

Abstract
Chromosomal instability is a hallmark of human cancer cells, but its role in carcinogenesis remains poorly resolved. Insights into this role have emerged from studies on the tumour suppressor BRCA2, whose inactivation in human cancers causes chromosomal instability through the loss of essential functions of the BRCA2 protein in the normal mechanisms responsible for the replication, repair and segregation of DNA during cell division. Humans who carry heterozygous germline mutations in the BRCA2 gene are highly predisposed to cancers of the breast, ovary, pancreas, prostate and other tissues. Here, we review recent studies that describe genetically engineered mouse models (GEMMs) for pancreatic cancer associated with BRCA2 mutations. These studies not only surprisingly show that BRCA2 does not follow the classical Knudson “two hit” paradigm for tumour suppression, but also highlight features of the interplay between TP53 inactivation and carcinogenesis in the context of BRCA2 deficiency. Thus, the models reveal novel aspects of cancer evolution in carriers of germline BRCA2 mutations, provide new insights into the tumour suppressive role of BRCA2, and establish valuable new preclinical settings for testing approaches to pancreatic cancer therapy; together, these features emphasize the value of GEMMs in cancer research.
Keywords: Pancreatic cancer, BRCA2, Genetically engineered mouse model, Chromosomal instability, Hereditary cancer predisposition
Highlights
The mechanisms underlying pancreatic carcinogenesis in germline BRCA2 mutation carriers remain unclear.
Recent studies on transgenic mouse models surprisingly show that even heterozygous BRCA2 mutations promote pancreatic cancer.
They also reveal the interplay between TP53 inactivation and pancreatic carcinogenesis in the context of BRCA2 deficiency.
We review insights into tumour evolution arising from these models, and highlight their value in testing new therapies.
Aberrations in the number and structure of chromosomes are a hallmark of cells derived from solid tumours. These aberrations not only include structural anomalies such as gross chromosomal rearrangements (including translocations, large deletions or inversions) and gene copy number variations, which have been connected to defective DNA replication or repair, but also anomalous chromosome number triggered by defective segregation between daughter cells during mitosis. Whereas much recent work has extensively documented the mechanisms underlying normal DNA replication, repair and segregation, what happens when these mechanisms are perturbed by the genetic alterations associated with cancer remains far less studied. Here, we will review this question from the perspective of recent work using transgenic models designed to recapitulate a specific human cancer involving mutations affecting the tumour suppressor gene BRCA2, associated with hereditary predisposition to breast, ovarian, pancreatic and other cancers. There is by now clear evidence to implicate the large, 3418 residue BRCA2 protein (3328 residues in the mouse) in the maintenance of chromosome integrity during cell division (Venkitaraman, 2009). BRCA2‐deficient cells accumulate structural chromosome aberrations as they divide, and also become aneuploid through losses or gains in whole chromosomes (Gretarsdottir et al., 1998; Patel et al., 1998; Tutt et al., 1999; Yu et al., 2000). A large body of evidence connects the chromosomal instability observed in BRCA2‐deficient cells to essential biological functions of the BRCA2 protein in DNA repair by homologous recombination (Connor et al., 1997; Patel et al., 1998; Moynahan et al., 2001), in progression through the S and G2/M phases of the cell cycle (Lomonosov et al., 2003; Ayoub et al., 2009; Menzel et al., 2011; Schlacher et al., 2011) and in mitotic cell division by cytokinesis (Daniels et al., 2004; Mondal et al., 2012). Thus, murine models that recapitulate the effect of cancer‐associated mutations in human BRCA2 on tissue‐specific carcinogenesis provide an important opportunity to dissect the complex roles played by chromosomal instability during human carcinogenesis.
1. Modelling human pancreatic cancer associated with BRCA2 inactivation
Pancreatic ductal adenocarcinoma (PDAC) represents the fourth leading cause of cancer mortality worldwide, with an incidence of approximately 217,000 new cases each year nearly matched by 213,000 deaths (Parkin et al., 2001). Several of the most frequent genetic events underlying the initiation and progression of human pancreatic cancer have been identified (Hezel et al., 2006; Maitra and Hruban, 2008). These include activating mutations in the KRAS proto‐oncogene, which occur in >90% of PDAC (Caldas and Kern, 1995) and are considered as a key driver for pancreatic carcinogenesis, and mutations inactivating the TP53 gene, which occur in 50–75% of patients (Redston et al., 1994).
Moreover, several lines of evidence implicate mutations inactivating the BRCA2 tumour suppressor in an estimated 5–20% of familial PDAC (Hahn et al., 2003; Couch et al., 2007). Germline carriers of deleterious BRCA2 mutations that commonly truncate the encoded protein exhibit an increased lifetime risk of developing PDAC, in addition to their well‐known predisposition to cancers of the breast and ovary (Breast Cancer Linkage Consortium, 1999). Within high‐risk pancreatic cancer kindreds, inherited mutations in BRCA2 represent the most frequently encountered germline genetic alteration (Hahn et al., 2003). The incidence of germline BRCA2 mutations in apparently sporadic pancreatic cancers may be as high as in breast or ovarian cancer (Goggins et al., 1996). More recently, PALB2, which encodes a BRCA2‐interacting protein also essential for homology‐directed DNA repair, has emerged as a pancreatic cancer susceptibility allele (Jones et al., 2009).
Three new transgenic models for pancreatic adenocarcinoma associated with BRCA2 inactivation have recently been described (Skoulidis et al., 2010; Feldmann et al., 2011; Rowley et al., 2011) [Figure 1]. One of these models does not incorporate activation of the Kras oncogene (Feldmann et al., 2011). In contrast, the other two models (Skoulidis et al., 2010; Rowley et al., 2011) use a conditional gene‐targeted allele developed by Tuveson, Jacks and colleagues (Jackson et al., 2001; Johnson et al., 2001), in which tissue‐specific activation of oncogenic KrasG12D is driven on a single allele by loxP‐CRE mediated recombination, mimicking a genetic event that frequently triggers Kras activation in human cancers. CRE recombinase expression is controlled by the PDX1 promoter, which is expressed at E8.5 and required for organogenesis of the pancreas, whereby loss of the gene is associated with an absence of pancreatic formation (Jonsson et al., 1994; Offield et al., 1996). The expression of the PDX1‐CRE transgene therefore occurs throughout the pancreatic cellular compartment, albeit in a stochastic manner, to trigger Kras activation (Hingorani et al., 2003).
Figure 1.
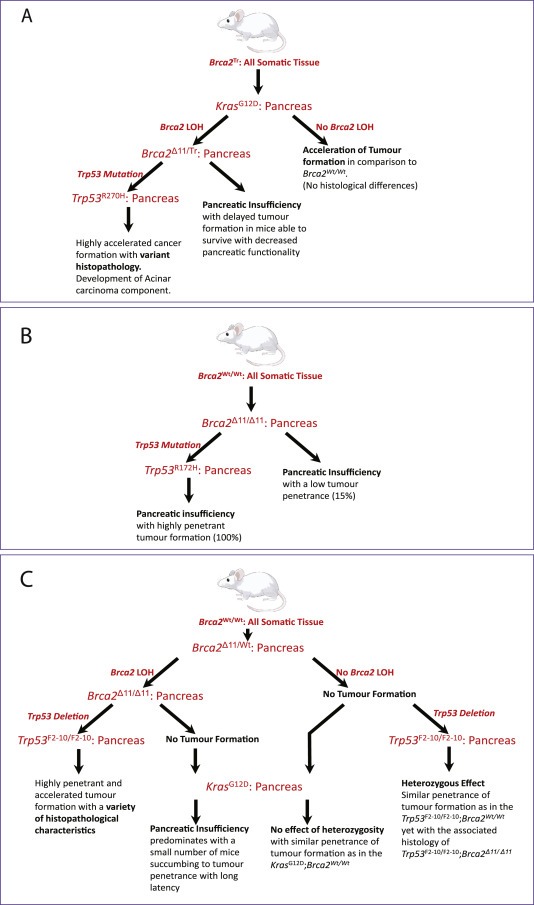
Modelling the role of Brca2 in PDAC development. Key features and findings from the three genetically engineered mouse models (GEMMs) are summarized here, and described in the main text. (A) Skoulidis et al. uniquely employ a truncated Brca2Tr allele mimicking germline mutations in human mutation carriers expressed in all somatic tissues. In contrast, (B) Feldmann et al. and (C) Rowley et al. conditionally delete Brca2 in the pancreas alone. All three GEMMs introduce tissue‐specific alleles activating Kras or inactivating Trp53. It is important to note that the Brca2 alleles as well as the Trp53 alleles used in each model are distinct.
Patients who carry germline mutations affecting BRCA2 harbour the germline mutant allele in all somatic tissues, whereas the second BRCA2 allele is wildtype (Wooster et al., 1994). It has been widely believed that loss of the second, wild‐type BRCA2 allele in nascent cancer cells (termed ‘loss of heterozygosity’ or LOH) is necessary for the emergence of tumours in germline mutation carriers. The pancreatic cancer model developed in our laboratory (Skoulidis et al., 2010) mimics this presumed sequence of events. It carries in all somatic tissues a truncated allele of murine Brca2 (Brca2 Tr), which truncates the gene in an evolutionarily conserved and functionally critical region encoded by exon 11, resembling deleterious germline mutations found in human carriers (Friedman et al., 1998). The second Brca2 allele, Brca2 F11 (Jonkers et al., 2001) can be conditionally disrupted to remove exon 11 by loxP‐CRE recombination in the pancreas. This event is driven by PDX1‐CRE, and therefore occurs in the same tissues which undergo Kras activation. There is evidence that both the Brca2 Tr and Brca2 F11 alleles can express a truncated protein product (Patel et al., 1998; Choi et al., 2012), a point we will later return to. Notably, the models developed by Rowley et al. and Feldmann et al. both exclusively use a strain homozygous for the conditional Brca2 F11 allele; thus, germline heterozygosity for BRCA2 is not modelled in their experiments.
All three of the new pancreatic cancer models incorporate conditional alleles that inactivate Tp53 in the pancreas, to mimic the frequent loss of this tumour suppressor in human pancreatic cancers. Somewhat different Tp53 alleles are used by each group, an important distinction given that the nature of Tp53 mutations is thought to affect pancreatic cancer development (Olive et al., 2004). Two of the studies employ gain‐of‐function mutants affecting a single allele. These are either the structural mutant Tp53R172H (Feldmann et al., 2011) or the contact mutant Tp53R270H (Skoulidis et al., 2010). Both these Tp53 mutants are associated with the development of carcinomas (Olive et al., 2004). In contrast, the third study uses a null allele of Tp53, wherein exons 2–10 are deleted by PDX1‐CRE activation (Rowley et al., 2011), in a manner that less faithfully represents human cancer‐associated mutations.
Thus, it should be clear from the foregoing that these three recently‐published models for pancreatic carcinogenesis associated with BRCA2 inactivation harbour important differences not only in the tissue‐specificity, nature and timing of mutant Tp53 and Brca2 alleles, but also in the presence of mutant Kras. We believe that these distinctions are vital to understanding the marked differences in pancreatic carcinogenesis observed in the studies, the key findings from which are highlighted in Table 1.
Table 1.
Comparison of the major phenotypes associated with Brca2‐deficient PDAC in three GEMMs. Each model employs distinct Brca2 alleles, in the context of different initiating lesions. Possible mechanisms underlying the variations in phenotype are discussed in the main text.
| Study | Cohort | Associated phenotype |
|---|---|---|
| Skoulidis et al., 2010 | KrasG12D | 15% tumour penetrance with long latency; 100% PDAC |
| KrasG12D, Brca2Tr/Wt | Accelerated tumourigenesis with an increase in tumour penetrance at 30%; 100% PDAC | |
| KrasG12D, Brca2Tr/F11 | Pancreatic insufficiency; some tumours develop but with long latency; 100% PDAC | |
| KrasG12D, Tp53R270H, Brca2Wt/Wt | Highly penetrant tumour formation with a median survival of 168 days; 100% PDAC | |
| KrasG12D, Tp53R270H, Brca2Tr/Wt | Accelerated tumourigenesis, median survival 143 days, in comparison to Brca2Wt; 100% PDAC | |
| KrasG12D, Tp53R270H, Brca2Tr/F11 | Further acceleration of tumourigenesis, median survival 84 days, all tumours showed regions of PDAC development with noted regions of Acinar histology in 18% of cases | |
| Rowley et al., 2011 | KrasG12D | 61% tumour penetrance with median survival 406 days |
| KrasG12D, Brca2F11/Wt | 66% tumour penetrance with median survival 366 days‐ similar to Brca2Wt | |
| KrasG12D, Brca2F11/F11 | Pancreatic insufficiency; 13% tumour penetrance with long latency | |
| Brca2Wt/Wt | No tumour formation | |
| Brca2F11/Wt | No tumour formation | |
| Brca2F11/F11 | No tumour formation | |
| Tp53F2‐10/F2‐10, Brca2Wt/Wt | Low tumour penetrance with acinar histology | |
| Tp53F2‐10/F2‐10, Brca2F11/Wt | Similar penetrance of tumour formation to Brca2Wt but with the associated histology of Brca2F11/F11 | |
| Tp53F2‐10/F2‐10, Brca2F11/F11 | Increased tumour penetrance and acceleration of tumour formation in comparison to Brca2Wt of Brca2F11/Wt Mixed histology: PDAC 40%, Acinar 15%, high‐grade undifferentiated 35%, remainder mucinous tumours. Median survival 300 days. | |
| Feldmann et al., 2011 | Brca2F11/F11 | Pancreatic insufficiency; development of PDAC but with incomplete penetrance (∼15%) at 15 months. Median survival 454 days. |
| Tp53R172H, Brca2F11/F11 | Pancreatic insufficiency; highly penetrant PDAC formation (100%) at 15 months. Median survival 375 days. |
2. Brca2Tr heterozygosity suffices for pancreatic carcinogenesis driven by mutant Kras
BRCA2 has been believed to follow the classical ‘two‐hit’ paradigm for tumour suppression (Smith et al., 1992; Collins et al., 1995; Rahman and Stratton, 1998). Initial studies soon after the discovery of BRCA2 reported consistent inactivation of the wild‐type BRCA2 allele through loss‐of‐heterozygosity (LOH) in breast or ovarian cancer cells from mutation carriers (Collins et al., 1995; Gudmundsson et al., 1995), engendering the widely accepted view that BRCA2 LOH is an essential event in carcinogenesis. A few notes of dissent have emerged in later studies (King et al., 2007; Willems et al., 2008), but they have not gained widespread attention.
In this context, it is notable that the studies reported in Skoulidis et al. unexpectedly reveal that BRCA2 heterozygosity promotes pancreatic cancer development in mice and men. In both the Tp53 wildtype and Tp53 R270H cohorts from the murine model, heterozygosity for Brca2 (through the Brca2 Tr/Wt genotype) acts with Kras G12D to accelerate the progression and development of PDAC. A similar conclusion is reached from studies on a small number of human pancreatic cancer samples from carriers of the Icelandic founder mutation in BRCA2, the allele BRCA2 999Del5, which is 5 bp deletion in exon 9 that causes a frame‐shift leading to the expression of a very short and unstable protein product (Mikaelsdottir et al., 2004). Three of the 4 cases tested do not exhibit LOH.
How heterozygosity for Brca2 Tr may promote tumourigenesis remains uncertain. One possibility is that this genotype causes a mutator phenotype, owing to defects in DNA repair arising from a known role of BRCA2 in homologous DNA recombination (Patel et al., 1998; Moynahan et al., 2001; Tutt et al., 2001). However, previous studies on murine embryo fibroblasts (MEFs) heterozygous for Brca2 Tr reveal no statistically significant effects on sensitivity to genotoxic agents (Patel et al., 1998; Yu et al., 2000). Neither Brca2 Tr/WT mice (Friedman et al., 1998), nor strains heterozygous for other Brca2 truncation mutants (Connor et al., 1997; Sharan et al., 1997; Jonkers et al., 2001; Yan et al., 2004), exhibit cancer predisposition. Notably, a lacZ mutation reporter gene (Boerrigter et al., 1995) incorporated into the germline of mice heterozygous for a Brca2 truncation similar but not identical to Brca2 Tr (Tutt et al., 2002) reveals no evident mutator phenotype. On the other hand, MEFs from this strain showed a mild alteration in DNA repair kinetics during recovery from 4Gy of ionizing radiation. Thus, there is little convincing evidence that heterozygosity for these Brca2 mutant alleles creates a DNA repair defect that could explain heightened cancer predisposition, although the possibility has not yet been conclusively excluded.
In this connection, it is important to note that these cellular approaches do not yet account for the cooperative effect of mutant Kras on pancreatic carcinogenesis associated with Brca2 heterozygosity, as suggested by the murine model developed by Skoulidis et al. Even a subtle increase in mutational load induced by Brca2 heterozygosity in mutant Kras expressing cells – which might be undetectable in cellular experiments, but significant in vivo – could plausibly accelerate the progression of pre‐malignant pancreatic intra‐epithelial (PanIN) lesions (which occur frequently even in apparently normal pancreatic parenchyma (Hruban et al., 2008)) to overt malignancy. Further studies addressing this issue in murine models are clearly warranted.
Whether different BRCA2 alleles behave in a manner similar to Brca2 Tr is not clear. Like Brca2 Tr, heterozygosity for BRCA2 999Del5 apparently suffices to predispose human carriers to pancreatic carcinogenesis. However, the instability of the truncated protein encoded by BRCA2 999Del5 (Mikaelsdottir et al., 2004) suggests that haploinsufficiency for BRCA2 (as opposed to any trans‐dominant effect of a mutant BRCA2 protein) accounts for the phenotypic effects of heterozygosity in patients who carry this Icelandic founder mutation. In contrast, Rowley et al. describe no heterozygous effect in any of their Brca2 F11/Wt cohorts despite the presence of mutant Kras G12D. Interpretation of this difference is not straightforward, since the Brca2 F11 allele engenders Brca2 loss only in the cells expressing PDX1‐CRE, unlike Brca2 Tr, which is expressed in all somatic cells. This raises the possibility that non‐cell autonomous effects of Brca2 F11 heterozygosity – for example on stromal cells rather than the nascent cancer cells – may account for the cancer‐predisposing effect of the Brca2 Tr allele.
Mitotic functions have also been ascribed to BRCA2, and interestingly, defects in G2 checkpoint function (Menzel et al., 2011), mitotic checkpoint enforcement (Choi et al., 2012) and the completion of cell division by cytokinesis (Daniels et al., 2004; Shive et al., 2010; Jonsdottir et al., 2012; Mondal et al., 2012) have been reported in BRCA2‐deficient cells. Whether or not these roles for BRCA2 may explain the effect of heterozygosity in tumour development is yet to be explored. Heterozygosity for the Brca2 Tr allele is enough to trigger cytokinetic defects in MEFs (Daniels et al., 2004), but it is unclear whether the other mitotic functions are perturbed by BRCA2 heterozygosity.
Importantly, recent data from human studies further support that BRCA2 heterozygosity is enough to promote carcinogenesis. In breast cancers, incomplete loss of the remaining wild‐type allele has been observed using techniques more sensitive than those applied in the original studies (King et al., 2007). Importantly large‐scale, unbiased genomic sequencing of high‐grade serous ovarian carcinomas highlighted the retention of the wild‐type allele in end stage disease from ∼25% of germline Brca2 carriers (Atlas, 2011). Furthermore, a detailed study of prostate tumour progression in BRCA2 germline mutation carriers uncovered no LOH in high‐grade prostatic intraepithelial neoplasias, considered precursor lesions to the development of prostate adenocarcinoma, and up to 55% of the malignant tumours analysed (Willems‐Jones et al., 2012). Collectively, these data suggest that cancers arising in germline BRCA2 mutation carriers frequently fail to exhibit loss of the wildtype allele, and that failure to exhibit LOH occurs in BRCA2‐mutant cancers from several different tissues. Thus, BRCA2 may not follow the classical Knudson “two hit” paradigm for tumour suppression.
Interestingly, these conclusions can be set against the emerging backdrop of ongoing studies on tissue samples from patients with familial forms of pancreatic cancer. A study of 58 pancreatic intra‐epithelial neoplasms and intraductal papillary mucinous neoplasms reveals that somatic losses in BRCA2 copy number are infrequent (Hong et al., 2012). However, definitive evidence addressing the extent to which the lessons from GEMMs of Brca2‐deficient pancreatic cancers can be applied to human neoplasia awaits the results of more extensive genome sequencing studies on pancreatic cancer samples from patients harbouring germline BRCA2 mutations.
3. Pancreatic cancer histopathology and BRCA2 genotype
Murine pancreatic cancers emerging in Brca2 Tr/F11 strains in which both Brca2 alleles are inactivated in PDX1‐CRE expressing cells exhibit a preponderance of acinar cell carcinoma histology. Correspondingly, 3 of the four human pancreatic cancers from BRCA2 999Del5 mutation carriers that exhibited LOH were also of the acinar type (Skoulidis et al., 2010), which normally accounts for only 1–2%% of human pancreatic cancers (Hruban, 2007). This raises the possibility that these genotypes promote the evolution of acinar cell carcinomas rather than PDAC. Rowley et al. (2011) also observe differences in the histopathological spectrum of pancreatic malignancies from mice in which Brca2 as well as Tp53 had been inactivated, when compared to Tp53 deficiency alone. These observations raise the possibility that the nature of Brca2 mutations, their timing, or their coincidence with alterations with Tp53 may alter the histopathological evolution of pancreatic cancers in mice. However, these observations remain too limited to allow firm conclusions to be drawn, and we draw attention to them here simply to emphasize the need for further studies.
4. Checkpoint inactivation, Tp53 mutations, and the evolution of cancers following Brca2 inactivation
We and others have shown (Patel et al., 1998; Lee et al., 1999; Tutt et al., 1999) that the genome‐wide DNA damage that follows homozygous inactivation of BRCA2 leads to checkpoint activation and cell cycle arrest, rather than the unrestrained cellular proliferation typical of cancer. We have previously proposed (Venkitaraman, 2009) that checkpoint inactivation may therefore be an essential pre‐requisite for homozygous BRCA2 inactivation through LOH during carcinogenesis. The work of Skoulidis et al. provides strong in vivo evidence for this hypothesis, supported by the observations of Rowley et al. In both murine models, bi‐allelic Brca2 inactivation by itself leads to a loss of exocrine pancreatic parenchyma, a concomitant increase in adipose tissue, and progressive loss of organ functionality. Skoulidis et al. further demonstrate that pancreatic insufficiency is preceded by the widespread occurrence of DNA double‐strand breakage marked by γH2AX staining in cells lacking both copies of Brca2. Moreover, both Skoulidis et al. and Rowley et al. find that the concomitant inactivation of Tp53 function prevents pancreatic insufficiency, and allows rapid PDAC development, in the pancreas of mice carrying bi‐allelic mutations inactivating Brca2. When the observations from these studies are synthesized, a picture emerges wherein BRCA2 heterozygosity in germline mutation carriers may suffice to allow the development of Kras‐driven PDAC. Later inactivation of Tp53 or other checkpoint genes may then allow eventual loss of the second BRCA2 allele: although LOH is not an obligate step, it may promote the emergence of advanced cancers. Indeed, inferences from a very small study of just 5 samples from human pancreatic cancer patients support such a scenario, although it remains to be firmly established.
5. Mouse models for PDAC associated with BRCA2 inactivation: lessons for cancer therapy
The work of Skoulidis et al. has implications for cancer therapy. As discussed above, our results suggest that Brca2 heterozygosity suffices for PDAC formation driven by mutant Kras in mice and men. However, the rationale for the use of targeted agents such as PARP1 inhibitors (PARP1i) in BRCA2‐deficient cancers is contingent upon bi‐allelic BRCA2 inactivation in the tumour cells (Bryant et al., 2005; Farmer et al., 2005). Therefore, as confirmed in our work (Skoulidis et al., 2010), PDAC cells that retain a functional Brca2 allele are resistant to PARPi such as the AstraZeneca compound Olaparib. Thus, PARP1 inhibitors should be reserved for clinical use when BRCA2 LOH can be verified in the tumour, assessment of which emerges as a critical requirement in the design of human clinical trials for the treatment of BRCA2‐deficient cancers.
These findings exemplify how the new generation of GEMMs for PDAC may represent valuable surrogate models for preclinical tests of therapeutic efficacy in patients. Importantly, such models not only allow in vivo proof of new therapeutic concepts, but may also provide a platform to assess the pharmacodynamic and pharmacokinetic properties of new agents, although species‐specific differences may limit such interpretations. The models also provide a flexible method to assess the impact of therapy on tumour progression using adapted multimodal imaging and drug bioavailability (including tissue drug penetrance) analyses. An important feature that determines if a particular GEMM is useful as a preclinical platform is if the model recapitulates a similar clinical response to standard therapy agents in clinical use in human. For instance, the KPC mouse model is relatively unaffected by gemcitabine similar to the small clinical benefit from this agent in the advanced pancreatic cancer setting in humans (Olive et al., 2009).
Each GEMM can be likened to a patient with a particular tumour type, and hence, can be enrolled into preclinical trial of novel agents (Eklund et al., 2013; Guerra and Barbacid, 2013). Such trials are facilitated by the use of adapted imaging techniques to monitor for tumour development and progression. Such utility is beginning to have an impact in the clinical setting. In humans, early phase clinical trials have shown promise for the combination of nanoparticle albumin‐linked paclitaxel (nab‐paclitaxel) and gemcitabine in advanced PDAC. Frese and colleagues have used the KPC mouse model of pancreatic cancer to provide a mechanistic understanding of the synergistic effect of this combination. Paclitaxel appears to inhibit the breakdown of gemcitabine through modulation of a degradative enzyme, cytidine deaminase through a reactive oxygen species‐dependent pathway (Frese et al., 2012). Such mechanistic analyses may help to rationalize our clinical strategy of using such drug combination. For instance, the nab‐paclitaxel can be used as an inducting agent followed by gemcitabine to enhance the tumouricidal effect of the latter.
Rational clinical trials in man are likely to benefit from the incorporation of an in vivo component that provides relatively rapid feedback of the predicted response to new agents. The preclinical assessment using GEMMs can either be used to screen potentially useful therapeutic agents before progressing to clinical trials, or alternatively, to critically assess the mechanisms of action in vivo once an agent has been found to be effective in a small‐scale trial, before progressing to larger Phase III clinical trial. One potential advantage of such GEMMs is that unlike human trials, they will allow sequential sampling of appropriate tumour tissues to assess the pharmacodynamic impact of a particular agent. In pancreatic cancer, several novel agents targeting a diverse range of molecular pathways have been tested in GEMMs to complement early phase clinical trials (Olive et al., 2009; Plentz et al., 2009; Cook et al., 2012; Jacobetz et al., 2012). The results of these trials will in due course affirm or refute the value of PDAC GEMMs as a predictive tool for clinical efficacy in human cancers.
The potential value of PDAC GEMMs as surrogates for the preclinical testing of new therapies is critically dependent on how closely these models mimic human PDAC. Several studies (Hingorani et al., 2003; Tuveson and Hingorani, 2005; Olive et al., 2009; Plentz et al., 2009; Cook et al., 2012) have emphasized the similarities in histopathology, cancer progression, clinical behaviour and even drug pharmacodynamics between PDAC GEMMs and human PDACs. However, it remains unclear whether the spectrum of genetic alterations is similar. Initial observations suggest that murine KPC PDACs bear resemblance to the human disease insofar as they exhibit a high degree of genomic instability, evident from multiple non‐reciprocal chromosomal translocations (Hingorani et al., 2005). However, with emerging data from large‐scale sequencing of human PDAC tumours (Biankin et al., 2012), it is now imperative that we further validate the GEMMs at the genomic level to compare the genomic landscapes of murine and human tumours.
Because PDAC GEMMs incorporate high‐penetrance genetic events such as initiating oncogenes or inactivated tumour suppressor genes from an early stage in a large number of susceptible cells, the resulting stereotypy of the malignancies arising therein may not reflect the heterogeneity likely to be present in human cancers. Importantly, the genetic heterogeneity of human cancers may give rise to differing therapeutic responses to any particular agent due to the differing genetic and epigenetic signatures of the constituent cells. It is conceivable that individual tumours can take differing genetic ‘routes’ to achieve tumoural progression, depending on the type of initiating genetic lesions and secondary genetic hits that occur stochastically. It is presumed that through inactivation of genes involved in maintaining genomic stability (e.g. BRCA2 in models of pancreatic cancer) may promote the stochastic acquisition of genetic and consequent morphologic heterogeneity due to the expected increase in mutation rate. However, this point remains to be established in future studies, and also has important implications for the potential value of GEMM models in testing new therapeutic approaches against PDAC.
Acknowledgements
LC was supported by a Cancer Research UK Molecular Pathology Studentship, and SL, by a Clinician‐Scientist Fellowship from the UK Medical Research Council (MRC). Work in ARV's laboratory is funded by the MRC.
Cassidy Liam D., Liau Siong-Seng and Venkitaraman Ashok R., (2014), Chromosome instability and carcinogenesis: Insights from murine models of human pancreatic cancer associated with BRCA2 inactivation, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.10.005.
References
- Atlas, T.C.G., 2011. Integrated genomic analyses of ovarian carcinoma. Nature. 474, (7353) 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub, N., Rajendra, E., 2009. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr. Biol.. 19, (13) 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin, A.V., Waddell, N., 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 491, (7424) 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerrigter, M.E., Dolle, M.E., 1995. Plasmid-based transgenic mouse model for studying in vivo mutations. Nature. 377, (6550) 657–659. [DOI] [PubMed] [Google Scholar]
- Breast Cancer Linkage Consortium, T, 1999. The breast cancer Linkage Consortium: cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst.. 91, (15) 1310–1316. [DOI] [PubMed] [Google Scholar]
- Bryant, H.E., Schultz, N., 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 434, (7035) 913–917. [DOI] [PubMed] [Google Scholar]
- Caldas, C., Kern, S.E., 1995. K-ras mutation and pancreatic adenocarcinoma. Int. J. Pancreatol.. 18, (1) 1–6. [DOI] [PubMed] [Google Scholar]
- Choi, E., Park, P.G., 2012. BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation. Dev. Cell. 22, (2) 295–308. [DOI] [PubMed] [Google Scholar]
- Collins, N., McManus, R., 1995. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12-13. Oncogene. 10, (8) 1673–1675. [PubMed] [Google Scholar]
- Connor, F., Bertwistle, D., 1997. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat. Genet.. 17, (4) 423–430. [DOI] [PubMed] [Google Scholar]
- Cook, N., Frese, K.K., 2012. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J. Exp. Med.. 209, (3) 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch, F.J., Johnson, M.R., 2007. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol. Biomarkers Prev.. 16, (2) 342–346. [DOI] [PubMed] [Google Scholar]
- Daniels, M.J., Wang, Y., 2004. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 306, (5697) 876–879. [DOI] [PubMed] [Google Scholar]
- Eklund, L., Bry, M., 2013. Mouse models for studying angiogenesis and lymphangiogenesis in cancer. Mol. Oncol.. 7, (2) 259–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, H., McCabe, N., 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434, (7035) 917–921. [DOI] [PubMed] [Google Scholar]
- Feldmann, G., Karikari, C., 2011. Inactivation of Brca2 cooperates with Trp53(R172H) to induce invasive pancreatic ductal adenocarcinomas in mice: a mouse model of familial pancreatic cancer. Cancer Biol. Ther.. 11, (11) 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese, K.K., Neesse, A., 2012. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov.. 2, (3) 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, L.S., Thistlethwaite, F.C., 1998. Thymic lymphomas in mice with a truncating mutation in Brca2. Cancer Res.. 58, (7) 1338–1343. [PubMed] [Google Scholar]
- Goggins, M., Schutte, M., 1996. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res.. 56, (23) 5360–5364. [PubMed] [Google Scholar]
- Gretarsdottir, S., Thorlacius, S., 1998. BRCA2 and p53 mutations in primary breast cancer in relation to genetic instability. Cancer Res.. 58, (5) 859–862. [PubMed] [Google Scholar]
- Gudmundsson, J., Johannesdottir, G., 1995. Different tumor types from BRCA2 carriers show wild-type chromosome deletions on 13q12-q13. Cancer Res.. 55, (21) 4830–4832. [PubMed] [Google Scholar]
- Guerra, C., Barbacid, M., 2013. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol. Oncol.. 7, (2) 232–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, S.A., Greenhalf, B., 2003. BRCA2 germline mutations in familial pancreatic carcinoma. J. Natl. Cancer Inst.. 95, (3) 214–221. [DOI] [PubMed] [Google Scholar]
- Hezel, A.F., Kimmelman, A.C., 2006. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev.. 20, (10) 1218–1249. [DOI] [PubMed] [Google Scholar]
- Hingorani, S.R., Petricoin, E.F., 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 4, (6) 437–450. [DOI] [PubMed] [Google Scholar]
- Hingorani, S.R., Wang, L., 2005. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 7, (5) 469–483. [DOI] [PubMed] [Google Scholar]
- Hong, S.M., Vincent, A., 2012. Genome-wide somatic copy number alterations in low-grade PanINs and IPMNs from individuals with a family history of pancreatic cancer. Clin. Cancer Res.. 18, (16) 4303–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban, R., Pitman, M., Klimstra, D.S., 2007. Tumors of the Pancreas, AFIP ATLAS of Tumour Pathology, Series IV. [Google Scholar]
- Hruban, R.H., Maitra, A., 2008. Update on pancreatic intraepithelial neoplasia. Int. J. Clin. Exp. Pathol.. 1, (4) 306–316. [PMC free article] [PubMed] [Google Scholar]
- Jackson, E.L., Willis, N., 2001. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev.. 15, (24) 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobetz, M.A., Chan, D.S., 2013. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut.. 62, (1) 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, L., Mercer, K., 2001. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 410, (6832) 1111–1116. [DOI] [PubMed] [Google Scholar]
- Jones, S., Hruban, R.H., 2009. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 324, (5924) 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers, J., Meuwissen, R., 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet.. 29, (4) 418–425. [DOI] [PubMed] [Google Scholar]
- Jonsdottir, A.B., Stefansson, O.A., 2012. Tetraploidy in BRCA2 breast tumours. Eur. J. Cancer. 48, (3) 305–310. [DOI] [PubMed] [Google Scholar]
- Jonsson, J., Carlsson, L., 1994. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 371, (6498) 606–609. [DOI] [PubMed] [Google Scholar]
- King, T.A., Li, W., 2007. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann. Surg. Oncol.. 14, (9) 2510–2518. [DOI] [PubMed] [Google Scholar]
- Lee, H., Trainer, A.H., 1999. Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol. Cell. 4, (1) 1–10. [DOI] [PubMed] [Google Scholar]
- Lomonosov, M., Anand, S., 2003. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev.. 17, (24) 3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra, A., Hruban, R.H., 2008. Pancreatic cancer. Annu. Rev. Pathol.. 3, 157–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel, T., Nahse-Kumpf, V., 2011. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep.. 12, (7) 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikaelsdottir, E.K., Valgeirsdottir, S., 2004. The Icelandic founder mutation BRCA2 999del5: analysis of expression. Breast Cancer Res.. 6, (4) R284–R290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal, G., Rowley, M., 2012. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev. Cell. 23, (1) 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan, M.E., Pierce, A.J., 2001. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell. 7, (2) 263–272. [DOI] [PubMed] [Google Scholar]
- Offield, M.F., Jetton, T.L., 1996. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 122, (3) 983–995. [DOI] [PubMed] [Google Scholar]
- Olive, K.P., Jacobetz, M.A., 2009. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 324, (5933) 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive, K.P., Tuveson, D.A., 2004. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 119, (6) 847–860. [DOI] [PubMed] [Google Scholar]
- Parkin, D.M., Bray, F.I., 2001. Cancer burden in the year 2000. The global picture. Eur. J. Cancer. 37, (Suppl. 8) S4–S66. [DOI] [PubMed] [Google Scholar]
- Patel, K.J., Yu, V.P., 1998. Involvement of Brca2 in DNA repair. Mol. Cell. 1, (3) 347–357. [DOI] [PubMed] [Google Scholar]
- Plentz, R., Park, J.S., 2009. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 136, (5) 1741–1749 e1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, N., Stratton, M.R., 1998. The genetics of breast cancer susceptibility. Annu. Rev. Genet.. 32, 95–121. [DOI] [PubMed] [Google Scholar]
- Redston, M.S., Caldas, C., 1994. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res.. 54, (11) 3025–3033. [PubMed] [Google Scholar]
- Rowley, M., Ohashi, A., 2011. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology. 140, (4) 1303–1313, e1301–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher, K., Christ, N., 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 145, (4) 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan, S.K., Morimatsu, M., 1997. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 386, (6627) 804–810. [DOI] [PubMed] [Google Scholar]
- Shive, H.R., West, R.R., 2010. brca2 in zebrafish ovarian development, spermatogenesis, and tumorigenesis. Proc. Natl. Acad. Sci. U. S. A.. 107, (45) 19350–19355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis, F., Cassidy, L.D., 2010. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell. 18, (5) 499–509. [DOI] [PubMed] [Google Scholar]
- Smith, S.A., Easton, D.F., 1992. Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat. Genet.. 2, (2) 128–131. [DOI] [PubMed] [Google Scholar]
- Tutt, A., Bertwistle, D., 2001. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J.. 20, (17) 4704–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutt, A., Gabriel, A., 1999. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr. Biol.. 9, (19) 1107–1110. [DOI] [PubMed] [Google Scholar]
- Tutt, A.N., van Oostrom, C.T., 2002. Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation. EMBO Rep.. 3, (3) 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson, D.A., Hingorani, S.R., 2005. Ductal pancreatic cancer in humans and mice. Cold Spring Harb Symp. Quant. Biol.. 70, 65–72. [DOI] [PubMed] [Google Scholar]
- Venkitaraman, A.R., 2009. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu. Rev. Pathol.. 4, 461–487. [DOI] [PubMed] [Google Scholar]
- Willems-Jones, A., Kavanagh, L., 2012. High grade prostatic intraepithelial neoplasia does not display loss of heterozygosity at the mutation locus in BRCA2 mutation carriers with aggressive prostate cancer. BJU Int.. 110, (11 Pt C) E1181–E1186. [DOI] [PubMed] [Google Scholar]
- Willems, A.J., Dawson, S.J., 2008. Loss of heterozygosity at the BRCA2 locus detected by multiplex ligation-dependent probe amplification is common in prostate cancers from men with a germline BRCA2 mutation. Clin. Cancer Res.. 14, (10) 2953–2961. [DOI] [PubMed] [Google Scholar]
- Wooster, R., Neuhausen, S.L., 1994. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 265, (5181) 2088–2090. [DOI] [PubMed] [Google Scholar]
- Yan, D.H., Wen, Y., 2004. A delayed chemically induced tumorigenesis in Brca2 mutant mice. Oncogene. 23, (10) 1896–1901. [DOI] [PubMed] [Google Scholar]
- Yu, V.P., Koehler, M., 2000. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev.. 14, (11) 1400–1406. [PMC free article] [PubMed] [Google Scholar]