

Abstract
Osteoarthritis (OA) is a whole joint disease, in which thinning and disappearance of cartilage is a critical determinant in OA progression. The rupture of cartilage homeostasis whatever its cause: aging, genetic predisposition, trauma or metabolic disorder, induces profound phenotypic modifications of chondrocytes, which then promote the synthesis of a subset of factors that induce cartilage damage and target other joint tissues. Interestingly, among these factors are numerous components of the inflammatory pathways. Chondrocytes produce cytokines, chemokines, alarmins, prostanoids and adipokines and express numerous cell surface receptors for cytokines and chemokines, as well as toll-like receptors. These receptors activate intracellular signaling pathways involved in inflammatory and stress responses of chondrocytes in OA joints. This review focuses on mechanisms responsible for the maintenance of cartilage homeostasis and highlights the role of inflammatory processes in OA progression.
Keywords: Chondrocytes, Homeostatic mechanisms, Articular, Cartilage, Osteoarthritis, Inflammation, Mechanical stress, Homeostasis, Cartilage matrix degradation, Alarmins, Toll-like receptors, Chemokines, Adipokines, Mechanotransduction
Introduction
Although osteoarthritis (OA) is considered a disease of the whole joint as an organ, the articular cartilage is altered to some extent in all affected joints with OA. In addition to the development of cartilage changes with aging, cartilage degeneration may occur in response to inappropriate mechanical stress and low-grade local or systemic inflammation associated with trauma, obesity, metabolic syndrome, and genetic predisposition, which are major risk factors of OA development and progression [1, 2]. However, strong functional interactions among the cartilage, synovium, and subchondral bone impact on cartilage function in such a way that it is difficult to know where and when pathological changes begin. Nevertheless, the knowledge we have gained from studies of cartilage derived from the clinic and from animal models has uncovered many important biological factors that impinge on chondrocytes, the cellular component of cartilage, in a temporal and spatial manner to produce pathological changes.
Structure and organization of the articular cartilage
Cartilage is a connective tissue comprising chondrocytes as the unique cell type, which are embedded within an extracellular matrix. Chondrocytes maintain the matrix components under normal, low-turnover conditions in which the glycosaminoglycans on proteoglycans and other non-collagen molecules can be replaced. In normal adult cartilage in the resting, non-stressed steady state, chondrocytes are quiescent cells and there is very little turnover of the collagen network, as the half-life of type II collagen has been calculated as 120 years, whereas that of aggrecan is 120 days.
In spite of this apparently simple structure, articular cartilage is a complex tissue showing different matrix composition and cellular organization ranging from the superficial zone through subchondral bone (Fig. 1). The thickest part of articular cartilage consists of non-mineralized tissue, whereas the thinnest and deepest layer is calcified cartilage. Within the non-mineralized cartilage can be defined three distinct zones, the superficial, the intermediate, and the radial, each characterized by the distinct extracellular matrix composition and organization, as well as by different phenotypic and the gene expression patterns of the resident chondrocytes [3]. Another level of complexity is revealed by differences in matrix constituents between the interterritorial region containing the collagen network and aggrecan as major components, and the pericellular matrix, containing such proteins as collagen VI, fibromodulin, and matrilin 3, but little or no type II collagen.
Figure 1. Schematic representation of cartilage organization in healthy joint.
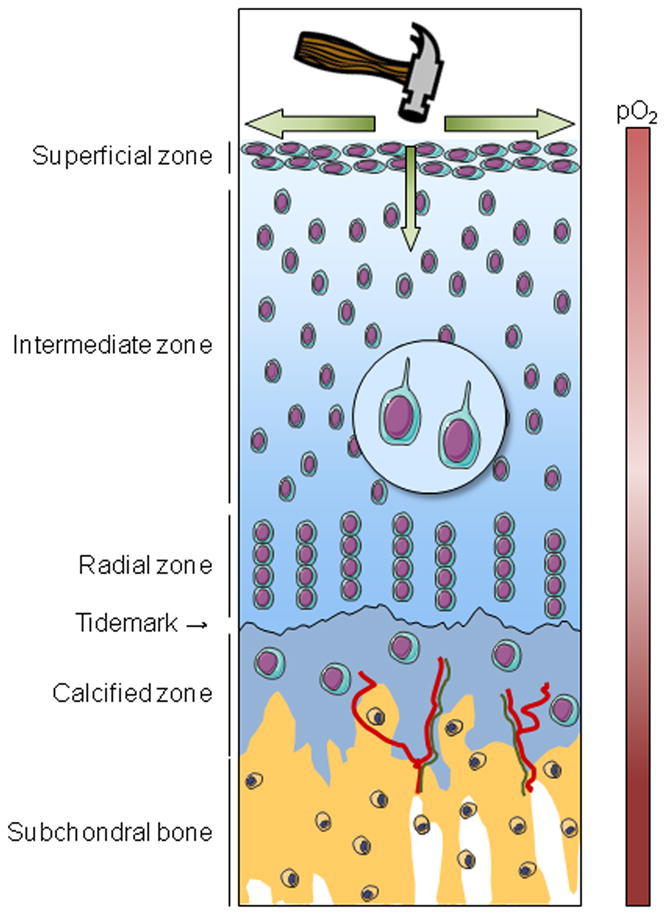
Healthy articular cartilage comprises four different areas: the superficial, intermediate, radial and calcified zones. Each is characterized by a peculiar chondrocyte phenotype and by distinctive extracellular matrix organization and composition. The calcified zone differs from the three other zones by the mineralization of its extracellular matrix, by the presence of vessels (red) and by nerve fibers (green) that originate from the subchondral bone. The calcified zone interfaces with the non-mineralized cartilage, from which it is separated by the tidemark, and the subchondral bone. Due to the absence of vessels within cartilage, chondrocytes live in a hypoxic environment. Hypoxia is important for chondrocyte function and viability. Oxygen and nutrients come from the vascular supply in the synovium and the subchondral bone. A main function of cartilage is the absorption and the dissipation of mechanical load, which is necessary to maintain cartilage homeostasis. The primary cilium plays a crucial role in cartilage homeostasis, especially in the perception of mechanical load due to the presence of integrins and ion channels.
The calcified cartilage interfaces the non-mineralized articular cartilage and the subchondral bone, although direct interactions between the non-calcified cartilage and the subchondral bone have also been described [4]. The tidemark, a thin line revealed after hematoxylin staining, marks the mineralization front between the calcified and non-calcified articular cartilage [5]. The tidemark displays a peculiar matrix composition [6] and also comprises matrix vesicles [7]. The calcified cartilage has unique matrix composition with chondrocytes that express markers of hypertrophy. With aging, blood vessels and nerves can be observed in the calcified cartilage arising from the subchondral bone [8], whereas the non-calcified cartilage is normally avascular and aneural (Fig. 1).
Cartilage functions and homeostasis
Cartilage provides a smooth surface with a very low coefficient of friction allowing for an efficient gliding motion during joint movement. This is facilitated by a boundary layer of lubricants on the articular surface provided by lubricin and hyaluronic acid produced by both chondrocytes and synovial cells [9,10]. A main function of cartilage is the absorption and dissipation of mechanical load. This is allowed by the spatial organization of the matrix components in the superficial layer and by the high content of proteoglycans. Mechanical load is necessary for cartilage homeostasis (Fig. 1). It induces fluid movement between the cartilage and the synovial fluid, playing an important role in the diffusion of molecules across cartilage and thus facilitating its nutrition [11].
Numerous in vivo studies show that immobilization leads to joint damage [12]. Notably, a loss of proteoglycan content associated with increased MMP-3 (matrix metalloproteinase 3) and ADAMTS-5 (A Disintegrin and Metalloproteinase with Thrombospondin Motifs 5) is observed in rodents after hind limb immobilization, whereas joint movement prevents protease increase and proteoglycan loss [13]. Moreover, mechanical stimulation has opposite effects on anabolism and catabolism, increasing aggrecan and a decreasing MMP-3 expression in human chondrocytes [14]. In addition, in vitro low-intensity cyclic mechanical loading of chondrocytes inhibits interleukin 1 (IL-1)- and tumor necrosis factor α (TNF-α)-induced inflammatory and catabolic responses [12]. Mechanical sensors in chondrocytes include integrin, syndecan, and ion channels. The primary cilium is a non-motile organelle that projects from cells in almost all vertebrate cells and acts as a mechanical sensor 15]. In Tg737orpk mutant chondrocytes, which do not express the protein polaris required for ciliary assembly, compression-induced Ca2+ signaling is lost [16]. Mechanical sensors, including calcium channels and integrins, are found in the primary cilium [15, 17], especially in chondrocytes [18]. In the load-bearing area of horse cartilage, the primary cilium is aligned in different orientations in the superficial and radial zones, whereas this organization is lost in the non-load-bearing areas [19].
The primary cilium has roles in cartilage homeostasis that exceed its involvement solely as a mechanical sensor, as suggested by observations of skeletal defects in mutant mice without primary cilia (Fig. 1) [20]. In the absence of primary cilia, marked changes in the cellular organization and defects in matrix component deposition are observed in the growth plate and the articular cartilage of mice [21-23]. Hedgehog (Hh) signaling plays an important role in chondrogenesis, hind limb formation and growth plate organization. Recent studies have shown that the primary cilium is a crucial component of Hh signaling [24]. Moreover, in the Col2aCre;Ift88fl/fl mouse strain, in which the polaris protein is specifically deleted in chondrocytes, the defect in articular cartilage structure is associated with increased Hh signaling [21]. A recent study proposed that increased Hh signaling due to the loss of primary cilia is involved in chondrosarcoma development [25]. In addition to mechanotransduction and Hh signaling, the primary cilium can also be a partner in inflammatory pathways. The induction of prostaglandin (PG) E2 and nitric oxide (NO) release by IL-1β in chondrocytes is indeed prevented in Tg737orpk-derived chondrocytes [16].
Since cartilage is an avascular tissue, chondrocytes live in a hypoxic environment (Fig. 1). Oxygen and nutrients come from the vascular supply in the joint capsule, synovium and subchondral bone. Hypoxia is therefore the normal environment for chondrocytes, which synthesize and accumulate higher amounts of type II collagen and aggrecan when cultured under hypoxia rather than normoxia [26]. In addition, hypoxia displays a protective effect on cartilage, since the basal synthesis and release of MMP-1 and MMP-13, as well as generation of type II collagen cleavage fragments, are lower under hypoxia than in normoxia [26]. Similarly, the production of PGE2 and NO by porcine chondrocytes in response to IL-1α and TNF-α is decreased when cells are cultured in hypoxia [27]. Hypoxia inducible factor 1 (HIF-1) is a heterodimeric (α/β) transcription factor, whose protein levels are regulated by oxygen. Under normoxia, the cell content of HIF-1 is low due to the hydroxylation of the α-subunit on specific proline residues by prolyl-hydroxylases. The proline-hydroxylated form of HIF-1α promotes interaction with the von Hippel-Landau tumor suppressor protein, an E3 ubiquitin ligase, and proteolytic inactivation by the proteasome. In contrast, under hypoxia, prolyl-hydroxylase activity is reduced and sustained amounts of HIF-1α can be measured. The gene encoding HIF-1α, Epas1, is expressed in cartilage. Several studies have provided evidence that HIF-1α is an important factor promoting chondrocyte function and survival [28-30]. The specific deletion of the Epas1 gene in the cartilaginous growth plate is associated with chondrocyte apoptosis [30]. The intraarticular injection of 2-methoxyestradiol, an inhibitor of HIF-1, in Balb/C mice promotes OA lesions with cartilage degradation and osteophyte formation [31].
Cartilage changes in osteoarthritis
In OA, early changes in cartilage appear at the joint surface in areas where mechanical forces such as shear stress are greatest [32]. The normally quiescent chondrocytes undergo a phenotypic shift and become “activated”, characterized by cell proliferation, cluster formation, and increased production of both matrix proteins and matrix-degrading enzymes (Fig. 2) [33]. Disruption of the normal resting state of chondrocytes may be viewed as an injury response involving the recapitulation of developmental programs, leading to matrix remodeling, inappropriate hypertrophy-like maturation, and cartilage calcification [10, 33, 34]. This increased cartilage calcification is associated with tidemark advancement, or duplication, and vascular penetration from the subchondral bone. Whether these events precede, or not, the early changes appearing at the surface remains controversial [35].
Figure 2. Schematic representation of cartilage alterations in OA.
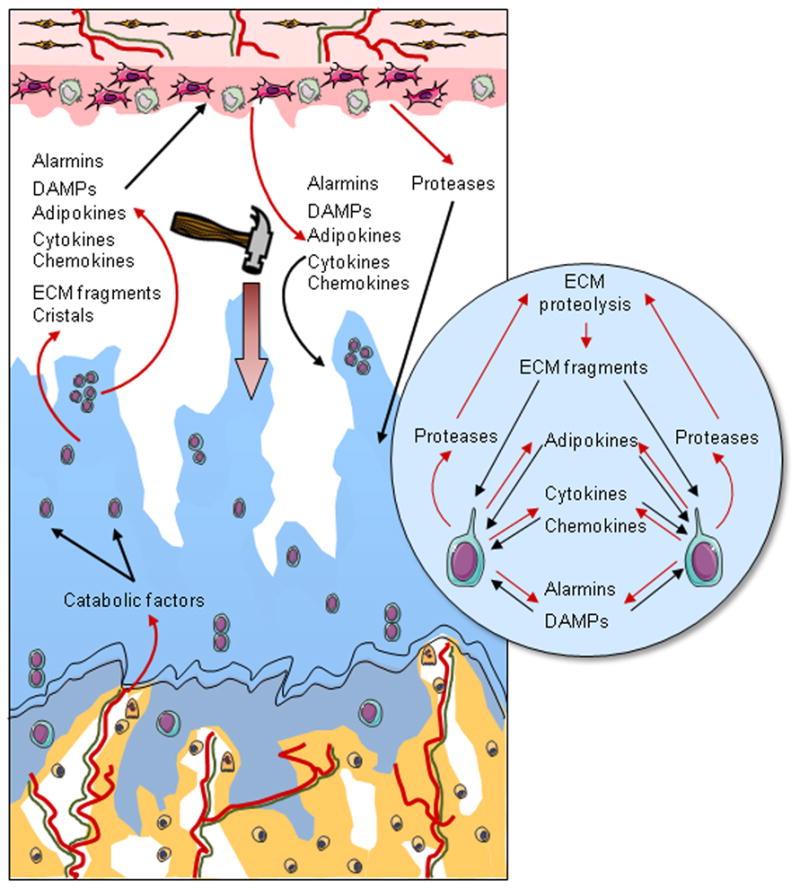
In OA, there is a progressive disappearance of cartilage associated with chondrocyte loss and phenotypic modifications, including the formation of clusters, the activation of a catabolic phenotypic and hypertrophic differentiation. In addition to cartilage damage, remodeling of the subchondral bone occurs with the development of vessels (red) located in structures called vascular channels, which also contain osteoblasts, osteoclasts, and sensory nerves (green). Vascular channels are supposed to facilitate biochemical communication between the bone and the cartilage. In response to several stimuli, including inappropriate mechanical loading and catabolic factors coming from the subchondral bone, chondrocytes modify their phenotype and express a subset of factors, such as cytokines, chemokines, alarmins, DAMPs, and adipokines. All of these mediators act as paracrine factors and initiate a vicious circle of cartilage degradation but also reach the synovium and provoke an inflammatory process with the production by synovial macrophages and fibroblasts of factors, which both promote inflammation in the synovium and participate in cartilage damage.
Hypertrophic chondrocytes express the genes encoding Runx2, MMP-13, and type X collagen, which can all be detected in OA cartilage. Interestingly, increased mRNA expression of these markers is observed in the articular cartilage of Col2a-Cre;Ift88fl/fl mice, whose chondrocytes are devoid of primary cilium [21], suggesting that disruption of cartilage development due to loss of primary cilia may play a role in OA development in adult mice. Paradoxically, the number of primary cilia observed on chondrocytes in bovine OA tissues seems to increase with OA progression[36], suggesting that any disruption of the normal pattern of primary cilia in cartilage results in loss of homeostasis.
At the osteochondral junction, vessels are found in structures called vascular channels [37], which also contain osteoblasts and osteoclasts (Fig. 2) [38]. Interestingly, soluble mediators secreted by these bone cells could cross the osteochondral junction inducing deleterious phenomenon in cartilage (Fig. 2) [39]. Osteoclast-derived TGF-β1 is activated in subchondral bone in response to altered mechanical loading in an anterior cruciate ligament transection (ACLT) mouse model of osteoarthritis [40]. Osteoblast-derived 14-3-3ε dose-dependently induces the release of catabolic factors by chondrocytes [41]. These vascular channels also contain sensory nerve terminations [42]. The presence of sensory nerve terminations within vascular channels and the positive association between the number of vascular channels and the clinical disease activity [42] suggest a link between the remodeling of the osteochondral junction and OA pain [42]. Moreover, the application of a specific angiogenesis inhibitor PPI-2458 in a rat OA model limits joint damage and pain, in addition to osteochondral angiogenesis [43].
Consequences of cartilage matrix degradation
The main cartilage matrix degrading enzymes are zinc-dependent metalloproteinases belonging to MMP and ADAMTS families. MMPs include the collagenases MMP-1 and MMP-13, the latter being highly efficient against type II collagen as a substrate, and MMP-3, which is a potent aggrecanase and MMP activator. The other major aggrecanases in cartilage are ADAMTS-4 and ADAMTS-5. In addition, several serine and cysteine proteases are found in OA joint, including cathepsins K [44].
Chondrocytes sense mechanical stress and changes in the pericellular matrix largely through receptors for extracellular matrix (ECM) components. The patterns of integrin receptors, for example, change in response to mechanical or inflammatory stimuli, resulting in upregulation of aggrecanases and collagenases. However, the receptors on the resting chondrocyte are protected from interacting with certain matrix components by the unique composition of the pericellular matrix. The type II collagen-containing network in the interterritorial region is normally not accessible to degradation because it is coated with proteoglycans. The importance of proteoglycan depletion in cartilage erosion was demonstrated in Adamts5 knockout mice, which are protected against progression in the surgical OA model [45]. However, aggrecan depletion, by itself, does not drive OA progression, as suggested by studies in Mmp13 knockout mice showing that MMP-13 deficiency inhibits cartilage erosion, but does not prevent aggrecan depletion [46]. Once the collagen network begins to degrade, this marks progression to irreversible cartilage degradation.
Mechanical stimulation induces the expression of MMPs by chondrocytes (Fig. 2) [47]. In addition, recent studies suggest that biomechanical stress may initiate the disruption of the pericellular matrix through the serine proteinase, High Temperature Requirement A1 (HTRA1) [48]. The receptor tyrosine kinase, discoidin domain receptor 2 (DDR2) is then exposed to its ligand, native type II collagen, becomes activated, and preferentially induces and activates MMP-13 [49]. Syndecan-4, a trans-membrane heparan sulfate proteoglycan involved in the maintenance of homeostasis, is a positive effector of ADAMTS-5 activation through controlling the synthesis of the MMP-3 [50].
As articular cartilage matrix proteins are degraded, activation of certain receptors stimulates the production of matrix-degrading proteinases and inflammatory cytokines and chemokines, either as initiating or feedback amplification events. Fragments of matrix proteins are produced which can interact with integrin receptors and stimulate or feedback amplify further matrix destruction (Fig. 2). Fragments found in OA cartilage include fibronectin [51, 52], small leucine-rich proteoglycans [53], and collagen [54]. Fibronectin and collagen fragments, in turn, can stimulate the production of inflammatory cytokines, chemokines, and MMPs [10,51, 55, 56].
Cartilage matrix degradation products may activate innate immune responses. Members of the small leucine-rich proteoglycan (SLRP) family such as fibromodulin and decorin may target the classic complement pathway and enhance or inhibit its activation [57]. COMP, on the other hand, is a potent activator of the alternative complement pathway and complexes of COMP and C3b may be found in OA synovial fluids [58]. Interestingly, expression levels of inflammatory and degradative molecules are lower in chondrocytes from destabilized joints from C5-deficient mice than C5-sufficient mice, and the membrane attack complex induces production of these molecules in cultured chondrocyte [59]. Many of the receptors discussed above may be present in synovial cells accounting for amplification of their downstream responses and perpetuation of the “vicious” cycle.
Pro-inflammatory signals and mechanotransduction
Although overt inflammatory processes are not present except locally in OA joints, abnormal mechanical and oxidative stresses are probably involved in the induction of inflammatory mediators, including cytokines, chemokines, cyclooxygenase (COX)-2, microsomal PGE synthase-1 (mPGES-1), soluble phospholipase A2 (sPLA2), and inducible nitric oxide synthase (NOS2), which contribute to the dysregulation of the chondrocyte function and the exacerbation of the cartilage erosion and loss of function (Fig. 2). Chondrocytes in OA cartilage, especially those in clonal clusters, express receptors that may respond to cytokines and chemokines produced in the synovium and other periarticular joint tissues and detected in OA synovial fluid [60]. Although IL-1β mRNA may be induced in chondrocytes, and the inflammasome complex, including NALP-3 and the IL-1β activator caspase-1, is expressed in OA cartilage, active IL-1β is not produced and secreted by OA chondrocytes suggesting that cartilage may be degraded independently of inflammasome activity [61]. Many studies have shown that inflammatory cytokines stimulate expression of MMP-3, - 9, and -13, which co-localize with type II collagen cleavage epitopes in regions of matrix depletion in OA cartilage. The regulation of ADAMTS-4 and -5 by inflammatory stimuli in cartilage may be species-specific [62-64].
High magnitude, injurious mechanical stress, which results in the subsequent release of cartilage matrix degradation products, is a major risk factor for OA onset and progression, in part because it triggers the same signaling pathways as those induced by inflammatory cytokines. Along with NF-κB activation and translocation [65-67], the activation of cell surface mechano-receptors by mechanical forces also induces mitogen activated protein kinase (MAPK) signaling [68, 69], thus controlling the expression of downstream target genes such as MMP13, NOS2, COX2, ADAMTS, and IL1B genes. Since these pathways are also known to both induce and amplify the expression of cytokine and chemokine genes, it therefore remains controversial whether inflammatory mediators are primary or secondary regulators of cartilage damage and defective repair mechanisms in OA (for review, see [33, 70]). Activation of the extracellular-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 MAPK signaling cascades coordinates phosphorylation events that result in the activation of transcription factors such as AP-1 (cFos/cJun), ETS, Runx2, HIF-2α, and C/EBPβ, which together with NF-κB, regulate expression of genes involved in catabolic and inflammatory events [60, 62, 71-76].
During mechanical loading and the development of OA, there is evidence for the loss of cells starting in the superficial zone of cartilage [77], which is associated with an age-related decrease in HMGB2 [78]. Increased production of ROS mediated by mechanical injury or in response to cytokines and matrix fragments may also contribute to cell death [77]. Caspase inhibitors block cell death and result in decreased severity of cartilage lesions in a rabbit model of post-traumatic OA [79]. Autophagy, which serves as a protective mechanism used by cells under stress, also declines with increasing OA severity [80], and rapamycin acting through the mTOR signaling pathway activates autophagy and reduces the severity of experimental murine osteoarthritis [10,81-83].
Chemokines
Chondrocytes also express chemokine receptors including CXCR3, CXCR4, CXCR5, CCR1, CCR3, CCR5 and CCR6 and numerous chemokines, including IL-8, MIP-1α, GROαβγ, MCP-1, eotaxin-1 and RANTES, which may play important roles in activating catabolic pathways and chondrocyte hypertrophy [56, 84-89]. Chemokines can act as chemoattractants and they play a crucial role in tissue homeostasis, especially in the immune system. Inappropriate activation of the chemokine network is associated with inflammatory arthritis and other autoimmune and inflammatory conditions. Many chemokines are produced in joint tissues of patients with OA and after joint injury [90-92]. The synovia of patients at an early stage OA have a unique synovial chemokine signature, with expression of CCL19 and its receptor CCR7 associated with increased symptoms [92]. Elevated levels of CCL5 and CCL19 have been detected in synovial fluids from patients with both RA and OA [93, 94].
Toll-like receptors and alarmins
Molecules released from the damaged cartilage matrix into the synovial fluid have been implicated in promoting the release of proteolytic enzymes by synovial cells and recruitment of inflammatory cells to the joint (Fig. 2) [95-97]. These secreted damage-associated molecular patterns (DAMPs) or alarmins, act as ligands of Toll-like receptors (TLR) or Receptor for Advanced Glycation Endproducts (RAGE) to activate inflammatory and catabolic events in articular cartilage and other joint tissues [98-100]. Chondrocytes express TLRs, whose expression is increased in OA cartilage and induced by inflammatory stimuli [101-104]. TLR-2 and 4 levels are increased in areas of cartilage near OA lesions. The activation by the TLR-2 and 4 ligands, peptidoglycan and LPS, respectively, leads to increased expression of downstream inflammation-related genes including MMPs and NOS2 via NF-κB signaling [105]. Plasma proteins present in OA synovial fluid may function as DAMPs and thereby contribute to a low-grade inflammatory state [106].
Both hydroxyapatite crystals and calcium pyrophosphate crystals, associated with calcification of the articular cartilage and meniscus, or chondrocalcinosis, are common in the joints of older adults with knee OA [107, 108]. Calcium pyrophosphate crystals may stimulate TLRs present on chondrocytes and synovial cells to promote production of inflammatory mediators, including nitric oxide [109]. Activation of the NLRP3 inflammasome by hydroxyapatite crystals may stimulate production of inflammatory mediators, including IL-1 and IL-18 [110].
The alarmins, S100A4, A8, A9, and A11, along with high mobility group box (HMGB) protein 1, also signal through RAGE and TLRs to drive matrix catabolism and increase reactive oxygen species (ROS) through upregulating cytokines and chemokines [71, 111-113]. The RAGE ligand S100A11 can drive inflammation-associated chondrocyte hypertrophy and matrix catabolism [114, 115]. HMGB1 acts on articular chondrocytes [116] and osteoarthritic synoviocytes [116] primarily by potentiating the responses to other alarmins. Together with TLR ligands, HMGB1 acts as a cytokine-like signal of innate immunity to induce a hypertrophy-like phenotypic shift in OA chondrocytes [105]. In addition, S100 proteins, including S100A4, S100A8 and S100A9 have been implicated in enhancing chondrocyte catabolism, indicating that the release of DAMPs by surrounding tissues may contribute to cartilage destruction [112, 118, 119].
Adipokines
Although obesity may result in overloading of joints and subsequent OA, the ‘low-grade inflammatory state’ of obese subjects is also considered an important risk factor for OA, partly based on the identification of adipokines in white adipose tissue as an endocrine organ that can contribute to immunity and inflammation (for review, see [120, 121]). Adipokines can also be produced by joint cells including chondrocytes and act locally in cartilage homeostasis and destruction (Fig. 2). Although leptin levels are increased in OA cartilage compared to normal articular cartilage, this adipokines has biphasic effects, contributing to degradation at concentrations and promoting anabolism at lower concentrations, whereas adiponectin may have a protective role against OA. Chondrocyte expression of adipokines can be induced by inflammatory stimuli [122], and stimulation of articular chondrocytes with leptin, adiponectin, visfatin/nampt or resistin, alone or in combination with other inflammatory cytokines, can induce and enhance the expression of, MMPs, NOS2, and cytokines themselves [123-127]. Whether the different adipokines are protective, possibly induced as a feedback mechanism, or detrimental in vivo is unclear, as there is limited information from in vivo models [128-130].
Conclusions
Cartilage is a highly specialized connective tissue, whose damage is the main feature of OA. Whatever the primary determinant of OA: aging, genetic predisposition, metabolic syndrome or trauma, an activation of the inflammatory pathways occurs in cartilage [131]. Chondrocytes express numerous cytokine and chemokine receptors as well as TLRs. Chondrocytes produce inflammatory mediators able to drive cartilage damage and adjacent joint tissue alterations, thus establishing a vicious cycle leading to the progression of OA. Therefore, it appears of primary importance to better define the key components of inflammatory pathways in order to discover disease-modifying OA drugs in the future.
Acknowledgments
Research related to this work was supported by French state funds managed by the ANR within the Investissements d'Avenir programme under reference ANR11-IDEX-0004-02 (to FB and XH), and by National Institutes of Health grants R01-AG022021 and RC4-AR060546 (to MBG).
References
- 1.Blagojevic M, Jinks C, Jeffery A, Jordan KP. Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2010;18(1):24–33. doi: 10.1016/j.joca.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Felson DT, Lawrence RC, Dieppe PA, et al. Osteoarthritis: new insights Part 1: the disease and its risk factors. Ann Intern Med. 2010;133(8):635–646. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 3.Mahjoub M, Berenbaum F, Houard X. Why subchondral bone in osteoarthritis? The importance of the cartilage bone interface in osteoarthritis. Osteoporos Int. 2012;23(Suppl 8):841–846. doi: 10.1007/s00198-012-2161-0. [DOI] [PubMed] [Google Scholar]
- 4.Lyons TJ, McClure SF, Stoddart RW, McClure J. The normal human chondro-osseous junctional region: evidence for contact of uncalcified cartilage with subchondral bone and marrow spaces. BMC Musculoskelet Disord. 2006;7:52. doi: 10.1186/1471-2474-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fawns HT, Landells JW. Histochemical studies of rheumatic conditions. I. Observations on the fine structures of the matrix of normal bone and cartilage. Ann Rheum Dis. 1953;12(2):105–113. doi: 10.1136/ard.12.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyons TJ, Stoddart RW, McClure SF, McClure J. The tidemark of the chondro-osseous junction of the normal human knee joint. J Mol Histol. 2005;36(3):207–215. doi: 10.1007/s10735-005-3283-x. [DOI] [PubMed] [Google Scholar]
- 7.Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab Invest. 2010;90(11):1549–1557. doi: 10.1038/labinvest.2010.152. [DOI] [PubMed] [Google Scholar]
- 8.Lane LB, Villacin A, Bullough PG. The vascularity and remodelling of subchondrial bone and calcified cartilage in adult human femoral and humeral heads An age- and stress-related phenomenon. J Bone Joint Surg Br. 1977;59(3):272–278. doi: 10.1302/0301-620X.59B3.893504. [DOI] [PubMed] [Google Scholar]
- 9.Greene GW, Banquy X, Lee DW, et al. Adaptive mechanically controlled lubrication mechanism found in articular joints. Proc Natl Acad Sci U S A. 2011;108(13):5255–5259. doi: 10.1073/pnas.1101002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Hara BP, Urban JP, Maroudas A. Influence of cyclic loading on the nutrition of articular cartilage. Ann Rheum Dis. 1990;49(7):536–539. doi: 10.1136/ard.49.7.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leong DJ, Hardin JA, Cobelli NJ, Sun HB. Mechanotransduction and cartilage integrity. Ann N Y Acad Sci. 2011;1240:32–37. doi: 10.1111/j.1749-6632.2011.06301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leong DJ, Gu XI, Li Y, et al. Matrix metalloproteinase-3 in articular cartilage is upregulated by joint immobilization and suppressed by passive joint motion. Matrix Biol. 2010;29(5):420–426. doi: 10.1016/j.matbio.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Millward-Sadler SJ, Wright MO, Davies LW, Nuki G, Salter DM. Mechanotransduction via integrins and interleukin-4 results in altered aggrecan and matrix metalloproteinase 3 gene expression in normal, but not osteoarthritic, human articular chondrocytes. Arthritis Rheum. 2000;43(9):2091–2099. doi: 10.1002/1529-0131(200009)43:9<2091::AID-ANR21>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 15.Praetorius HA, Praetorius J, Nielsen S, Frokiaer J, Spring KR. Beta1-integrins in the primary cilium of MDCK cells potentiate fibronectin-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2004;287(5):F969–978. doi: 10.1152/ajprenal.00096.2004. [DOI] [PubMed] [Google Scholar]
- 16.Wann AK, Knight MM. Primary cilia elongation in response to interleukin-1 mediates the inflammatory response. Cell Mol Life Sci. 2012;69(17):2967–2977. doi: 10.1007/s00018-012-0980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 18.McGlashan SR, Jensen CG, Poole CA. Localization of extracellular matrix receptors on the chondrocyte primary cilium. J Histochem Cytochem. 2006;54(9):1005–1014. doi: 10.1369/jhc.5A6866.2006. [DOI] [PubMed] [Google Scholar]
- 19.Farnum CE, Wilsman NJ. Orientation of primary cilia of articular chondrocytes in three-dimensional space. Anat Rec (Hoboken) 2011;294(3):533–549. doi: 10.1002/ar.21330. [DOI] [PubMed] [Google Scholar]
- 20.Moyer JH, Lee-Tischler MJ, Kwon HY, et al. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science. 1994;264(5163):1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- 21.Chang CF, Ramaswamy G, Serra R. Depletion of primary cilia in articular chondrocytes results in reduced Gli3 repressor to activator ratio, increased Hedgehog signaling, and symptoms of early osteoarthritis. Osteoarthritis Cartilage. 2012;20(2):152–161. doi: 10.1016/j.joca.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaushik AP, Martin JA, Zhang Q, Sheffield VC, Morcuende JA. Cartilage abnormalities associated with defects of chondrocytic primary cilia in Bardet-Biedl syndrome mutant mice. J Orthop Res. 2009;27(8):1093–1099. doi: 10.1002/jor.20855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGlashan SR, Haycraft CJ, Jensen CG, Yoder BK, Poole CA. Articular cartilage and growth plate defects are associated with chondrocyte cytoskeletal abnormalities in Tg737orpk mice lacking the primary cilia protein polaris. Matrix Biol. 2007;26(4):234–246. doi: 10.1016/j.matbio.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Roy S. Cilia and Hedgehog: when and how was their marriage solemnized? Differentiation. 2012;83(2):S43–48. doi: 10.1016/j.diff.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Ho L, Ali SA, Al-Jazrawe M, et al. Primary cilia attenuate hedgehog signalling in neoplastic chondrocytes. Oncogene. 2012 doi: 10.1038/onc.2012.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strobel S, Loparic M, Wendt D, et al. Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Res Ther. 2010;12(2):R34. doi: 10.1186/ar2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cernanec J, Guilak F, Weinberg JB, Pisetsky DS, Fermor B. Influence of hypoxia and reoxygenation on cytokine-induced production of proinflammatory mediators in articular cartilage. Arthritis Rheum. 2002;46(4):968–975. doi: 10.1002/art.10213. [DOI] [PubMed] [Google Scholar]
- 28.Duval E, Leclercq S, Elissalde JM, et al. Hypoxia-inducible factor 1alpha inhibits the fibroblast-like markers type I and type III collagen during hypoxia-induced chondrocyte redifferentiation: hypoxia not only induces type II collagen and aggrecan, but it also inhibits type I and type III collagen in the hypoxia-inducible factor 1alpha-dependent redifferentiation of chondrocytes. Arthritis Rheum. 2009;60(10):3038–3048. doi: 10.1002/art.24851. [DOI] [PubMed] [Google Scholar]
- 29.Pfander D, Cramer T, Schipani E, Johnson RS. HIF-1alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci. 2003;116(Pt 9):1819–1826. doi: 10.1242/jcs.00385. [DOI] [PubMed] [Google Scholar]
- 30.Schipani E, Ryan HE, Didrickson S, et al. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15(21):2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfander D, Gelse K. Hypoxia and osteoarthritis: how chondrocytes survive hypoxic environments. Curr Opin Rheumatol. 2007;19(5):457–462. doi: 10.1097/BOR.0b013e3282ba5693. [DOI] [PubMed] [Google Scholar]
- 32.Andriacchi TP, Mundermann A, Smith RL, et al. A framework for the in vivo pathomechanics of osteoarthritis at the knee. Ann Biomed Eng. 2004;32(3):447–457. doi: 10.1023/b:abme.0000017541.82498.37. [DOI] [PubMed] [Google Scholar]
- 33.Goldring MB, Otero M, Plumb DA, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cell Mater. 2011;21:202–220. doi: 10.22203/ecm.v021a16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage. 2012;20(3):223–232. doi: 10.1016/j.joca.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Ko FC, Dragomir C, Plumb DA, et al. In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheum. 2013;65(6):1569–1578. doi: 10.1002/art.37906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGlashan SR, Cluett EC, Jensen CG, Poole CA. Primary cilia in osteoarthritic chondrocytes: from chondrons to clusters. Dev Dyn. 2008;237(8):2013–2020. doi: 10.1002/dvdy.21501. [DOI] [PubMed] [Google Scholar]
- 37.Clark JM. The structure of vascular channels in the subchondral plate. J Anat. 1990;171:105–115. [PMC free article] [PubMed] [Google Scholar]
- 38.Shibakawa A, Yudoh K, Masuko-Hongo K, et al. The role of subchondral bone resorption pits in osteoarthritis: MMP production by cells derived from bone marrow. Osteoarthritis Cartilage. 2005;13(8):679–687. doi: 10.1016/j.joca.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 39.Guevremont M, Martel-Pelletier J, Massicotte F, et al. Human adult chondrocytes express hepatocyte growth factor (HGF) isoforms but not HgF: potential implication of osteoblasts on the presence of HGF in cartilage. J Bone Miner Res. 2003;18(6):1073–1081. doi: 10.1359/jbmr.2003.18.6.1073. [DOI] [PubMed] [Google Scholar]
- 40.Zhen G, Wen C, Jia X, et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19(6):704–712. doi: 10.1038/nm.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Priam S, Bougault C, Houard X, et al. Identification of soluble 14-3-3 as a novel subchondral bone mediator involved in cartilage degradation in osteoarthritis. Arthritis Rheum. 2013;65(7):1831–1842. doi: 10.1002/art.37951. [DOI] [PubMed] [Google Scholar]
- 42.Suri S, Gill SE, Massena de Camin S, et al. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann Rheum Dis. 2007;66(11):1423–1428. doi: 10.1136/ard.2006.063354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashraf S, Mapp PI, Walsh DA. Contributions of angiogenesis to inflammation, joint damage, and pain in a rat model of osteoarthritis. Arthritis Rheum. 2011;63(9):2700–2710. doi: 10.1002/art.30422. [DOI] [PubMed] [Google Scholar]
- 44.Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta. 2011;1824(1):133–145. doi: 10.1016/j.bbapap.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glasson SS, Askew R, Sheppard B, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434(7033):644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- 46.Little CB, Barai A, Burkhardt D, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60(12):3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gosset M, Berenbaum F, Levy A, et al. Mechanical stress and prostaglandin E2 synthesis in cartilage. Biorheology. 2008;45(3-4):301–320. [PubMed] [Google Scholar]
- 48.Polur I, Lee PL, Servais JM, Xu L, Li Y. Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol Histopathol. 2010;25(5):599–608. doi: 10.14670/hh-25.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu H, Raynal N, Stathopoulos S, et al. Collagen binding specificity of the discoidin domain receptors: binding sites on collagens II and III and molecular determinants for collagen IV recognition by DDR1. Matrix Biol. 2010;30(1):16–26. doi: 10.1016/j.matbio.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Echtermeyer F, Bertrand J, Dreier R, et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med. 2009;15(9):1072–1076. doi: 10.1038/nm.1998. [DOI] [PubMed] [Google Scholar]
- 51.Homandberg GA, Wen C, Hui F. Cartilage damaging activities of fibronectin fragments derived from cartilage and synovial fluid. Osteoarthritis Cartilage. 1998;6(4):231–244. doi: 10.1053/joca.1998.0116. [DOI] [PubMed] [Google Scholar]
- 52.Zack MD, Arner EC, Anglin CP, et al. Identification of fibronectin neoepitopes present in human osteoarthritic cartilage. Arthritis Rheum. 2006;54(9):2912–2922. doi: 10.1002/art.22045. [DOI] [PubMed] [Google Scholar]
- 53.Melrose J, Fuller ES, Roughley PJ, et al. Fragmentation of decorin, biglycan, lumican and keratocan is elevated in degenerate human meniscus, knee and hip articular cartilages compared with age-matched macroscopically normal and control tissues. Arthritis Res Ther. 2008;10(4):R79. doi: 10.1186/ar2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bank RA, Krikken M, Beekman B, et al. A simplified measurement of degraded collagen in tissues: application in healthy, fibrillated and osteoarthritic cartilage. Matrix Biol. 1997;16(5):233–243. doi: 10.1016/s0945-053x(97)90012-3. [DOI] [PubMed] [Google Scholar]
- 55.Fichter M, Korner U, Schomburg J, et al. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J Orthop Res. 2006;24(1):63–70. doi: 10.1002/jor.20001. [DOI] [PubMed] [Google Scholar]
- 56.Pulai JI, Chen H, Im HJ, et al. NF-kappa B mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J Immunol. 2005;174(9):5781–5788. doi: 10.4049/jimmunol.174.9.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heinegard D, Saxne T. The role of the cartilage matrix in osteoarthritis. Nat Rev Rheumatol. 2011;7(1):50–56. doi: 10.1038/nrrheum.2010.198. [DOI] [PubMed] [Google Scholar]
- 58.Happonen KE, Saxne T, Aspberg A, et al. Regulation of complement by cartilage oligomeric matrix protein allows for a novel molecular diagnostic principle in rheumatoid arthritis. Arthritis Rheum. 2010;62(12):3574–3583. doi: 10.1002/art.27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59**.Wang Q, Rozelle AL, Lepus CM, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17(12):1674–1679. doi: 10.1038/nm.2543. A clear demonstration of the role of inflammation in OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berenbaum F. Signaling transduction: target in osteoarthritis. Curr Opin Rheumatol. 2004;16(5):616–622. doi: 10.1097/01.bor.0000133663.37352.4a. [DOI] [PubMed] [Google Scholar]
- 61.Bougault C, Gosset M, Houard X, et al. Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum. 2012;64(12):3972–3981. doi: 10.1002/art.34678. [DOI] [PubMed] [Google Scholar]
- 62.Gabay O, Sanchez C, Salvat C, et al. Stigmasterol: a phytosterol with potential anti-osteoarthritic properties. Osteoarthritis Cartilage. 2010;18(1):106–116. doi: 10.1016/j.joca.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 63.Rogerson FM, Chung YM, Deutscher ME, Last K, Fosang AJ. Cytokine-induced increases in ADAMTS-4 messenger RNA expression do not lead to increased aggrecanase activity in ADAMTS-5-deficient mice. Arthritis Rheum. 2010;62(11):3365–3373. doi: 10.1002/art.27661. [DOI] [PubMed] [Google Scholar]
- 64.Song RH, Tortorella MD, Malfait AM, et al. Aggrecan degradation in human articular cartilage explants is mediated by both ADAMTS-4 and ADAMTS-5. Arthritis Rheum. 2007;56(2):575–585. doi: 10.1002/art.22334. [DOI] [PubMed] [Google Scholar]
- 65.Dossumbekova A, Anghelina M, Madhavan S, et al. Biomechanical signals inhibit IKK activity to attenuate NF-kappaB transcription activity in inflamed chondrocytes. Arthritis Rheum. 2007;56(10):3284–3296. doi: 10.1002/art.22933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Knobloch TJ, Madhavan S, Nam J, Agarwal S, Jr, Agarwal S. Regulation of chondrocytic gene expression by biomechanical signals. Crit Rev Eukaryot Gene Expr. 2008;18(2):139–150. doi: 10.1615/critreveukargeneexpr.v18.i2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nam J, Aguda BD, Rath B, Agarwal S. Biomechanical thresholds regulate inflammation through the NF-kappaB pathway: experiments and modeling. PLoS One. 2009;4(4):e5262. doi: 10.1371/journal.pone.0005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fanning PJ, Emkey G, Smith RJ, et al. Mechanical regulation of mitogen-activated protein kinase signaling in articular cartilage. J Biol Chem. 2003;278(51):50940–50948. doi: 10.1074/jbc.M305107200. [DOI] [PubMed] [Google Scholar]
- 69.Fitzgerald JB, Jin M, Chai DH, et al. Shear- and compression-induced chondrocyte transcription requires MAPK activation in cartilage explants. J Biol Chem. 2008;283(11):6735–6743. doi: 10.1074/jbc.M708670200. [DOI] [PubMed] [Google Scholar]
- 70.Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets. 2010;11(5):599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu FC, Hung LF, Wu WL, et al. Chondroprotective effects and mechanisms of resveratrol in advanced glycation end products-stimulated chondrocytes. Arthritis Res Ther. 2010;12(5):R167. doi: 10.1186/ar3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishitani K, Ito H, Hiramitsu T, et al. PGE2 inhibits MMP expression by suppressing MKK4-JNK MAP kinase-c-JUN pathway via EP4 in human articular chondrocytes. J Cell Biochem. 2010;109(2):425–433. doi: 10.1002/jcb.22421. [DOI] [PubMed] [Google Scholar]
- 73.Tetsunaga T, Nishida K, Furumatsu T, et al. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthritis Cartilage. 2011;19(2):222–232. doi: 10.1016/j.joca.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 74.Thirunavukkarasu K, Pei Y, Moore TL, et al. Regulation of the human ADAMTS-4 promoter by transcription factors and cytokines. Biochem Biophys Res Commun. 2006;345(1):197–204. doi: 10.1016/j.bbrc.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 75.Thirunavukkarasu K, Pei Y, Wei T. Characterization of the human ADAMTS-5 (aggrecanase-2) gene promoter. Mol Biol Rep. 2007;34(4):225–231. doi: 10.1007/s11033-006-9037-3. [DOI] [PubMed] [Google Scholar]
- 76*.Yang S, Kim J, Ryu JH, et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16(6):687–693. doi: 10.1038/nm.2153. This article examplifies the role of HIF-2α in cartilage degradation. [DOI] [PubMed] [Google Scholar]
- 77.Del Carlo M, Jr, Loeser RF. Cell death in osteoarthritis. Curr Rheumatol Rep. 2008;10(1):37–42. doi: 10.1007/s11926-008-0007-8. [DOI] [PubMed] [Google Scholar]
- 78.Taniguchi N, Carames B, Ronfani L, et al. Aging-related loss of the chromatin protein HMGB2 in articular cartilage is linked to reduced cellularity and osteoarthritis. Proc Natl Acad Sci U S A. 2009;106(4):1181–1186. doi: 10.1073/pnas.0806062106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.D'Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54(6):1814–1821. doi: 10.1002/art.21874. [DOI] [PubMed] [Google Scholar]
- 80*.Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801. doi: 10.1002/art.27305. Convincing data on the role of autophagy in cartilage degradation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carames B, Hasegawa A, Taniguchi N, et al. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71(4):575–581. doi: 10.1136/annrheumdis-2011-200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carames B, Taniguchi N, Seino D, et al. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012;64(4):1182–1192. doi: 10.1002/art.33444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lotz M, Carames B. Autophagy: a new therapeutic target in cartilage injury and osteoarthritis. J Am Acad Orthop Surg. 2012;20(4):261–262. doi: 10.5435/JAAOS-20-04-261. [DOI] [PubMed] [Google Scholar]
- 84.Alaaeddine N, Olee T, Hashimoto S, Creighton-Achermann L, Lotz M. Production of the chemokine RANTES by articular chondrocytes and role in cartilage degradation. Arthritis Rheum. 2001;44(7):1633–1643. doi: 10.1002/1529-0131(200107)44:7<1633::AID-ART286>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 85.Chauffier K, Laiguillon MC, Bougault C, et al. Induction of the chemokine il-8/kc by the articular cartilage: possible influence on osteoarthritis. Joint Bone Spine. 2012;79(6):604–609. doi: 10.1016/j.jbspin.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 86.Hsu YH, Hsieh MS, Liang YC, et al. Production of the chemokine eotaxin-1 in osteoarthritis and its role in cartilage degradation. J Cell Biochem. 2004;93(5):929–939. doi: 10.1002/jcb.20239. [DOI] [PubMed] [Google Scholar]
- 87.Mazzetti I, Magagnoli G, Paoletti S, et al. A role for chemokines in the induction of chondrocyte phenotype modulation. Arthritis Rheum. 2004;50(1):112–122. doi: 10.1002/art.11474. [DOI] [PubMed] [Google Scholar]
- 88.Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171(8):4406–4415. doi: 10.4049/jimmunol.171.8.4406. [DOI] [PubMed] [Google Scholar]
- 89.Sandell LJ, Xing X, Franz C, et al. Exuberant expression of chemokine genes by adult human articular chondrocytes in response to IL-1beta. Osteoarthritis Cartilage. 2008;16(12):1560–1571. doi: 10.1016/j.joca.2008.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cuellar JM, Scuderi GJ, Cuellar VG, Golish SR, Yeomans DC. Diagnostic utility of cytokine biomarkers in the evaluation of acute knee pain. J Bone Joint Surg Am. 2009;91(10):2313–2320. doi: 10.2106/JBJS.H.00835. [DOI] [PubMed] [Google Scholar]
- 91.Endres M, Andreas K, Kalwitz G, et al. Chemokine profile of synovial fluid from normal, osteoarthritis and rheumatoid arthritis patients: CCL25, CXCL10 and XCL1 recruit human subchondral mesenchymal progenitor cells. Osteoarthritis Cartilage. 2010;18(11):1458–1466. doi: 10.1016/j.joca.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 92.Scanzello CR, McKeon B, Swaim BH, et al. Synovial inflammation in patients undergoing arthroscopic meniscectomy: molecular characterization and relationship to symptoms. Arthritis Rheum. 2011;63(2):391–400. doi: 10.1002/art.30137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pickens SR, Chamberlain ND, Volin MV, et al. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum. 2011;63(4):914–922. doi: 10.1002/art.30232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang MH, Wu FX, Xie CM, et al. Expression of CC chemokine ligand 5 in patients with rheumatoid arthritis and its correlation with disease activity and medication. Chin Med Sci J. 2009;24(1):50–54. doi: 10.1016/s1001-9294(09)60059-6. [DOI] [PubMed] [Google Scholar]
- 95.Bondeson J, Blom AB, Wainwright S, et al. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 2010;62(3):647–657. doi: 10.1002/art.27290. [DOI] [PubMed] [Google Scholar]
- 96.Scanzello CR, Plaas A, Crow MK. Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol. 2008;20(5):565–572. doi: 10.1097/BOR.0b013e32830aba34. [DOI] [PubMed] [Google Scholar]
- 97.Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6(11):625–635. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- 98.Geurts J, van den Brand BT, Wolf A, et al. Toll-like receptor 4 signalling is specifically TGF-beta-activated kinase 1 independent in synovial fibroblasts. Rheumatology (Oxford) 2011;50(7):1216–1225. doi: 10.1093/rheumatology/ker021. [DOI] [PubMed] [Google Scholar]
- 99.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15(7):774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 100.Sofat N. Analysing the role of endogenous matrix molecules in the development of osteoarthritis. Int J Exp Pathol. 2009;90(5):463–479. doi: 10.1111/j.1365-2613.2009.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bobacz K, Sunk IG, Hofstaetter JG, et al. Toll-like receptors and chondrocytes: the lipopolysaccharide-induced decrease in cartilage matrix synthesis is dependent on the presence of toll-like receptor 4 and antagonized by bone morphogenetic protein 7. Arthritis Rheum. 2007;56(6):1880–1893. doi: 10.1002/art.22637. [DOI] [PubMed] [Google Scholar]
- 102.Haglund L, Bernier SM, Onnerfjord P, Recklies AD. Proteomic analysis of the LPS-induced stress response in rat chondrocytes reveals induction of innate immune response components in articular cartilage. Matrix Biol. 2008;27(2):107–118. doi: 10.1016/j.matbio.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 103.Kim HA, Cho ML, Choi HY, et al. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006;54(7):2152–2163. doi: 10.1002/art.21951. [DOI] [PubMed] [Google Scholar]
- 104.Zhang Q, Hui W, Litherland GJ, et al. Differential Toll-like receptor-dependent collagenase expression in chondrocytes. Ann Rheum Dis. 2008;67(11):1633–1641. doi: 10.1136/ard.2007.079574. [DOI] [PubMed] [Google Scholar]
- 105.Liu-Bryan R, Terkeltaub R. Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum. 2010;62(7):2004–2012. doi: 10.1002/art.27475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sohn DH, Sokolove J, Sharpe O, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res Ther. 2012;14(1):R7. doi: 10.1186/ar3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ea HK, Nguyen C, Bazin D, et al. Articular cartilage calcification in osteoarthritis: insights into crystal-induced stress. Arthritis Rheum. 2011;63(1):10–18. doi: 10.1002/art.27761. [DOI] [PubMed] [Google Scholar]
- 108.Musacchio E, Ramonda R, Perissinotto E, et al. The impact of knee and hip chondrocalcinosis on disability in older people: the ProVA Study from northeastern Italy. Ann Rheum Dis. 2011;70(11):1937–1943. doi: 10.1136/ard.2011.150508. [DOI] [PubMed] [Google Scholar]
- 109.Liu-Bryan R, Pritzker K, Firestein GS, Terkeltaub R. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J Immunol. 2005;174(8):5016–5023. doi: 10.4049/jimmunol.174.8.5016. [DOI] [PubMed] [Google Scholar]
- 110.Jin C, Frayssinet P, Pelker R, Cwirka D, et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc Natl Acad Sci U S A. 2011;108(36):14867–14872. doi: 10.1073/pnas.1111101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rasheed Z, Akhtar N, Haqqi TM. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-kappaB in human osteoarthritis chondrocytes. Rheumatology (Oxford) 2011;50(5):838–851. doi: 10.1093/rheumatology/keq380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yammani RR, Carlson CS, Bresnick AR, Loeser RF. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum. 2006;54(9):2901–2911. doi: 10.1002/art.22042. [DOI] [PubMed] [Google Scholar]
- 113.Zreiqat H, Belluoccio D, Smith MM, et al. S100A8 and S100A9 in experimental osteoarthritis. Arthritis Res Ther. 2010;12(1):R16. doi: 10.1186/ar2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cecil DL, Johnson K, Rediske J, et al. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J Immunol. 2005;175(12):8296–8302. doi: 10.4049/jimmunol.175.12.8296. [DOI] [PubMed] [Google Scholar]
- 115.Cecil DL, Terkeltaub R. Transamidation by transglutaminase 2 transforms S100A11 calgranulin into a procatabolic cytokine for chondrocytes. J Immunol. 2008;180(12):8378–8385. doi: 10.4049/jimmunol.180.12.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heinola T, Kouri VP, Clarijs P, et al. High mobility group box-1 (HMGB-1) in osteoarthritic cartilage. Clin Exp Rheumatol. 2010;28(4):511–518. [PubMed] [Google Scholar]
- 117.Garcia-Arnandis I, Guillen MI, Gomar F, et al. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1beta in osteoarthritic synoviocytes. Arthritis Res Ther. 2010;12(4):R165. doi: 10.1186/ar3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van Lent PL, Grevers L, Blom AB, et al. Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann Rheum Dis. 2008;67(12):1750–1758. doi: 10.1136/ard.2007.077800. [DOI] [PubMed] [Google Scholar]
- 119.van Lent PL, Grevers LC, Blom AB, et al. Stimulation of chondrocyte-mediated cartilage destruction by S100A8 in experimental murine arthritis. Arthritis Rheum. 2008;58(12):3776–3787. doi: 10.1002/art.24074. [DOI] [PubMed] [Google Scholar]
- 120.Conde J, Scotece M, Gomez R, et al. Adipokines: biofactors from white adipose tissue. A complex hub among inflammation, metabolism, and immunity. Biofactors. 2011;37(6):413–420. doi: 10.1002/biof.185. [DOI] [PubMed] [Google Scholar]
- 121.Pottie P, Presle N, Terlain B, et al. Obesity and osteoarthritis: more complex than predicted! Ann Rheum Dis. 2006;65(11):1403–1405. doi: 10.1136/ard.2006.061994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Conde J, Gomez R, Bianco G, et al. Expanding the adipokine network in cartilage: identification and regulation of novel factors in human and murine chondrocytes. Ann Rheum Dis. 2011;70(3):551–559. doi: 10.1136/ard.2010.132399. [DOI] [PubMed] [Google Scholar]
- 123.Jacques C, Holzenberger M, Mladenovic Z, et al. Proinflammatory actions of visfatin/nicotinamide phosphoribosyltransferase (Nampt) involve regulation of insulin signaling pathway and Nampt enzymatic activity. J Biol Chem. 2012;287(18):15100–15108. doi: 10.1074/jbc.M112.350215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kang EH, Lee YJ, Kim TK, et al. Adiponectin is a potential catabolic mediator in osteoarthritis cartilage. Arthritis Res Ther. 2010;12(6):R231. doi: 10.1186/ar3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koskinen A, Juslin S, Nieminen R, et al. Adiponectin associates with markers of cartilage degradation in osteoarthritis and induces production of proinflammatory and catabolic factors through mitogen-activated protein kinase pathways. Arthritis Res Ther. 2011;13(6):R184. doi: 10.1186/ar3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Koskinen A, Vuolteenaho K, Nieminen R, Moilanen T, Moilanen E. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from OA patients. Clin Exp Rheumatol. 2011;29(1):57–64. [PubMed] [Google Scholar]
- 127.Zhang Z, Xing X, Hensley G, et al. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010;62(7):1993–2003. doi: 10.1002/art.27473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen TH, Chen L, Hsieh MS, et al. Evidence for a protective role for adiponectin in osteoarthritis. Biochim Biophys Acta. 2006;1762(8):711–718. doi: 10.1016/j.bbadis.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 129.Griffin TM, Fermor B, Huebner JL, et al. Diet-induced obesity differentially regulates behavioral, biomechanical, and molecular risk factors for osteoarthritis in mice. Arthritis Res Ther. 2010;12(4):R130. doi: 10.1186/ar3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Griffin TM, Huebner JL, Kraus VB, Guilak F. Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum. 2009;60(10):2935–2944. doi: 10.1002/art.24854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!) Osteoarthritis Cartilage. 2013;21(1):16–21. doi: 10.1016/j.joca.2012.11.012. [DOI] [PubMed] [Google Scholar]