoma generation, antigen microarrays and a convenient method to identify plasmids encoding functional recombinant antibodies, we have addressed two major bottlenecks that prevented increasing the scale of a method for the parallel selection of high-quality monoclonal antibodies.
Acknowledgments
This work was supported by grants from the National Institutes of Health [RO1 NS063400] and the Wellcome Trust [098051]. We would like to thank David Millrine, Alla Fane-Dremucheva and Shahnaz Yusaf for their contributions to this project.
Contributor Information
Nicole Staudt, Email: ns8@sanger.ac.uk.
Nicole Müller-Sienerth, Email: nms@sanger.ac.uk.
Gavin J. Wright, Email: gw2@sanger.ac.uk.
References
- 1.Buss N.A., Henderson S.J., McFarlane M., Shenton J.M., de Haan L. Monoclonal antibody therapeutics: history and future. Curr. Opin. Pharmacol. 2012;12:615–622. doi: 10.1016/j.coph.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Kohler G., Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 3.Tomita M., Tsumoto K. Hybridoma technologies for antibody production. Immunotherapy. 2011;3:371–380. doi: 10.2217/imt.11.4. [DOI] [PubMed] [Google Scholar]
- 4.McCafferty J., Griffiths A.D., Winter G., Chiswell D.J. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 5.Geyer C.R., McCafferty J., Dubel S., Bradbury A.R., Sidhu S.S. Recombinant antibodies and in vitro selection technologies. Methods Mol. Biol. 2012;901:11–32. doi: 10.1007/978-1-61779-931-0_2. [DOI] [PubMed] [Google Scholar]
- 6.Wrammert J., Smith K., Miller J., Langley W.A., Kokko K., Larsen C., Zheng N.Y., Mays I., Garman L., Helms C., James J., Air G.M., Capra J.D., Ahmed R., Wilson P.C. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lightwood D.J., Carrington B., Henry A.J., McKnight A.J., Crook K., Cromie K., Lawson A.D. Antibody generation through B cell panning on antigen followed by in situ culture and direct RT-PCR on cells harvested en masse from antigen-positive wells. J. Immunol. Methods. 2006;316:133–143. doi: 10.1016/j.jim.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Love J.C., Ronan J.L., Grotenbreg G.M., van der Veen A.G., Ploegh H.L. A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat. Biotechnol. 2006;24:703–707. doi: 10.1038/nbt1210. [DOI] [PubMed] [Google Scholar]
- 9.Jin A., Ozawa T., Tajiri K., Obata T., Kondo S., Kinoshita K., Kadowaki S., Takahashi K., Sugiyama T., Kishi H., Muraguchi A. A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nat. Med. 2009;15:1088–1092. doi: 10.1038/nm.1966. [DOI] [PubMed] [Google Scholar]
- 10.Crosnier C., Staudt N., Wright G.J. A rapid and scalable method for selecting recombinant mouse monoclonal antibodies. BMC Biol. 2010;8:76. doi: 10.1186/1741-7007-8-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bushell K.M., Sollner C., Schuster-Boeckler B., Bateman A., Wright G.J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Res. 2008;18:622–630. doi: 10.1101/gr.7187808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin S., Sollner C., Charoensawan V., Adryan B., Thisse B., Thisse C., Teichmann S., Wright G.J. Construction of a large extracellular protein interaction network and its resolution by spatiotemporal expression profiling. Mol. Cell. Proteomics. 2010;9:2654–2665. doi: 10.1074/mcp.M110.004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sollner C., Wright G.J. A cell surface interaction network of neural leucine-rich repeat receptors. Genome Biol. 2009;10:R99. doi: 10.1186/gb-2009-10-9-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerr J.S., Wright G.J. Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor–ligand interactions. J. Vis. Exp. 2012 doi: 10.3791/3881. (e3881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y., Gallagher-Jones M., Barker C., Wright G.J. A benchmarked protein microarray-based platform for the identification of novel low-affinity extracellular protein interactions. Anal. Biochem. 2012;424:45–53. doi: 10.1016/j.ab.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durocher Y., Perret S., Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartholdson S.J., Bustamante L.Y., Crosnier C., Johnson S., Lea S., Rayner J.C., Wright G.J. Semaphorin-7A is an erythrocyte receptor for P. falciparum merozoite-specific TRAP homolog, MTRAP. PLoS Pathog. 2012;8:e1003031. doi: 10.1371/journal.ppat.1003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Tao S.C., Zhu H., Schneck J.P. High-throughput lectin microarray-based analysis of live cell surface glycosylation. Curr. Protoc. Protein Sci. 2011 doi: 10.1002/0471140864.ps1209s63. (Chapter 12, Unit12 19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Powell G.T., Wright G.J. Jamb and jamc are essential for vertebrate myocyte fusion. PLoS Biol. 2011;9:e1001216. doi: 10.1371/journal.pbio.1001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charoensawan V., Adryan B., Martin S., Sollner C., Thisse B., Thisse C., Wright G.J., Teichmann S.A. The impact of gene expression regulation on evolution of extracellular signaling pathways. Mol. Cell. Proteomics. 2010;9:2666–2677. doi: 10.1074/mcp.M110.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizuno T., Kawasaki M., Nakahira M., Kagamiyama H., Kikuchi Y., Okamoto H., Mori K., Yoshihara Y. Molecular diversity in zebrafish NCAM family: three members with different VASE usage and distinct localization. Mol. Cell. Neurosci. 2001;18:119–130. doi: 10.1006/mcne.2001.1007. [DOI] [PubMed] [Google Scholar]
- 22.Jensen B.C., Swigart P.M., Simpson P.C. Ten commercial antibodies for alpha-1-adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch. Pharmacol. 2009;379:409–412. doi: 10.1007/s00210-008-0368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeong J.S., Jiang L., Albino E., Marrero J., Rho H.S., Hu J., Hu S., Vera C., Bayron-Poueymiroy D., Rivera-Pacheco Z.A., Ramos L., Torres-Castro C., Qian J., Bonaventura J., Boeke J.D., Yap W.Y., Pino I., Eichinger D.J., Zhu H., Blackshaw S. Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.016253. (O111 016253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu S., Li Y., Liu G., Song Q., Wang L., Han Y., Zhang Y., Song Y., Yao X., Tao Y., Zeng H., Yang H., Wang J., Zhu H., Chen Z.N., Wu L. A protein chip approach for high-throughput antigen identification and characterization. Proteomics. 2007;7:2151–2161. doi: 10.1002/pmic.200600923. [DOI] [PubMed] [Google Scholar]
- 25.De Masi F., Chiarella P., Wilhelm H., Massimi M., Bullard B., Ansorge W., Sawyer A. High throughput production of mouse monoclonal antibodies using antigen microarrays. Proteomics. 2005;5:4070–4081. doi: 10.1002/pmic.200401279. [DOI] [PubMed] [Google Scholar]