

Graphical abstract
Tyrosinase from walnut leaves (Juglans regia) corresponding to the known jrPPO1 sequence was purified and characterized. Two major tyrosinase forms differing only in their C-termini were identified. The first form (jrPPO1(Asp101 → Pro444)) is one amino acid shorter than the second form (jrPPO1(Asp101 → Arg445)).

Keywords: Juglans regia, Type-3 copper protein, Polyphenol oxidase, Tyrosinase, Catechol oxidase, Laccase
Abbreviations: ACN, acetonitrile; CT, charge transfer; ESI-MS, Electrospray Ionisation Mass Spectrometry; FPLC, Fast Protein Liquid Chromatography; IEF, isoelectric focusing; Jr, Juglans regia; nanoESI-QTOF, nanoElectrospray ionisation quadrupole-time-of-flight mass spectrometry; LC–MS/MS, liquid chromatography–mass spectrometry/mass spectrometry; MS, mass spectrometry; nanoUHPLC–ESI-MS/MS, nanoUltra high performance liquid chromatography–electrospray tandem mass spectrometry; PEG-4000, polyethylene glycol 4000; PMSF, phenylmethylsulfonyl fluoride; PPO, polyphenol oxidase; SDS, sodium dodecyl sulfate; SDS–PAGE, sodium dodecyl sulfate–polyacrylamid gel electrophoresis; UV/Vis spectroscopy, ultraviolet/visible spectroscopy
Highlights
-
•
A tyrosinase from walnut leaves (Juglans regia) was purified.
-
•
The Juglans regia enzyme was determined to have a mass of 39,047 Da and a pI of 5.2.
-
•
Addition of 2 equivalent H2O2 leads to oxytyrosinase.
-
•
The proteolytically activated tyrosinase was clearly identified as PPO1 Juglans regia.
Abstract
Polyphenol oxidase (PPO) is a type-3 copper enzyme catalyzing the oxidation of phenolic compounds to their quinone derivates, which are further converted to melanin, a ubiquitous pigment in living organisms. In this study a plant originated tyrosinase was isolated from walnut leaves (Juglans regia) and biochemically characterized. It was possible to isolate and purify the enzyme by means of an aqueous two-phase extraction method followed by chromatographic purification and identification. Interestingly, the enzyme showed a rather high monophenolase activity considering that the main part of plant PPOs with some exceptions solely possess diphenolase activity. The average molecular mass of 39,047 Da (Asp101 → Arg445) was determined very accurately by high resolution mass spectrometry. This proteolytically activated tyrosinase species was identified as a polyphenol oxidase corresponding to the known jrPPO1 sequence by peptide sequencing applying nanoUHPLC–ESI-MS/MS. The polypeptide backbone with sequence coverage of 96% was determined to start from Asp101 and not to exceed Arg445.
Introduction
Polyphenol oxidases (PPO) are metalloenzymes containing a type-3 copper center occurring in many organisms including plants, fungi and bacteria (Mayer, 2006; Selinheimo et al., 2007). Representatives of this class are catechol oxidases, tyrosinases and laccases. Tyrosinases, a class of bifunctional PPOs, use molecular oxygen to catalyze the oxidation of various monophenols to o-diphenols (cresolase/monophenolase activity; EC 1.14.18.1), and the subsequent oxidation of o-diphenols to the corresponding o-quinones (catecholase/diphenolase activity; EC 1.10.3.1). Catechol oxidase catalyzes exclusively the oxidation of o-diphenols to o-quinones, lacking the hydroxylation reaction (Mayer, 2006; Mayer and Harel, 1979). Laccases (EC 1.10.3.2) can oxidize a wide range of compounds including aminophenols, monophenols, o- and p-diphenols by removing single electrons from the reducing group of the substrate and generate free radicals (Mayer and Harel, 1979; Sanchez-Amat and Solano, 1997).
Melanines generated by polymerisation of quinones are dying-compounds which are responsible for the damage-induced browning of many fruits and vegetables (Martinez and Whitaker, 1995; Yoruk and Marshall, 2003). While the physiological function of PPOs in many plants is still not clear, there is strong evidence that PPOs play a key role in parasite and pathogen resistance in some species. Protective effects of PPOs have generally been attributed to the generation of reactive quinones (Steffens et al., 1994; Van Gelder et al., 1997; Vaughn et al., 1988).
Type-3 copper proteins contain two copper ions, each coordinated by three histidine residues. During the catalytic reaction, the type-3 copper center of tyrosinase exists in three different states. The reduced deoxy state [Cu(I)–Cu(I)] binds molecular oxygen and results in the oxy state [Cu(II)–O22−–Cu(II)]. In the oxy state, peroxide is bound in a μ–η2:η2 bridging mode (Kitajima et al., 1989). The met state [Cu(II)–Cu(II)] is assumed as the resting state of the copper site, where Cu(II) ions are bridged by a water molecule or hydroxyl ion (Solomon et al., 1996).
The functional importance of the PPO-activity in walnut hull was first described in 1991 (Piffaut and Metche, 1991). Walnut (Juglans regia) PPO was attributed to possess a putative pathogenic resistance as early as 1911 (Cook et al., 1911). Walnut leaves have a high content of various polyphenols, some of which might be important in pathogenic resistance (Colaric et al., 2005; Solar et al., 2006). PPO from walnut leaves has been poorly studied in the past (Escobar et al., 2008; Piffaut and Metche, 1991). Escobar et al. (2008) demonstrated that PPO of walnut is encoded by a single gene, jrPPO1, which is constitutively expressed in all green, herbaceous tissues of walnut.
In this work a tyrosinase from walnut leaves (J. regia) is extracted and purified by means of fast protein liquid chromatography (FPLC) and characterized by molecular mass determination (SDS–PAGE, nanoESI-QTOF), isoelectrical focusing (IEF), UV/Vis spectroscopy and sequence analysis (nanoUHPLC–ESI-MS/MS). The purification method described in this paper allows a very efficient isolation of two forms from walnut tyrosinase, identified and characterized by mass spectrometry based methodology providing sequence information and the highly accurate mass.
Results and discussion
Extraction and purification of tyrosinase from J. regia
The applied extraction method was based on a technique described for a latent tyrosinase from mushrooms (Agaricus bisporus) and had proven to be effective (Mauracher et al., 2014). Several consecutive aqueous two-phase separations using triton X-114 and PEG-4000 (polyethylene glycol) resulted in a quantitative removal of hydrophobic dyes, non-target proteins and other hydrophobic compounds. Hence, the obtained polyphenol free and clear protein solution was very well suitable for the subsequent chromatographic purification steps.
Using a cation exchange column (SP-Sepharose) as a first purification step proved being very effective in terms of removing a major part of non-target protein. The target PPO eluted late in the sodium chloride gradient together with one of in total three co-eluted heme-proteins. This is clearly shown in Fig. 1A by following the absorption at 410 nm characteristic for prosthetic heme groups. Implying some characterization experiments (data not shown) it can be assumed that the interfering protein is a peroxidase. This was also observed by Trémolières and Bieth (1984), Rompel et al. (1999a, 2012). Fractions showing highest tyrosinase activity were pooled and loaded onto the second cation exchange column (MonoS) where two major forms of the tyrosinase, which are named after the chromatographic elution order, forms 1 and 2, were eluted consecutively but efficiently separated early in the gradient (see Fig. 1B). On the cation exchange column (MonoS) the remaining heme protein (peroxidase) could be very effectively separated from the tyrosinase (see Fig. 1B). For polishing reasons fractions of the two tyrosinase forms were separately applied to the same cation exchange column (MonoS) resulting in a very high purity of the two tyrosinase species (see Figs. 1C/D and 2).
Fig. 1.

Chromatographic runs (FPLC). (A) Cation exchange chromatography on SP-Sepharose. (B) Cation exchange chromatography on MonoS. (C) Cation exchange chromatography on MonoS jrPPO1(Asp101 → Pro444). (D) Cation exchange chromatography on MonoS jrPPO1(Asp101 → Arg445). Legend:  , UV absorbance at 280 nm [mAU];
, UV absorbance at 280 nm [mAU];  , UV absorbance at 410 nm [mAU];
, UV absorbance at 410 nm [mAU];  , monophenolase activity [U/ml];
, monophenolase activity [U/ml];  , gradient [% buffer B];
, gradient [% buffer B];  , conductivity [mS/cm] (f = ∼1.5).
, conductivity [mS/cm] (f = ∼1.5).
Fig. 2.

Analytic one- and two-dimensional SDS–PAGEs. (A) Purified jrPPO1(Asp101 → Pro444) and jrPPO1(Asp101 → Arg445). Staining: Coomassie Brilliant Blue. Mw marker [kDa] in the middle lane. (B) In-gel activity staining by l-tyrosine. Mw marker [kDa] in the middle lane. (C) Coomassie-stained two-dimensional polyacrylamide gel of jrPPO1(Asp101 → Pro444) and jrPPO1(Asp101 → Arg445), which were separated in the first dimension on an immobilized pH gradient strip (pH 4.0–7.0) and in the second dimension on a 12% polyacrylamide gel.
Often isoforms of polyphenol oxidase were found during isolation and purification. Currently six amino acid sequences (PPO1 to 6) are known for a polyphenol oxidase originating from Agaricus bisporus (Li et al., 2011; Mauracher et al., 2014; Weijn et al., 2013; Wichers et al., 2003; Wu et al., 2010). Eicken et al. (1998) isolated two catechol oxidases with different amino acid sequences from sweet potato (Ipomoea batatas) having molecular masses of 39 and 40 kDa, respectively. Four PPO isoforms were purified from coats and pods of green bean (Phaseolus vulgaris L.), and their molecular weights were estimated to be 57.5, 54, 46 and 39 kDa, respectively (Guo et al., 2009). In this manuscript it is demonstrated that two forms of walnut tyrosinase, arising from the same gene (jrPPO1, Escobar et al., 2008), but proteolytic cleaved on different positions were purified and characterized.
Electrophoresis study
Purity of the two tyrosinase forms was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE). Whereas no bands corresponding to a non-target protein were visible, both forms showed a single band at around 39 kDa (see Fig. 2A). The in-gel activity was measured following non reducing SDS–PAGE as the reduction is detrimental to the activity of the enzyme. In this non reduced form the enzyme displays a higher electrophoretic mobility, which is probably due to its more compact conformation. Under non-reducing conditions the intramolecular disulfide bridges are intact and can therefore stabilize the enzyme’s conformation (see Fig. 2B). Moreover, the IEF (both samples applied) resulted in two spots at a pI of 5.1 and 5.2, respectively (see Fig. 2C). This matches with the theoretically calculated pI of 5.25 (ProtParam, ExPASy.org) corresponding to the identified amino acid sequence jrPPO1(Asp101–Pro444) and pI of 5.35 for jrPPO1(Asp101–Arg445) (UniProt.: COLU17) as described below.
Protein identification and sequence confirmation
The isolated and purified two forms of tyrosinases were clearly identified as PPO1 J. regia (UniProt.: COLU17, Escobar et al., 2008) by means of nanoUHPLC–ESI-MS/MS yielding a maximum sequence coverage of 96% (jrPPO1(Asp101 → Pro444)) and 96% (jrPPO1(Asp101 → Arg445)) yellow highlighted in Fig. 3. 91 (jrPPO1(Asp101 → Pro444)) and 71 (jrPPO1(Asp101 → Arg445)) tryptic peptides were identified and listed in Table 1 (jrPPO1(Asp101 → Pro444)) and Table 2 (jrPPO1(Asp101 → Arg445)). Hence, the two separated and purified tyrosinase forms distinguish themselves in possessing or lacking the terminal amino acid Arg445. This was confirmed as well by the mass determination of the intact protein by nanoESI-QTOF.
Fig. 3.

Sequence of jrPPO1. Highlighted are:  , copper coordinating histidines;
, copper coordinating histidines;  , Peptides identified by nanoUHPLC–ESI-MS/MS are highlighted yellow. Red lines (
, Peptides identified by nanoUHPLC–ESI-MS/MS are highlighted yellow. Red lines ( ) indicates the start/end of the jrPPO1(Asp101 → Pro444) and jrPPO1(Asp101 → Arg445) sequence, respectively, as deduced from matching with the molecular mass determined for the isolated protein by nanoESI-QTOF. Black thin lines (
) indicates the start/end of the jrPPO1(Asp101 → Pro444) and jrPPO1(Asp101 → Arg445) sequence, respectively, as deduced from matching with the molecular mass determined for the isolated protein by nanoESI-QTOF. Black thin lines ( ) indicate the cleavage positions of the minor sub-species (A, B) only found in jrPPO1(Asp101 → Pro444).
) indicate the cleavage positions of the minor sub-species (A, B) only found in jrPPO1(Asp101 → Pro444).
Table 1.
List of peptides found by nanoUHPLC–ESI-MS/MS protein identification experiments of jrPPO1(Asp101 → Pro444). Sequence coverage: 96%.
| Start–End | Sequence | Modifications | XCorr | Delta Mass [ppm] | Enzyme for digestion | Peptide mass |
|---|---|---|---|---|---|---|
| 101–108 | DPVSAPEL | 1.5 | −0.93 | Chymotrypsin | 826.406 | |
| 101–110 | DPVSAPELTL | 1.97 | 0.46 | Chymotrypsin | 1040.539 | |
| 101–131 | DPVSAPELTLcSEADLPAGALPVNccPPTSK | C11(Carbamidomethyl); C25(Carbamidomethyl); C26(Carbamidomethyl) | 6.51 | −1.72 | Trypsin | 3265.524 |
| 111–136 | cSEADLPAGALPVNccPPTSKKIKDF | C1(Carbamidomethyl); C15(Carbamidomethyl); C16(Carbamidomethyl) | 5.5 | −1.18 | Chymotrypsin | 2874.368 |
| 122–136 | PVNccPPTSKKIKDF | C4(Carbamidomethyl); C5(Carbamidomethyl) | 3.4 | 0.91 | Chymotrypsin | 1789.887 |
| 132–146 | KIKDFVLPSQNTPLR | 4.86 | −2 | Trypsin | 1755.000 | |
| 133–146 | IKDFVLPSQNTPLR | 5.05 | −2.46 | Trypsin | 1626.905 | |
| 135–146 | DFVLPSQNTPLR | 3.06 | −118 | Trypsin | 1385728 | |
| 139–153 | PSQNTPLRVRPAAHL | 1.62 | 2.05 | Chymotrypsin | 1655.925 | |
| 146–153 | RVRPAAHL | 2.03 | −1.21 | Chymotrypsin | 918.550 | |
| 146–158 | RVRPAAHLVDNDY | 3 | 1.99 | Chymotrypsin | 1524.782 | |
| 146–162 | RVRPAAHLVDNDYIAKY | 4.22 | 0 | Chymotrypsin | 2000.059 | |
| 147–161 | VRPAAHLVDNDYIAK | 5.17 | −1.54 | Trypsin | 1680.892 | |
| 154–162 | VDNDYIAKY | 2.51 | 0.48 | Chymotrypsin | 1099.519 | |
| 162–170 | YNKGIELmK | M8(Oxidation) | 2.17 | −1.16 | Trypsin | 1110.573 |
| 163–168 | NKGIEL | 1.85 | −0.4 | Chymotrypsin | 672.380 | |
| 163–180 | NKGIELmKSLPADDPRSF | M7(Oxidation) | 5.07 | −1.8 | Chymotrypsin | 2033.021 |
| 163–180 | NKGIELMKSLPADDPRSF | 4.73 | −0.13 | Chymotrypsin | 2017.030 | |
| 169–180 | mKSLPADDPRSF | M1(Oxidation) | 3.53 | −0.13 | Chymotrypsin | 1378.654 |
| 169–180 | MKSLPADDPRSF | 2.91 | −1.08 | Chymotrypsin | 1362.658 | |
| 171–178 | SLPADDPR | 1.7 | −1.54 | Trypsin | 869.422 | |
| 173–180 | PADDPRSF | 1.65 | −0.44 | Chymotrypsin | 903.408 | |
| 181–190 | TQQANVHcAY | C8(Carbamidomethyl) | 2.21 | −1.43 | Chymotrypsin | 1190.512 |
| 181–195 | TQQANVHcAYcDGAY | C8(Carbamidomethyl); C11(Carbamidomethyl) | 2.5 | −1.49 | Chymotrypsin | 1756.690 |
| 196–203 | TQVGFPDL | 2.03 | 0.15 | Chymotrypsin | 875.439 | |
| 196–205 | TQVGFPDLSL | 2.18 | 0.34 | Chymotrypsin | 1075.555 | |
| 219–226 | YYVYFFEK | 2.61 | −2.57 | Trypsin | 1157.540 | |
| 219–230 | YYVYFFEKILGK | 2.12 | −1.23 | Trypsin | 1568.825 | |
| 224–231 | FEKILGKL | 2.68 | 0.42 | Chymotrypsin | 946.585 | |
| 225–237 | EKILGKLIGDPTF | 3.38 | −0.64 | Chymotrypsin | 1429.817 | |
| 229–237 | GKLIGDPTF | 2.45 | −0.14 | Chymotrypsin | 946.512 | |
| 231–268 | LIGDPTFALPFWNWDSPPGmQLPSLYAVSNSAIYDPLR | M20(Oxidation) | 6.57 | 0.27 | Trypsin | 4264.099 |
| 231–268 | LIGDPTFALPFWNWDSPPGMQLPSLYAVSNSAIYDPLR | 4.55 | −1.96 | Trypsin | 4248.094 | |
| 245–256 | DSPPGmQLPSLY | M6(Oxidation) | 2.26 | 1.09 | Chymotrypsin | 1319.608 |
| 245–256 | DSPPGMQLPSLY | 2.72 | 0.57 | Chymotrypsin | 1303.612 | |
| 257–267 | AVSNSAIYDPL | 1.69 | −0.12 | Chymotrypsin | 1148.571 | |
| 257–280 | AVSNSAIYDPLRNANHQPPTIIDL | 2.94 | 1.12 | Chymotrypsin | 2618.348 | |
| 265–280 | DPLRNANHQPPTIIDL | 3.09 | −1.55 | Chymotrypsin | 1812.945 | |
| 268–280 | RNANHQPPTIIDL | 2.32 | 0.19 | Chymotrypsin | 1487.784 | |
| 268–299 | RNANHQPPTIIDLDYGETSESTTTTDQVPSNL | 7.47 | −3.7 | Chymotrypsin | 3513.636 | |
| 269–300 | NANHQPPTIIDLDYGETSESTTTTDQVPSNLK | 7.62 | −0.96 | Trypsin | 3485.640 | |
| 281–299 | DYGETSESTTTTDQVPSNL | 2.95 | 2.68 | Chymotrypsin | 2043.881 | |
| 300–316 | KImYRQmVSGAKNPTLF | M3(Oxidation); M7(Oxidation) | 1.47 | 0.1 | Chymotrypsin | 2015.033 |
| 301–311 | ImYRQmVSGAK | M2(Oxidation); M6(Oxidation) | 2.21 | −0.22 | Trypsin | 1314.642 |
| 304–316 | RQmVSGAKNPTLF | M3(Oxidation) | 4.59 | 1.53 | Chymotrypsin | 1463.757 |
| 304–316 | RQMVSGAKNPTLF | 4.15 | −0.15 | Chymotrypsin | 1447.760 | |
| 304–317 | RQmVSGAKNPTLFF | M3(Oxidation) | 3.57 | 0.8 | Chymotrypsin | 1610.825 |
| 304–317 | RQMVSGAKNPTLFF | 3.47 | 0.88 | Chymotrypsin | 1594.830 | |
| 312–322 | NPTLFFGSPYR | 3.89 | −2.13 | Trypsin | 1297.642 | |
| 322–344 | RAGDEPDPGAGTIESTPHNNIHL | 6.32 | −0.98 | Chymotrypsin | 2397.128 | |
| 322–345 | RAGDEPDPGAGTIESTPHNNIHLW | 8.24 | −0.92 | Chymotrypsin | 2583.207 | |
| 345–361 | WTGDDTQPNIENmGNFY | M13(Oxidation) | 1.9 | 2.95 | Chymotrypsin | 2016.821 |
| 346–360 | TGDDTQPNIENmGNF | M12(Oxidation) | 3.18 | 0.09 | Chymotrypsin | 1667.673 |
| 346–360 | TGDDTQPNIENMGNF | 2.79 | 0.34 | Chymotrypsin | 1651.679 | |
| 346–361 | TGDDTQPNIENmGNFY | M12(Oxidation) | 3.34 | −0.1 | Chymotrypsin | 1830.736 |
| 346–361 | TGDDTQPNIENMGNFY | 3.3 | 2.27 | Chymotrypsin | 1814.745 | |
| 361–370 | YSAGRDPIFF | 2.84 | 1.15 | Chymotrypsin | 1171.567 | |
| 362–370 | SAGRDPIFF | 2.25 | 0.63 | Chymotrypsin | 1008.503 | |
| 362–380 | SAGRDPIFFAHHSNVDRmW | M18(Oxidation) | 4.74 | 0.12 | Chymotrypsin | 2258.044 |
| 366–378 | DPIFFAHHSNVDR | 4.07 | −2.16 | Trypsin | 1553.734 | |
| 366–389 | DPIFFAHHSNVDRmWTIWKTLGGK | M14(Oxidation) | 3.01 | −1.14 | Trypsin | 2871.424 |
| 371–380 | AHHSNVDRmW | M9(Oxidation) | 2.93 | −1.01 | Chymotrypsin | 1267.550 |
| 371–380 | AHHSNVDRMW | 3.42 | −0.36 | Chymotrypsin | 1251.556 | |
| 371–383 | AHHSNVDRmWTIW | M9(Oxidation) | 4 | −1.68 | Chymotrypsin | 1667.759 |
| 371–383 | AHHSNVDRMWTIW | 2.33 | 0.11 | Chymotrypsin | 1651.767 | |
| 371–386 | AHHSNVDRmWTIWKTL | M9(Oxidation) | 2.48 | −1.23 | Chymotrypsin | 2009.986 |
| 379–384 | mWTIWK | M1(Oxidation) | 1.88 | −2.53 | Trypsin | 879.429 |
| 379–384 | MWTIWK | 1.88 | −2.18 | Trypsin | 863.434 | |
| 379–389 | mWTIWKTLGGK | M1(Oxidation) | 2.36 | −1.39 | Trypsin | 1335.699 |
| 384–399 | KTLGGKRKDITDPDWL | 3.77 | −1.36 | Chymotrypsin | 1841.997 | |
| 387–399 | GGKRKDITDPDWL | 3.74 | −0.94 | Chymotrypsin | 1499.771 | |
| 387–403 | GGKRKDITDPDWLNSSF | 3.6 | −0.87 | Chymotrypsin | 1934.946 | |
| 390–414 | RKDITDPDWLNSSFFFYDENADPVR | 4.9 | −0.72 | Trypsin | 3046.407 | |
| 391–414 | KDITDPDWLNSSFFFYDENADPVR | 7.02 | −1.3 | Trypsin | 2890.304 | |
| 392–414 | DITDPDWLNSSFFFYDENADPVR | 6.48 | −1.21 | Trypsin | 2762.210 | |
| 392–416 | DITDPDWLNSSFFFYDENADPVRVK | 2.67 | −0.2 | Trypsin | 2989.376 | |
| 405–426 | FYDENADPVRVKVKDcVDNTKL | C16(Carbamidomethyl) | 6.06 | 2.94 | Chymotrypsin | 2624.298 |
| 406–426 | YDENADPVRVKVKDcVDNTKL | C15(Carbamidomethyl) | 7.26 | −2.45 | Chymotrypsin | 2477.216 |
| 406–428 | YDENADPVRVKVKDcVDNTKLRY | C15(Carbamidomethyl) | 7.6 | −1.21 | Chymotrypsin | 2796.383 |
| 407–426 | DENADPVRVKVKDcVDNTKL | C14(Carbamidomethyl) | 6.25 | −1.18 | Chymotrypsin | 2314.156 |
| 407–428 | DENADPVRVKVKDcVDNTKLRY | C14(Carbamidomethyl) | 6.73 | −0.72 | Chymotrypsin | 2633.321 |
| 415–425 | VKVKDcVDNTK | C6(Carbamidomethyl) | 1.48 | −0.38 | Trypsin | 1304.675 |
| 417–427 | VKDcVDNTKLR | C4(Carbamidomethyl) | 3.09 | 0.1 | Trypsin | 1346.697 |
| 427–437 | RYVYQDVEIPW | 2.86 | −0.83 | Chymotrypsin | 1466.718 | |
| 428–439 | YVYQDVEIPWLK | 4.01 | −0.47 | Trypsin | 1551.796 | |
| 428–444 | YVYQDVEIPWLKTKPTP | 3.39 | −0.83 | Trypsin | 2076.091 | |
| 429–437 | VYQDVEIPW | 1.91 | 0.01 | Chymotrypsin | 1147.554 | |
| 429–438 | VYQDVEIPWL | 1.72 | −0.64 | Chymotrypsin | 1260.638 | |
| 431–437 | QDVEIPW | 1.95 | −0.85 | Chymotrypsin | 885.422 | |
| 431–444 | QDVEIPWLKTKPTP | 3.47 | −0.39 | Chymotrypsin | 1650.897 | |
| 438–444 | LKTKPTP | 1.84 | −0.35 | Chymotrypsin | 783.485 |
Table 2.
List of peptides found by nanoUHPLC–ESI-MS/MS protein identification experiments of jrPPO1(Asp101 → Arg445). Sequence coverage: 96%.
| Start–End | Sequence | Modifications | XCorr | Delta Mass [ppm] | Enzyme for digestion | Peptide mass |
|---|---|---|---|---|---|---|
| 101–110 | DPVSAPELTL | 2.12 | −0.71 | Chymotrypsin | 1040.5378 | |
| 101–131 | DPVSAPELTLcSEADLPAGALPVNccPPTSK | C11(Carbamidomethyl); C25(Carbamidomethyl); C26(Carbamidomethyl) | 6.56 | −2.17 | Trypsin | 3265.5230 |
| 111–136 | cSEADLPAGALPVNccPPTSKKIKDF | C1(Carbamidomethyl); C15(Carbamidomethyl); C16(Carbamidomethyl) | 5.93 | 2.45 | Chymotrypsin | 2874.3781 |
| 122–136 | PVNccPPTSKKIKDF | C4(Carbamidomethyl); C5(Carbamidomethyl) | 2.56 | 0.8 | Chymotrypsin | 1789.8865 |
| 132–146 | KIKDFVLPSQNTPLR | 3.15 | −1.79 | Trypsin | 1755.0007 | |
| 133–146 | IKDFVLPSQNTPLR | 5.19 | −2.01 | Trypsin | 1626.9056 | |
| 135–146 | DFVLPSQNTPLR | 3.16 | −0.74 | Trypsin | 1385.7288 | |
| 146–153 | RVRPAAHL | 1.8 | −0.32 | Chymotrypsin | 918.5504 | |
| 146–158 | RVRPAAHLVDNDY | 3.16 | 0.21 | Chymotrypsin | 1524.7795 | |
| 146–162 | RVRPAAHLVDNDYIAKY | 4.25 | 1.41 | Chymotrypsin | 2000.0615 | |
| 147–161 | VRPAAHLVDNDYIAK | 5.28 | −1.87 | Trypsin | 1680.8911 | |
| 154–162 | VDNDYIAKY | 2.58 | 0.48 | Chymotrypsin | 1099.5186 | |
| 163–180 | NKGIELmKSLPADDPRSF | M7(Oxidation) | 4.69 | −2.97 | Chymotrypsin | 2033.0187 |
| 169–180 | mKSLPADDPRSF | M1(Oxidation) | 3.27 | −1.82 | Chymotrypsin | 1378.6521 |
| 169–180 | MKSLPADDPRSF | 2.88 | −0.9 | Chymotrypsin | 1362.6585 | |
| 171–178 | SLPADDPR | 1.84 | −3.57 | Trypsin | 869.4207 | |
| 181–190 | TQQANVHcAY | C8(Carbamidomethyl) | 1.95 | −0.09 | Chymotrypsin | 1190.5132 |
| 181–195 | TQQANVHcAYcDGAY | C8(Carbamidomethyl); C11(Carbamidomethyl) | 2.55 | −1.21 | Chymotrypsin | 1756.6907 |
| 196–205 | TQVGFPDLSL | 2.23 | −0.68 | Chymotrypsin | 1075.5537 | |
| 219–226 | YYVYFFEK | 2.67 | −3.41 | Trypsin | 1157.5389 | |
| 224–231 | FEKILGKL | 2.9 | 0.55 | Chymotrypsin | 946.5852 | |
| 225–237 | EKILGKLIGDPTF | 1.9 | 0.48 | Chymotrypsin | 1429.8183 | |
| 229–237 | GKLIGDPTF | 2.38 | 1.92 | Chymotrypsin | 946.5137 | |
| 231–268 | LIGDPTFALPFWNWDSPPGmQLPSLYAVSNSAIYDPLR | M20(Oxidation) | 7.3 | −2.7 | Trypsin | 4264.0861 |
| 231–268 | LIGDPTFALPFWNWDSPPGMQLPSLYAVSNSAIYDPLR | 4.64 | 0 | Trypsin | 4248.1027 | |
| 245–256 | DSPPGmQLPSLY | M6(Oxidation) | 2.67 | −1.87 | Chymotrypsin | 1319.6038 |
| 245–256 | DSPPGMQLPSLY | 2.09 | 0.48 | Chymotrypsin | 1303.6120 | |
| 257–267 | AVSNSAIYDPL | 1.87 | 0.2 | Chymotrypsin | 1148.5711 | |
| 257–280 | AVSNSAIYDPLRNANHQPPTIIDL | 2.56 | −2.31 | Chymotrypsin | 2618.3388 | |
| 265–280 | DPLRNANHQPPTIIDL | 2.99 | −0.64 | Chymotrypsin | 1812.9466 | |
| 268–280 | RNANHQPPTIIDL | 2.38 | −1.54 | Chymotrypsin | 1487.7817 | |
| 268–299 | RNANHQPPTIIDLDYGETSESTTTTDQVPSNL | 6.4 | −1.69 | Chymotrypsin | 3513.6432 | |
| 269–300 | NANHQPPTIIDLDYGETSESTTTTDQVPSNLK | 7.62 | −0.5 | Trypsin | 3485.6412 | |
| 281–299 | DYGETSESTTTTDQVPSNL | 2.5 | 0.95 | Chymotrypsin | 2043.8771 | |
| 301–311 | ImYRQmVSGAK | M2(Oxidation); M6(Oxidation) | 2.23 | 0.26 | Trypsin | 1314.6423 |
| 304–316 | RQmVSGAKNPTLF | M3(Oxidation) | 4.59 | 2.34 | Chymotrypsin | 1463.7584 |
| 304–316 | RQMVSGAKNPTLF | 4.25 | −3.56 | Chymotrypsin | 1447.7549 | |
| 304–317 | RQmVSGAKNPTLFF | M3(Oxidation) | 3.91 | 0.35 | Chymotrypsin | 1610.8240 |
| 304–317 | RQMVSGAKNPTLFF | 2.97 | 0.3 | Chymotrypsin | 1594.8290 | |
| 312–322 | NPTLFFGSPYR | 3.87 | −1.66 | Trypsin | 1297.6429 | |
| 322–344 | RAGDEPDPGAGTIESTPHNNIHL | 7.11 | −0.59 | Chymotrypsin | 2397.1290 | |
| 322–345 | RAGDEPDPGAGTIESTPHNNIHLW | 8.28 | −3.12 | Chymotrypsin | 2583.2017 | |
| 346–360 | TGDDTQPNIENmGNF | M12(Oxidation) | 3.01 | −1.67 | Chymotrypsin | 1667.6701 |
| 346–360 | TGDDTQPNIENMGNF | 2.75 | −2.03 | Chymotrypsin | 1651.6746 | |
| 346–361 | TGDDTQPNIENmGNFY | M12(Oxidation) | 3.17 | −1.57 | Chymotrypsin | 1830.7333 |
| 346–361 | TGDDTQPNIENMGNFY | 3.05 | −0.21 | Chymotrypsin | 1814.7409 | |
| 361–370 | YSAGRDPIFF | 2.72 | 0.52 | Chymotrypsin | 1171.5663 | |
| 362–370 | SAGRDPIFF | 2.18 | −1.06 | Chymotrypsin | 1008.5013 | |
| 362–380 | SAGRDPIFFAHHSNVDRmW | M18(Oxidation) | 4.16 | −0.72 | Chymotrypsin | 2258.0419 |
| 366–378 | DPIFFAHHSNVDR | 4.06 | −2.08 | Trypsin | 1553.7338 | |
| 371–380 | AHHSNVDRmW | M9(Oxidation) | 3.04 | −0.51 | Chymotrypsin | 1267.5505 |
| 371–383 | AHHSNVDRmWTIW | M9(Oxidation) | 2.42 | 0.27 | Chymotrypsin | 1667.7627 |
| 379–384 | mWTIWK | M1(Oxidation) | 1.92 | −2.6 | Trypsin | 879.4285 |
| 379–384 | MWTIWK | 1.78 | −1.26 | Trypsin | 863.4348 | |
| 384–399 | KTLGGKRKDITDPDWL | 3.14 | −0.9 | Chymotrypsin | 1841.9978 | |
| 387–399 | GGKRKDITDPDWL | 3.04 | −0.61 | Chymotrypsin | 1499.7719 | |
| 387–403 | GGKRKDITDPDWLNSSF | 3.94 | 0.36 | Chymotrypsin | 1934.9489 | |
| 391–414 | KDITDPDWLNSSFFFYDENADPVR | 7.56 | −1.24 | Trypsin | 2890.3046 | |
| 392–414 | DITDPDWLNSSFFFYDENADPVR | 7.01 | −1.48 | Trypsin | 2762.2091 | |
| 405–426 | FYDENADPVRVKVKDcVDNTKL | C16(Carbamidomethyl) | 6.25 | −0.33 | Chymotrypsin | 2624.2891 |
| 406–426 | YDENADPVRVKVKDcVDNTKL | C15(Carbamidomethyl) | 7.3 | −0.6 | Chymotrypsin | 2477.2201 |
| 406–428 | YDENADPVRVKVKDcVDNTKLRY | C15(Carbamidomethyl) | 5.94 | −0.41 | Chymotrypsin | 2796.3849 |
| 407–426 | DENADPVRVKVKDcVDNTKL | C14(Carbamidomethyl) | 6.03 | −0.71 | Chymotrypsin | 2314.1566 |
| 407–428 | DENADPVRVKVKDcVDNTKLRY | C14(Carbamidomethyl) | 6.48 | 2.13 | Chymotrypsin | 2633.3283 |
| 417–427 | VKDcVDNTKLR | C4(Carbamidomethyl) | 2.81 | −0.1 | Trypsin | 1346.6970 |
| 427–437 | RYVYQDVEIPW | 2.69 | −1 | Chymotrypsin | 1466.7174 | |
| 428–439 | YVYQDVEIPWLK | 4 | −2 | Trypsin | 1551.7937 | |
| 429–437 | VYQDVEIPW | 1.98 | −0.63 | Chymotrypsin | 1147.5537 | |
| 431–437 | QDVEIPW | 1.71 | −0.37 | Chymotrypsin | 885.4224 | |
| 431–445 | QDVEIPWLKTKPTPR | 3.9 | −0.39 | Chymotrypsin | 1806.9981 | |
| 438–445 | LKTKPTPR | 1.88 | 0.23 | Chymotrypsin | 939.5863 |
Whereas, an almost complete peptide coverage of the main core region was found the preceding transit peptide region (Met1–Ala100) as well as the C-terminal part (Lys446–Gly603) are missing (Flurkey and Inlow, 2008). Giving the fact that protease inhibition agents (PMSF, benzamidine hydrochloride, both serine-protease inhibitors) were used during extraction, it can be assumed that proteolytic removal of the enzymes’ C-terminal part happens in vivo or is caused by non-serine proteases affection. The peptide carrying the common thioether bridge (Gielens et al., 1997; Klabunde et al., 1998) found for all known eukaryotic PPOs in literature could not be detected. Cysteine–histidine thioether bridges are reported for the sequences of I. batatas catechol oxidase (Klabunde et al., 1998), Neurospora crassa tyrosinase (Lerch, 1982), Vitis vinifera polyphenol oxidase (Virador et al., 2010) and mushroom tyrosinases (Ismaya et al., 2011; Mauracher et al., 2014; Van Gelder et al., 1997).
Molecular mass determination
The mass spectrum in Fig. 4 shows the first species jrPPO1(Asp101 → Pro444) obtained by the nanoESI-QTOF instrument with resolution (FWHM) of 40,000 and a mass accuracy of better than 5 ppm. The charge state distributions shown in inset (a) of Fig. 4A (ranging from about 28 to up to more than 40 charges) indicates the presence of one major protein species C when assorting by increasing Mr and two minor abundant ones A, B. A zoomed-in section of this spectrum is shown in Fig. 4B. The magnified inset (b) shows unambiguously that each species peak has shoulders (Δm ≈ 16 Da or 32 Da) presumably due to the presence of oxidized and non-oxidized species. For the deconvolution of the charge state distribution shown in inset (a) (Fig. 4A), 15 distinct peaks were used for the major species C, and at least 10 peaks with sufficient signal to noise ratios for the species A and B. Assuming that these positive charge (z) states are solely caused by the attachment of z protons the average molecular masses for the major species C can be assessed as 38,890 Da (STD less than 0.1 Da) and A = 38,692 Da; B = 38,793 Da (STD less than 1 Da), respectively.
Fig. 4.
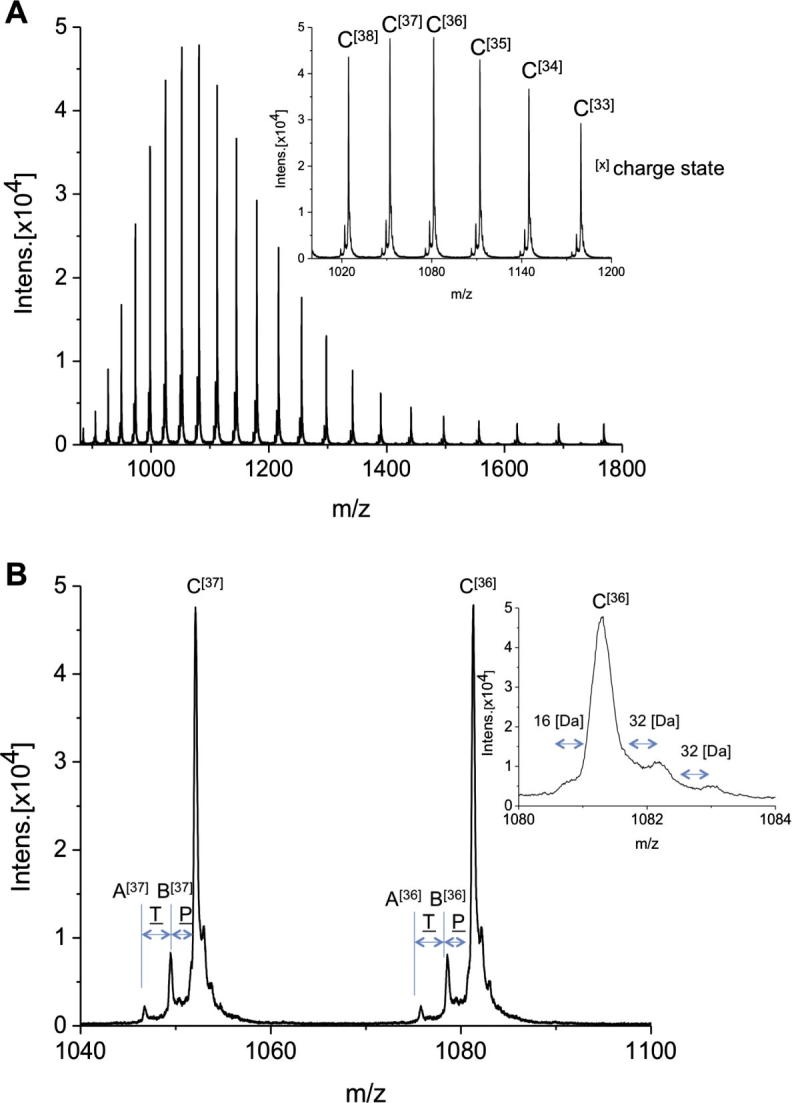
(A) nanoESI-QTOF mass spectra of jrPPO1(Asp101 → Pro444). Inset(A): Peaks of charge state [33] to [38] magnified. (B) Zoomed section of charge state [36] and [37]. Masses of species A–C: A = 38,692 Da, B = 38,793 Da; C = 38,890 Da; Mass differences between species fit to distinct amino acid composition indicated in amino acid letter code. Inset(B): Highly zoomed figure of charge state [36] showing clearly shoulders corresponding to mass differences of 16 or 32 Da (oxidation).
Fig. 5 shows the mass spectrum of jrPPO1(Asp101 → Arg445). The charge state distributions shown in inset (a) in Fig. 5A (ranging from about 28 to up to more than 40 charges) indicates the presence of one major protein species D, when assorting by increasing Mr and one minor protein species C. The average molecular masses for the major species D can be assessed as 39,047 Da (STD less than 0.1 Da) and C = 38,890 Da (STD less than 1 Da).
Fig. 5.
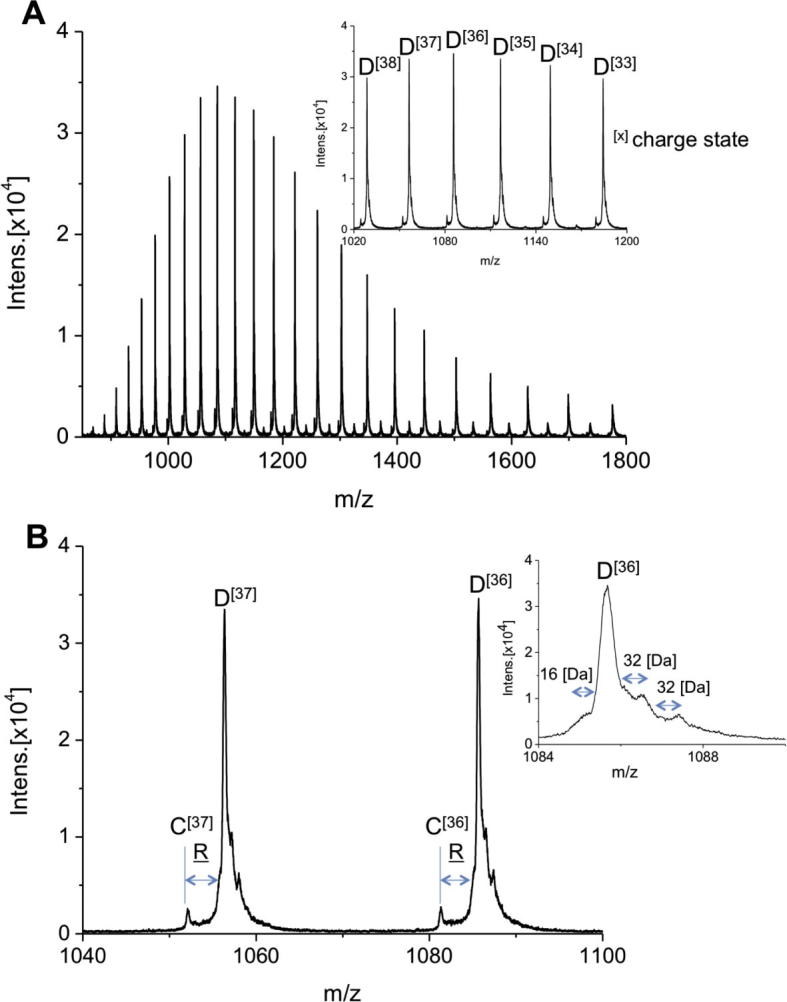
(A) nanoESI-QTOF mass spectra of jrPPO1(Asp101 → Arg445). Inset(A): Peaks of charge state [33] to [38] magnified. (B) Zoomed section of charge state [36] and [37]. Masses of species C–D: C = 38,890 Da, D = 39,047 Da; Mass difference between species fit to distinct amino acid composition indicated in amino acid letter code. Inset(B): Highly zoomed figure of charge state [36] showing clearly shoulders corresponding to mass differences of 16 or 32 Da (oxidation).
The nanoESI-QTOF-MS measurements gave evidence for the presence of two major species C (Asp101 → Pro444), D (Asp101 → Arg445) and two minor species A, B (occur together with C) that are reasonably deduced as protein forms being proteolytically cleaved at different positions of the main core polypeptide-chain-end as specified in Figs. 4B and 5B. The mass differences between these proteolytic species fit accurately to the amino acids Pro442, Thr443, Pro444, and Arg445 (see Fig. 3). This is additionally confirmed by the peptide mass analyses which found all kinds of possible main core end peptides (see Fig. 3). Thus, it can be assumed that either the proteolytic removal of the enzymes’ C-terminal part has no distinct preferential cleavage site but a region (Pro442–Arg445), or the distinct site is Arg445, however, C-terminal fringing is caused by C-terminal exo-peptidases in vivo or during extraction.
Kinetic parameters
Parameters obtained by kinetic Michaelis–Menten measurements are reported in Table 3. Giving a kcat value of 20.8 s−1 toward l-tyrosine and a kcat value of 199.3 s−1 toward l-dopa the enzyme possess a higher monophenolase- and diphenolase activity as enzymes in literature e.g. Agaricus bisporus tyrosinase with a kcat value of 7.9 s−1 (l-tyrosine) and 107.4 s−1 (l-dopa) (Espín et al., 2000) and Bacillus megaterium tyrosinase with a kcat value of 4.0 s−1 (l-tyrosine) and 44.1 s−1 (l-dopa) (Goldfeder et al., 2013). Notably, whereas tyrosinases from fungi, bacteria and mammals are kinetically well characterized, only catechol oxidases from plants received comparable attention in literature (Eicken et al., 1998; Rompel et al., 1999b). The relative high kcat values of the purified jrPPO1 results in a classification as a tyrosinase.
Table 3.
Kinetic parameters for the monophenolase and diphenolase activity of Juglans regia tyrosinase jrPPO1(Asp101 → Arg445).
| Substrate | λ [nm] | ε [M−1 cm−1] | Km [mM] | Vm [mM min−1] | Vm/Km [min−1] | kcat [s−1] | kcat/Km [mM−1 s−1] |
|---|---|---|---|---|---|---|---|
| l-tyrosine | 475 | 3600⁎ | 1.9 | 0.08 | 0.04 | 20.8 | 10.9 |
| l-dopa | 475 | 3600⁎ | 8.8 | 0.06 | 0.01 | 199.3 | 22.8 |
Values taken from Gandía-Herrero et al. (2005).
UV/Vis spectroscopic studies
The UV/Vis spectrum of the native jrPPO1(Asp101 → Pro444) is presented in Fig. 6. The absorption maximum occurs at 280 nm and a significant shoulder at 292 nm indicates the presence of several tryptophan residues in the amino acid sequence. 16 tyrosines, 18 phenylalanines, 8 tryptophans and 9 histidines are present in the mature jrPPO1(Asp101 → Pro444), which are also found in similar amounts in the catechol oxidase amino acid sequence from I. batatas (Eicken et al., 1998; Klabunde et al., 1998). For all type-3 copper enzymes a weak absorption maximum at 345 nm is observed, due to the charge transfer (CT) transition O22− (π∗σ) → Cu (II) (dx2 − y2). Addition of H2O2 causes an increasing absorption band at 345 nm (ε345 = 12,984 M−1 cm−1 per protein) and 580 nm (ε580 = 761 M−1 cm−1 per protein). The absorption band at 580 nm corresponds to the second O22− (π∗v) → Cu (II) (dx2 − y2) CT transition as found in oxy hemocyanin and oxy tyrosinase (Eickman et al., 1979; Himmelwright et al., 1980; Jolley et al., 1974; Solomon et al., 1992, 1994). The saturation of jrPPO1 is reached with the addition of two equivalents H2O2. This result is very similar to the catechol oxidase from Melissa officinalis and to the 40 kDa catechol oxidase from I. batatas where addition of two equivalents of H2O2 led to saturation (Eicken et al., 1998; Rompel et al., 2012).
Fig. 6.
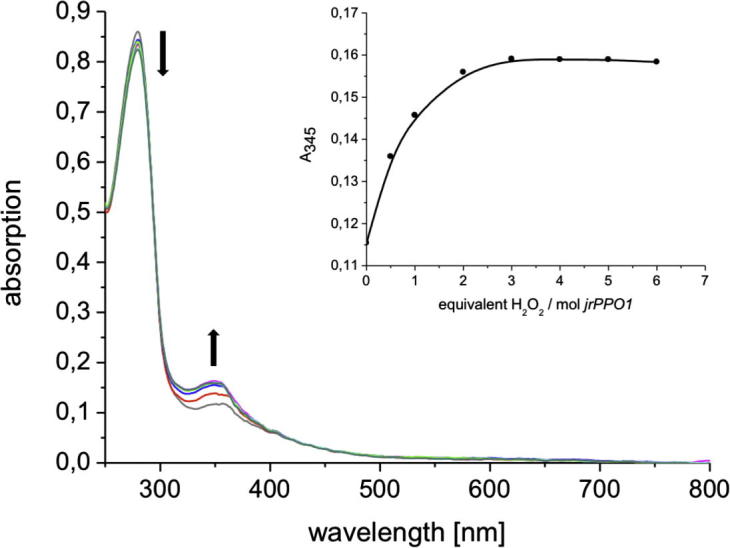
UV/Vis spectra of the native tyrosinase in 0.05 M HEPES pH 7 after treatment with 0 equivalents, 0.5, 1, 2, 3, 4, 5, and 6 eq. H2O2; T = 25 °C – Inset: absorption at 345 nm vs. equivalents H2O2.
Concluding remarks
By applying a modified and adjusted purification method published before from our group (Mauracher et al., 2014) two forms of walnut tyrosinase from the same gene (jrPPO1, Escobar et al., 2008), however proteolytically digested at different positions, were efficiently isolated and purified to identity. The accurate molecular mass was determined by means of high resolution mass spectrometry allowing the deduction of the enzymes polypeptide backbone. Thus, the in silico backbone masses of Asp101 → Pro444 and Asp101 → Arg445 matched with the determined molecular masses of 38,890 and 39,047 Da, respectively. This was also confirmed by mass spectrometric peptide analyses. The UV/Vis spectroscopic results verified that the tyrosinase from J. regia contains a type-3 copper center. After addition of two equivalents H2O2 the full oxy form of the tyrosinase is developed. Kinetic experiments classified the enzyme as a tyrosinase (EC. 1.14.18.1 and EC. 1.10.3.1) possessing comparable catalytic activity towards l-tyrosine as other well characterized tyrosinases.
Experimental
Plant material
Walnut leaves were harvested from several trees in the surroundings of Vienna (June–September 2012) and stored at −80 °C in a freezer until used.
Extraction of tyrosinase from J. regia
Tyrosinase was extracted as described by Mauracher et al. (2014) with some modifications. About 1 kg of frozen leaves were mixed and suspended in 2 L extraction buffer (125 mM sodium citrate, 4% (v/v) triton X-114, 0.5% (w/v) sodium ascorbate, 40 mM l-proline, 2 mM benzamidine hydrochloride, 1 mM phenylmethylsulfonyl fluoride (PMSF). The suspension was centrifuged at 14,500 rpm (Beckmann XP26, rotor: JLA 16.250) for 15 min at 4 °C. The supernatant was filtered through a coarse filter (cheesecloth) and treated with ammonium sulfate (15 g/L) to obtain a phase separation. This process was supported by centrifugation (14,500 rpm at 15 °C for 15 min) and allowed removal of the detergent-rich phase (lower phase). The supernatant containing soluble tyrosinase was brought to 30% (176 g/L) saturation with ammonium sulfate under continuous stirring at 4 °C. After 30 min of stirring the solution was centrifuged (14,500 rpm) for 30 min at 4 °C and the pellet was discarded. For further purification, PEG-4000 was dissolved to a concentration of 4% (w/v) in the supernatant at 4 °C. Following a centrifugation (14,500 rpm, 4 °C, 10 min) the generated PEG phase (upper phase) was discarded. This step was followed by the subsequent addition of 3.5% (w/v) PEG-4000 and centrifugation (14,500 rpm at 15 °C for 15 min) which was repeated twice. The purified and clear supernatant containing the soluble tyrosinase was brought to 80% (351 g/L) saturation with ammonium sulfate and stored overnight at 4 °C. The obtained protein pellet was then filtered through a bottle top filter and resuspended in 20 mM sodium acetate buffer, pH 4.5. The solution was then centrifuged at 14,500 rpm (4 °C) for 30 min and the supernatant was diluted with 20 mM sodium acetate buffer, pH 4.5 until the conductivity was below 9 mS/cm.
Purification of tyrosinase by Fast Protein Liquid Chromatography
The supernatant was loaded onto a cation exchange column (SP-Sepharose FF, GE Healthcare, length = 10 cm, diameter = 2.6 cm) and equilibrated with 20 mM sodium acetate, pH 4.5. Bound proteins were eluted with a linear gradient of sodium chloride (0–1 M) at a flow rate of 5 mL/min (see Fig. 1A). All collected fractions were tested photometrically for monophenolase and diphenolase activity. Fractions containing activity were pooled, ultra filtrated (size exclusion membrane of 10 kDa) and centrifuged (4000 rpm, 4 °C) to remove sodium chloride. The protein solution was then applied to a MonoS HR 5/50 Gl column (cation exchange column, GE Healthcare, length = 50 mm, diameter = 5 mm) and eluted with a linear gradient of sodium chloride (0–0.7 M) at a flow rate of 1 mL/min. Two forms were eluted at a conductivity of 13 and 16 mS/cm (see Fig. 1B), respectively. Fractions containing the first and the second eluted protein form were separately pooled and again loaded on the MonoS HR 5/50 Gl column under same conditions for removing further non-target proteins in a final polishing step (see Fig. 1C/D).
Enzyme activity assay during purification
The activity of the enzyme during the purification was monitored by spectrophotometric measurements (SHIMADZU UV-1800). Two different enzyme assays were performed. Monophenolase activity was determined by measuring the rate of increase absorbance at 305 nm and 25 °C. The reaction was performed in a 1 cm quartz cuvette using 1 mL of 10 mM sodium phosphate buffer, pH 6.5 containing 2.5 mM SDS, 0.033 mM l-tyrosine as substrate and 10 μL of enzyme solution. Diphenolase activity was determined through absorbance at 400 nm and 25 °C. The reaction mixture (1 mL) consisted of 125 mM sodium citrate buffer, pH 5.4, 5 mM 4-tert-butylcatechol as substrate and 1 μL of enzyme solution. One unit (U) of enzyme activity is defined as a change in 1 absorbance unit/min/ml.
Protein concentration
Protein concentration was determined by the method of Bradford (1976) using bovine serum albumin (BSA) as standard.
Enzyme kinetic analysis
The Michaelis–Menten constant (Km) and maximum reaction velocity (Vmax) were determined using two substrates (l-dopa and l-tyrosine) at five different concentrations (1.96, 1.30, 0.98, 0.49 and 0.29 mM). The substrates were dissolved in 50 mM sodium phosphate buffer pH 6.5. Data were plotted as 1/V and 1/[S] concentration according to the method of Lineweaver and Burk (1934).
Isoelectrical focusing
Isoelectrical focusing was performed using a PROTEAN IEF cell (Bio-Rad) and IPG-stripes with the pH range 4–7 as marker.
Molecular mass determination
The molecular mass of the purified enzyme was determined by denaturating SDS–PAGE. SDS–PAGE was performed according to the method of Laemmli (1970) using Precision Plus Protein Standard Dual Color (Bio-Rad) as molecular weight marker. Samples were applied to 10% polyacrylamide gels mixed with reduced loading dye. Gels were stained with Coomassie Brilliant Blue. The target protein band was cut out and used for protein identification. For in-gel tyrosinase activity staining a partially denaturating 10% SDS–PAGE was performed as described above, but without ß-mercaptoethanol in the loading dye, to preserve the enzyme activity. The gel was soaked with 10 mM sodium phosphate buffer pH 6.5 containing 0.033 mM l-tyrosine for activity staining. Imaging of the gels was done with Gel Doc™ XR of BIO-RAD.
Electrospray Ionisation Mass Spectrometry (ESI-MS) was performed on a nanoESI-QTOF mass spectrometer (maxis 4G UHR-TOF, Bruker, coupled to a nanospray robotic device, Nanomate, Advion Biosiences, voltage: 1.4 kV, dry gas: 6.0 l/min, dry heater 150 °C). Prior to MS measurements, the purified enzyme solution was ultra filtrated by centrifugation (14,000 rpm) and the buffer system was changed to 5 mM ammonium acetate pH 5.0, in order to reduce the salt concentration to a minimum. Afterward acetonitrile (ACN) and formic acid were added to a final concentration of 25% (v/v) ACN and 0.05% (v/v) formic acid.
UV/Vis spectroscopic studies
Visible and ultraviolet spectra were monitored by a 2-beam cuvette photometer (SHIMADZU UV-1800) with 10 mM sodium acetate buffer pH 5.0 as reference (220–800 nm). The samples were tested in a quartz cuvette with coat thickness of 1 cm path length. The peroxo complex was prepared by adding 0.50–6.00 eq. H2O2 to tyrosinase in a 50 mM HEPES buffer, pH 7.0.
Protein identification and sequence confirmation
2 μg of both protein forms were further purified for LC–MS/MS analysis by 1D SDS–PAGE. The proteins were visualized by Coomassie staining and the bands corresponding to the forms excised. The gel slices were destained with 30% acetonitrile/100 mM NH4HCO3 followed by cysteine carbamidomethylation applying dithiothreitol and iodoacetamide. The digestion was carried out with trypsin as well as chymotrypsin and the peptides eluted from the gel slices by ultrasonication. The peptide samples were completely dried by vacuum centrifugation and stored at −20 °C for LC–MS/MS analysis. The samples were solubilized in 5 μL 30% formic acid and diluted with 40 μL eluent A (97.9% H2O, 2% acetonitrile, 0.1% formic acid). Samples analysis was carried out by nanoUHPLC–ESI-MS/MS applying a high resolution orbitrap mass spectrometer (Dionex Ultimate 3000 RSLCnano, Q Exactive orbitrap, Thermo Scientific). The data analysis was performed with Proteome Discoverer 1.3.0.339 by searching against the J. regia fasta file from the UniProt database (COLU17). The peptide mass tolerance was 5 ppm with a maximum number of 2 missed cleavages, carbamidomethylation of cysteines was set as static modification whereas oxidation of methionine was the only dynamic modification. For high confidence of the MS data, the false discovery rate (FDR) of the peptide spectrum matches (PSM) was set to <0.01 (Proteome Discoverer).
Acknowledgments
The authors are grateful to the University of Vienna for financial support of the graduate training program entitled “Functional Molecules” (grant no. IK I041-N). Financial support by the “Fonds zur Förderung der wissenschaftlichen Forschung” (FWF) under P25217-N28 is gratefully acknowledged.
References
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Colaric M., Veberic R., Solar A., Hudina M., Stampar F. Phenolic acids, syringaldehyde, and juglone in fruits of different cultivars of Juglans regia L. J. Agric. Food Chem. 2005;53:6390–6396. doi: 10.1021/jf050721n. [DOI] [PubMed] [Google Scholar]
- Cook M.T., Bassett H.P., Thompson F., Taubenhaus J.J. Protective enzymes. Science. 1911;33:624–629. doi: 10.1126/science.33.851.624. [DOI] [PubMed] [Google Scholar]
- Eicken C., Zippel F., Büldt-Karentzopoulos K., Krebs B. Biochemical and spectroscopic characterization of catechol oxidase from sweet potatoes (Ipomoea batatas) containing a type-3 dicopper center. FEBS Lett. 1998;436:293–299. doi: 10.1016/s0014-5793(98)01113-2. [DOI] [PubMed] [Google Scholar]
- Eickman N.C., Himmelwright R.S., Solomon E.I. Geometric and electronic structure of oxyhemocyanin: spectral and chemical correlations to met apo, half met, met, and dimer active sites. Proc. Natl. Acad. Sci. 1979;76:2094–2098. doi: 10.1073/pnas.76.5.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar M.A., Shilling A., Higgins P., Uratsu S.L., Dandekar A.M. Characterization of polyphenol oxidase from walnut. J. Am. Soc. Hort. Sci. 2008;133:852–858. [Google Scholar]
- Espín J.C., Varón R., Fenoll L.G., Gilabert M., García-Ruíz P.A., Tudela J., García-Cánovas F. Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase. Eur. J. Biochem. 2000;267:1270–1279. doi: 10.1046/j.1432-1327.2000.01013.x. [DOI] [PubMed] [Google Scholar]
- Flurkey W.H., Inlow J.K. Proteolytic processing of polyphenol oxidase from plants and fungi. J. Inorg. Biochem. 2008;102:2160–2170. doi: 10.1016/j.jinorgbio.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Gandía-Herrero F., Jiménez-Atiénzar M., Cabanes J., García-Carmona F., Escribano J. Differential activation of a latent polyphenol oxidase mediated by sodium dodecyl sulfate. J. Agric. Food Chem. 2005;53:6825–6830. doi: 10.1021/jf050505e. [DOI] [PubMed] [Google Scholar]
- Gielens C., De Geest N., Xin X.-Q., Devreese B., Van Beeumen J., Préaux G. Evidence for a cysteine–histidine thioether bridge in functional units of molluscan haemocyanins and location of the disulfide bridges in functional units d and g of the βC-haemocyanin of Helix pomatia. Eur. J. Biochem. 1997;248:879–888. doi: 10.1111/j.1432-1033.1997.00879.x. [DOI] [PubMed] [Google Scholar]
- Goldfeder M., Kanteev M., Adir N., Fishman A. Influencing the monophenolase/diphenolase activity ratio in tyrosinase. Biochim. Biophys. Acta BBA – Proteins and Proteomics. 2013;1834:629–633. doi: 10.1016/j.bbapap.2012.12.021. [DOI] [PubMed] [Google Scholar]
- Guo L., Ma Y., Shi J., Xue S. The purification and characterisation of polyphenol oxidase from green bean (Phaseolus vulgaris L.) Food Chem. 2009;117:143–151. [Google Scholar]
- Himmelwright R.S., Eickman N.C., LuBien C.D., Solomon E.I., Lerch K. Chemical and spectroscopic studies of the binuclear copper active site of Neurospora tyrosinase: comparison to hemocyanins. J. Am. Chem. Soc. 1980;102:7339–7344. [Google Scholar]
- Ismaya W.T., Rozeboom H.J., Schurink M., Boeriu C.G., Wichers H., Dijkstra B.W. Crystallization and preliminary X-ray crystallographic analysis of tyrosinase from the mushroom Agaricus bisporus. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2011;67:575–578. doi: 10.1107/S174430911100738X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley R.L., Jr., Evans L.H., Makino N., Mason H.S. Oxytyrosinase. J. Biol. Chem. 1974;249:335–345. [PubMed] [Google Scholar]
- Kitajima N., Fujisawa K., Morooka Y., Toriumi K. μ-η2:η2-Peroxo binuclear copper complex, [Cu(HB(3, 5-iPr2pz)3)]2(O2) J. Am. Chem. Soc. 1989;111:8975–8976. [Google Scholar]
- Klabunde T., Eicken C., Sacchettini J.C., Krebs B. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat. Struct. Biol. 1998;5:1084–1090. doi: 10.1038/4193. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lerch K. Primary structure of tyrosinase from Neurospora crassa. II. Complete amino acid sequence and chemical structure of a tripeptide containing an unusual thioether. J. Biol. Chem. 1982;257:6414–6419. [PubMed] [Google Scholar]
- Li N., Cai W., Jin Q., Qin Q., Ran F. Molecular cloning and expression of polyphenoloxidase genes from the mushroom, Agaricus bisporus. Agric. Sci. China. 2011;10:185–194. [Google Scholar]
- Lineweaver H., Burk D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934;56:658–666. [Google Scholar]
- Martinez M.V., Whitaker J.R. The biochemistry and control of enzymatic browning. Trends Food Sci. Technol. 1995;6:195–200. [Google Scholar]
- Mauracher S.G., Molitor C., Michael C., Kragl M., Rizzi A., Rompel A. High level protein-purification allows the unambiguous polypeptide determination of latent isoform PPO4 of mushroom tyrosinase. Phytochemistry. 2014;99:14–25. doi: 10.1016/j.phytochem.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A.M. Polyphenol oxidases in plants and fungi: Going places? A review. Phytochemistry. 2006;67:2318–2331. doi: 10.1016/j.phytochem.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Mayer A.M., Harel E. Polyphenol oxidases in plants. Phytochemistry. 1979;18:193–215. [Google Scholar]
- Piffaut B., Metche M. Properties of peroxidase and polyphenol oxidase in natural complexes from walnuts (Juglans regia) and in active DL-DOPA copolymers. J. Sci. Food Agric. 1991;57:493–506. [Google Scholar]
- Rompel A., Büldt-Karentzopoulos K., Molitor C., Krebs B. Purification and spectroscopic studies on catechol oxidase from lemon balm (Melissa officinalis) Phytochemistry. 2012;81:19–23. doi: 10.1016/j.phytochem.2012.05.022. [DOI] [PubMed] [Google Scholar]
- Rompel A., Fischer H., Meiwes D., Büldt-Karentzopoulos K., Dillinger R., Tuczek F., Witzel H., Krebs B. Purification and spectroscopic studies on catechol oxidases from Lycopus europaeus and Populus nigra: Evidence for a dinuclear copper center of type 3 and spectroscopic similarities to tyrosinase and hemocyanin. JBIC J. Biol. Inorg. Chem. 1999;4:56–63. doi: 10.1007/s007750050289. [DOI] [PubMed] [Google Scholar]
- Rompel A., Fischer H., Meiwes D., Büldt-Karentzopoulos K., Magrini A., Eicken C., Gerdemann C., Krebs B. Substrate specificity of catechol oxidase from Lycopus europaeus and characterization of the bioproducts of enzymic caffeic acid oxidation. FEBS Lett. 1999;445:103–110. doi: 10.1016/s0014-5793(99)00106-4. [DOI] [PubMed] [Google Scholar]
- Sanchez-Amat A., Solano F. A pluripotent polyphenol oxidase from the melanogenic marine Alteromonas sp. shares catalytic capabilities of tyrosinases and laccases. Biochem. Biophys. Res. Commun. 1997;240:787–792. doi: 10.1006/bbrc.1997.7748. [DOI] [PubMed] [Google Scholar]
- Selinheimo E., NiEidhin D., Steffensen C., Nielsen J., Lomascolo A., Halaouli S., Record E., O’Beirne D., Buchert J., Kruus K. Comparison of the characteristics of fungal and plant tyrosinases. J. Biotechnol. 2007;130:471–480. doi: 10.1016/j.jbiotec.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Solar A., Colarič M., Usenik V., Stampar F. Seasonal variations of selected flavonoids, phenolic acids and quinones in annual shoots of common walnut (Juglans regia L.) Plant Sci. 2006;170:453–461. [Google Scholar]
- Solomon E.I., Baldwin M.J., Lowery M.D. Electronic structures of active sites in copper proteins: contributions to reactivity. Chem. Rev. 1992;92:521–542. [Google Scholar]
- Solomon E.I., Sundaram U.M., Machonkin T.E. Multicopper oxidases and oxygenases. Chem. Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- Solomon E.I., Tuczek F., Root D.E., Brown C.A. Spectroscopy of binuclear dioxygen complexes. Chem. Rev. 1994;94:827–856. [Google Scholar]
- Steffens J., Harel E., Hunt M. Polyphenol oxidase. In: Ellis B., Kuroki G., Stafford H., editors. Genetic Engineering of Plant Secondary Metabolism, Recent advances in Phytochemistry. Springer; US: 1994. pp. 275–312. [Google Scholar]
- Trémolières M., Bieth J.G. Isolation and characterization of the polyphenoloxidase from senescent leaves of black poplar. Phytochemistry. 1984;23:501–505. [Google Scholar]
- Van Gelder C.W., Flurkey W.H., Wichers H.J. Sequence and structural features of plant and fungal tyrosinases. Phytochemistry. 1997;45:1309–1323. doi: 10.1016/s0031-9422(97)00186-6. [DOI] [PubMed] [Google Scholar]
- Vaughn K.C., Lax A.R., Duke S.O. Polyphenol oxidase: the chloroplast oxidase with no established function. Physiol. Plant. 1988;72:659–665. [Google Scholar]
- Virador V.M., Reyes Grajeda J.P., Blanco-Labra A., Mendiola-Olaya E., Smith G.M., Moreno A., Whitaker J.R. Cloning, sequencing, purification, and crystal structure of grenache (Vitis vinifera) polyphenol oxidase. J. Agric. Food Chem. 2010;58:1189–1201. doi: 10.1021/jf902939q. [DOI] [PubMed] [Google Scholar]
- Weijn A., Bastiaan-Net S., Wichers H.J., Mes J.J. Melanin biosynthesis pathway in Agaricus bisporus mushrooms. Fungal Genet. Biol. 2013;55:42–53. doi: 10.1016/j.fgb.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Wichers H.J., Recourt K., Hendriks M., Ebbelaar C.E.M., Biancone G., Hoeberichts F.A., Mooibroek H., Soler-Rivas C. Cloning, expression and characterisation of two tyrosinase cDNAs from Agaricus bisporus. Appl. Microbiol. Biotechnol. 2003;61:336–341. doi: 10.1007/s00253-002-1194-2. [DOI] [PubMed] [Google Scholar]
- Wu J., Chen H., Gao J., Liu X., Cheng W., Ma X. Cloning, characterization and expression of two new polyphenol oxidase cDNAs from Agaricus bisporus. Biotechnol. Lett. 2010;32:1439–1447. doi: 10.1007/s10529-010-0329-2. [DOI] [PubMed] [Google Scholar]
- Yoruk R., Marshall M.R. Physicochemical properties and function of plant polyphenol oxidase: a review. J. Food Biochem. 2003;27:361–422. [Google Scholar]