

Abstract
Characterization of binding kinetics and affinity between a potential new drug and its receptor are key steps in the development of new drugs. Among the techniques available to determine binding affinities, surface plasmon resonance has emerged as the gold standard because it can measure binding and dissociation rates in real-time in a label-free fashion. Surface plasmon resonance is now finding applications in the characterization of molecules for treatment of neurodegenerative diseases, characterization of molecules associated with pathogenesis of neurodegenerative diseases and detection of neurodegenerative disease biomarkers. In addition it has been used in the characterization of a new class of natural autoantibodies that have therapeutic potential in a number of neurologic diseases. In this review we will introduce surface plasmon resonance and describe some applications of the technique that pertain to neurodegenerative disorders and their treatment.
Keywords: Surface plasmon resonance, surface modification, lipid bilayer membrane, antibodies, multiple sclerosis, neuromyelitisoptica, Alzheimer’s disease, amyloid-beta, biomarkers
Introduction
The term “neurodegenerative disorder” encompasses a wide range of medical conditions that primarily affect neuronal cells of the human brain. Neurodegenerative diseases take a sharp toll on millions of individuals and directly affect patients and indirectly their caregivers, families and communities. Most neurodegenerative diseases share a common course: uncontrolled neuronal death, which leads to a progressive decline of nervous system function (cognition or locomotor control). A few examples of neurodegenerative disorders are Alzheimer’s disease (AD) and other dementias, Parkinson’s disease (PD) and PD-related disorders, prion disease, motor neuron diseases (MND), Huntington’s disease (HD), spinocerebellar ataxia (SCA), spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS). In addition diseases with an inflammatory component such as multiple sclerosis (MS) or neuromyelitisoptica (NMO) also have a neurodegenerative component in that neurons and axons ultimately die from the destructive immune response. Both AD and PD are also characterized by intrinsic chronic inflammation mediated by microglial cells which is disputed to be the cause of disease. Non-steroid inhibitors of inflammation (NSAIDs) have since been clinically tested on AD and PD patients, although large-scale double-blind placebo-controlled clinical trials have not supported the use of NSAIDS in neither reducing the risk of AD [1]and PD [2] nor the treatment of AD.[3]
In neurodegenerative disorders, the stimuli for neuronal cell death are multi-factorial. These may include genetic predisposition, environmental factors and cellular stress (oxidative stress and free radical production). Cellular stress disrupts functioning of the endoplasmic reticulum (ER) leading to induction of the unfolded protein response (UPR). ER stress may contribute to neuronal loss in a range of neurodegenerative disorders such as AD, PD and HD. Other stimuli for neuronal cell death are glutamate-induced excitotoxicity, neuroinflammation, bioenergy failure, disruption of Ca2+-regulating systems, mitochondrial dysfunction and misfolded protein accumulation. Neurons rely on autophagy to clear and purge unwanted cytosolic proteins and damaged organelles. Neuronal autophagy may in some cases be defective and results in aggregation of proteins. Accordingly, defective neuronal autophagy was observed in several neurodegenerative conditions. In addition animal models with deficient autophagic processes recapitulate many aspects of neurodegenerative diseases. [5–8] Characterization of protein-protein aggregation strength and their association and dissociation kinetics would help further understand the disease pathogenesis. Moreover, determination of how potential drugs (a) modulate protein-protein interactions and (b) bind to target molecules will help devise new therapeutic approaches.
One such technique for investigation of molecular interactions is surface plasmon resonance (SPR). SPR is a well-established and widely used optical detection technique for measuring the strength and rate of biomolecular interactions.[9] SPR is advantageous because it is label-free and monitors molecular interactions in real time. Label-free detection eliminates the need for fluorescent reporter molecules or radioisotope tags, saving a labeling step, which can be time-consuming, expensive, and more importantly, could alter the molecular interactions depending on the nature of the tag. Real-time measurements allow calculation of the association and dissociation kinetic rate constants, kon and koff of the interaction, in addition to the equilibrium dissociation constant, KD. Another benefit of SPR experiments is that they only need small amounts of reagents compared to equilibrium assays, especially for low affinity binding interactions. [9]
The basic SPR experiment involves immobilizing the receptor on the sensing surface of a flow-cell, and then flowing an analyte solution over the receptors and monitoring the sensor signal change as the molecules bind (a typical setup can be seen in Fig. 1a). The flow-cells are microfluidic devices, usually with channels less than 1 mm wide. The sensing surface is a thin metal film, usually gold or silver, and may be flat or have nanometer-scale structures in it, such as arrays of holes or line gratings. [10–12] Light is directed onto the metal film, and photons of light couple to free electrons in the metal, generating charge oscillations (plasmons) at the surface of the metal, which resonate at certain wavelengths of light. The plasmons are confined to the metal surface and are sensitive to changes in the interfacial refractive index. As analyte binds to receptors at the surface, the refractive index changes, and the plasmon resonance shifts to larger reflection angles. The magnitude of this shift is in proportion to the amount of bound analyte.
Figure 1.

(a) Schematic of typical SPR device. Light is directed onto a gold film and the reflection is monitored with a detector. At a certain angle, the light does not reflect but generates surface plasmon resonance (SPR). In a flow cell above the film, receptors are linked to the film and analyte solution is flowed past the receptors. If the analyte binds to the receptors, the angle at which SPR is generated shifts, and this angle is tracked over time as the reaction progresses. (b) Simulated SPR binding curves of four different molecules (A,B,C,D) with identical KD values (10 nM), but different rate constants, kon and koff. Analyte concentration was 100 nM for all molecules. Units for kon are M−1s−1 and for koff, s−1. This plot demonstrates the significance of knowing the kinetic constants. Molecule A would bind quickly and dissociate quickly, whereas molecule D would bind slowly but dissociate very slowly. The half-lives (t1/2 = ln 2/koff) of the bound complexes are roughly (from A–D): 12 min, 2 hr, 20 hr, and 8 days. (a) is adapted from Cooper [9] and used with permission of Macmillan Publishers Ltd.
Obtaining kinetic rate constants is advantageous over endpoint-only equilibrium measurements such as ELISAs or radioisotope binding assays, in helping to choose which drug leads to follow. Some diseases may need the drug to quickly dissociate from its target receptor (high koff) to reduce toxicity. Other diseases may benefit from the drug staying bound to its target for a longer time (low koff) to prolong its biological effect and reduce necessary dosages. Also, a long residence time of the drug in its target receptor and short residence time in other receptors would indicate higher specificity and would likely translate into fewer side-effects.[13,14]
SPR is versatile and has been used to study molecular interactions between a wide variety of molecules (from simple proteins, carbohydrates, lipids, nucleic acids, and small molecule compounds, to larger protein complexes and even cells), across a wide range of affinities from picomolar to millimolar. [9] In addition, membrane proteins, such as the important therapeutic target G-protein coupled receptors (GPCRs), have been incorporated into SPR experiments, both in detergent solubilized form [15]and in lipid membranes, such as rhodopsin [16], nicotinic acetylcholine receptors [17], and aquaporin-4 [18]. Nevertheless, membrane proteins such as GPCRs are still a challenging system to study outside of cells due to their low level of expression, difficulty to get host systems to express them, and declining stability and activity when removed from their native lipid environment. However advances in protein engineering and SPR methodology are being been made which make the proteins more able to be studied by SPR. The addition of peptide tags to the C-terminus provide a way to uniformly capture the proteins from crude lysates of cells. SPR devices can then be used to screen for the best detergents which will solubilize the protein and still maintain structure and function {Navritolova 2005, 15797568}. This method was recently utilized to find novel high-affinity antagonists to wild-type β2 adrenergic receptor using fragment screening {Aristotelous}. Another protein engineering solution has been to create point mutations in GPCRs to increase their conformational homogeneity and thermostability. One disadvantage to this approach is that the modified proteins may not bind to ligands the same way as wild-type proteins.[19] SPR can also be used for epitope mapping, competition assays, analyte quantitation for applications such as biomarker detection, monitoring production of antibodies and cytokines, ligand fishing, and ADME (adsorption, distribution, metabolism, and excretion) characterization, and other applications, as has been reviewed elsewhere.[9,20] {Rich 2011}
While most SPR devices do not have the throughput of other systems and consist of only a few channels limiting the number of receptors that can be tested per chip, computer automated robotic systems can sample numerous analytes of multiple concentrations from multiwell plates, allowing for higher throughput sampling of analytes and hands free operation. For example, GE Healthcare’s latest system, Biacore 4000, is able to run unattended for 60 hours measuring up to 2400 interactions in 24 hours. Variations of SPR chip designs have been developed as well which allow detection from an array of spotted proteins, enabling high-throughput measurements. [11,12]
There are some limitations and challenges to the SPR technique. It is more difficult to detect the binding of small molecules, since small molecules induce smaller changes in the interfacial refractive index. However, advances in commercial SPR instruments have enabled small molecule detection down to 90 Da.[21] Also, there are challenges with it being a surface based technique, requiring immobilization of one of the binding partners, which can create several artifacts. However, these artifacts can be avoided for most binding pairs with careful experimental design. For example, too many receptors on the surface can lead to steric hindrance of binding sites or shielding by large analytes, reducing the expected binding site concentration, or can pull analytes out of solution too quickly reducing expected analyte concentration. Simply reducing the number of receptors can solve this. Controls are a very important aspect to rule out possible artifacts. Excellent advice on how to obtain high quality SPR data can be found in many of Rich and Myszka’s publications.[20,22–24]
To confirm that the surface-based SPR technology is comparable to traditional solution-based methods and can measure small molecules and immobilized enzymes accurately, Myszka and colleagues have conducted several global benchmark studies. In one study, 22 users using different commercial SPR instrument models, tested four small molecule inhibitors binding to an immobilized enzyme, carbonic anhydrase II, at 6 different temperatures. [25] The results demonstrated some challenges, but also the accuracy, of the instrument. Only a limited number of data sets (8) had useable data for every compound and temperature (about half had instrument artifacts). However, the useable data demonstrated good reproducibility, with a low standard error of the rate constants (less than 11%) and van’t (Is this correct spelling?) Hoff thermodynamic parameters (less than 13%), and KD’s that matched conventional solution-based isothermal titration calorimetry.
A simulation of a typical SPR experiment using four different analyte molecules (A–D, all at 100nM concentration) is shown in Fig. 1b to illustrate SPR curves and demonstrate the usefulness of obtaining binding kinetics information. The plot shows percent bound complexes, which corresponds to the SPR wavelength shift, as a function of time. The experiment consists of two main phases: an analyte injection phase (the association phase in which molecules bind and dissociate), and a washing buffer phase (the dissociation phase, in which molecules only dissociate). All four simulated analytes have the same equilibrium dissociation constant, KD (10 nM), but have different kinetic rate constants, kon (1×105 – 1×102 M−1s−1) and koff (1×10−3 – 1×10−6 s−1), illustrating very different binding profiles. The koff value, is inversely proportional to t1/2, the half-life of the complex (koff= ln(2)/t1/2). In this simulation, the half-lives of analytes (from A–D) are ~12 min (koff=10−3 s−1), ~2 hr (koff=10−4 s−1), ~20 hr (koff=10−5 s−1), and ~8 days (koff=10−6 s−1).
As a surface-sensitive technique, SPR requires receptors to be immobilized on the sensor surface. Since the development of SPR methods in the 1980s, [26] many research groups and companies have advanced surface chemistry to probe a wide variety of molecular interactions. Commonly employed surface chemistries rely upon the formation of self-assembled monolayers (SAMs) on gold surfaces [27]or layers of functionalized dextran.[28] The most well-known SAMs are based upon covalent bonding between gold and thiols, molecules with terminal –SH groups.[29] If a gold sensor chip is exposed to a solution containing an alkanethiol, decanethiol for example, the terminal thiol groups will react with the gold surface and the alkane portions of the molecule will point away from the surface. Eventually self-assembly of the molecules will result in a tightly packed monolayer.
If some of the alkanethiol in the surface-functionalization solution is replaced with bifunctional thiol terminated by a reactive group such as an amine or carboxylate, it is straightforward to link molecules to the sensor surface. For example, if 11-mercaptoundecanoic acid is included in the SAM-formation solution, a portion of the resultant SAM will be reactive to primary amines via well-established carbodiimide (EDC) or EDC/N-hydroxysuccinimide (NHS) chemistries.[30] Fig. 2 shows how these reactions can immobilize molecules like peptides that have free amines on amino acid residues such as asparagine, glutamine and lysine. Using other linkage chemistries receptor molecules with amine, aldehyde, thiol, hydroxyl and carboxyl functionality can also be immobilized on carboxy-terminated SAMs, making this surface-preparation method very versatile. [31] Commercially available sensor chips employ this immobilization scheme through a layer of carboxymethylated dextran on the surface of the gold sensor chip.[28] Most commercially available immobilization schemes rely upon surface-immobilized functionalized dextran matrices to immobilize receptors.
Figure 2.
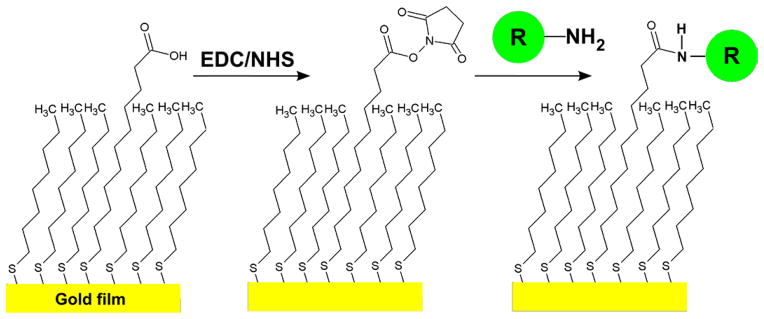
Immobilization of a receptor (R) with a primary amine on a self assembled monolayer (SAM) via EDC/NHS coupling. The carboxyl-terminated thiol is converted to a succinimide ester by reaction with carbodiimide (EDC) and N-hydroxylsuccinimide (NHS), making it reactive toward molecules with available primary amines, as shown by the circle marked “R”. The final product is a receptor (R) covalently linked to the surface with an amide bond.
Capturing receptors to the sensor surface through indirect biotin/streptavidin or antibody interactions is also possible.[32] In this method, a biotinylated SAM can be used to capture streptavidin, or streptavidin can be covalently linked to the sensor surface using one of the above methods. Then streptavidin is used to capture a biotinylated molecule, which can be used as a receptor in further SPR analysis. Like carboxylated sensor chips, commercial streptavidin chips are available.
Nearly half of the top 100-selling pharmaceuticals act on targets associated with cellular membranes.[33] Thus, technology to evaluate interactions between potential therapeutics and membranes or membrane-associated proteins is crucial. Commercially available options are liposome capture chips and hydrophobic SAM layers that allow the formation of a lipid monolayer on the sensor surface. Examples include the Biacore L1 and HPA chips, which capture liposomes or facilitate lipid monolayer formation, respectively. The liposome capture chips are effective and can be used with proteoliposomes, but there is some disagreement in the literature about the morphology of immobilized liposomes. Some groups report liposomes are captured intact, [34] while other groups report that the captured liposomes eventually rupture to form a planar lipid layer.[35] Moreover, the surface charge present on liposomes can alter the immobilization efficiency, which complicates comparisons between different types of liposomes.[36] To reduce uncertainty about the structure of the immobilized lipid membrane layer, a different approach can be taken for membrane functionalization of SPR sensors. Instead of liposomes or lipid monolayers, the sensors are covered with planar lipid bilayers. It is well established that SiO2 surfaces induce rupture of liposomes to form a planar lipid bilayer.[37] Our sensors typically have thin (10 – 20 nm) layers of SiO2 deposited on gold or silver films in order not to reduce the sensitivity. Liposomes adhere strongly to SiO2 and when a critical population of vesicles adsorb, they rupture to form a continuous lipid bilayer, typically known as a supported lipid bilayer (SLB).[38] Membrane-bound proteins can be incorporated into SLBs, but the conformation of transmembrane proteins can be altered by the presence of the supporting substrate.[39] To alleviate this effect a configuration of SPR with a SLB suspended over nanopores in a gold film can be employed to allow incorporation of trans-membrane proteins.[40] Beyond SPR, SLBs can be integrated into a number of different analytical techniques, including mass spectrometry, fluorescence imaging and electrochemical methods.[41] Planar SLBs on SPR chips are convenient mimics for cellular membranes, especially when the molecules under investigation are lipid-binding IgMs that induce remyelination in animal models of human MS (see below).
Multiple Sclerosis (MS)
Studies using imaging, serology, pathology and genetics, and patient response to anti-inflammatory treatments suggest that MS is primarily an inflammatory demyelinating disease of the central nervous system (CNS) with varied clinical presentations and heterogeneous histo-pathologic features. The disease has a peak onset between ages 20 and 40 years, [42] however it may develop in children and has been reported in individuals over 60 years. MS affects women approximately twice as often as men. [43,44] To date, the pathogenesis of MS still remains elusive and there is no definitive cause and no effective cure. Disease pathophysiology is complex and involves genetic susceptibility, environmental factors, and development of pathologic immune-mediated responses leading to focal myelin destruction, axonal loss, and local inflammatory infiltrates. One of the most efficacious drug in MS, but with most significant side effects is Natalizumab (Tysabri), [46,47] a humanized monoclonal antibody that binds to the cell adhesion molecule α4-integrin, a molecule that mediates adhesion and migration of immune cells to organs. Natalizumab reduces the ability of inflammatory immune cells to attach and blocks their ability to cross blood vessel walls. [48,49]
Remyelination in MS
Despite our increased understanding of the biology of glial cells and remyelination, no clear explanation exists for this lack of regenerative potential in CNS demyelinating diseases. Myelination is a key developmental stage in the human brain and continues for at least another 10 to 12 years after birth (precedes adulthood). In contrast, MS patients with detectable brain lesions are commonly, but not exclusively, adults. With respect to remyelination, oligodendrocyte progenitor cells (OPCs) from neonates seem to be 3–5-times more efficient than adult OPCs at colonizing OPC-depleted tissue. [50] This indicates the importance of the recipient’s age as well as the OPC donor’s age for effective remyelination. [51] Supporting evidence comes from lysolecithin-induced demyelination, where mice show less pronounced spontaneous remyelination with increasing age. [52]. This may suggest that age-related, increased deficits of MS patients are due to the decreased ability of OPCs to repopulate demyelinated areas. In addition, the risk of injury accumulates over time with “naked” demyelinated axons, particularly in a hostile environment that caused oligodendrocyte death. Axonal loss will terminate any chances of remyelination and functional recovery. In addition, demyelinated axons may become dysfunctional with loss of specific molecular signals to recruit oligodendrocytes and form protective, functional myelin sheaths. [53] Signals sent from oligodendrocytes to axons may be either inappropriate or inappropriately regulated. However, it is difficult to discern whether the specific villain in the failure of remyelination is the axon or the oligodendrocyte progenitor cell; it is also possible that both may contribute to the problem. In some situations, the axons are missing or dysfunctional, whereas in others, the axons may be normal in number and function but progenitor cells are missing. Both components (axons and progenitor cells) must be intact and able to accept “remyelination signals” for the process to ensue. In addition to age, remyelination failure may be due to a differentiation block of OPCs that are unable to maturate into myelinating oligodendrocytes (reviewed in [54]). It is of note that sufficient OPCs are present also during late stages of adulthood and within the demyelinated plaque. With age, OPCs may become less responsive to either environmental triggers or may express fewer cellular triggers to respond adequately to OPC maturation-stimulating factors.
SPR Characterization of Lipid-Binding Remyelinating IgMs
More than two decades ago, we observed that immunization with CNS homogenates or transfer of anti-serum to CNS homogenates promoted repair of demyelinated CNS lesions in a mouse model of MS. [55] Based on this finding, it was hypothesized that it might be possible to promote CNS repair through the regulation of immune system. As a proof-of-concept, myelin-binding antibodies in the sera of immunized mice preserved oligodendrocytes [56] and promoted remyelination in vivo. [57] Following the success of preliminary studies, to tempt the shift in therapeutics, Rodriguez and coworkers employed a novel strategy to identify human monoclonal antibodies that promote CNS protection and repair. These repair-promoting natural human antibodies are of IgM subtype and have characteristics of classic natural autoreactive antibodies. The antibodies were isolated from the sera of patients with monoclonal gammopathies, using selection criteria of monoclonal immunoglobulin concentration of 3 g/dL or greater and a lack of neurologic or antibody-associated pathologies. The screening strategy involved binding to live CNS tissue (cerebellar slices). [58] Two IgM antibodies were identified in the serum (sHIgM22 and sHIgM46) that promoted significant remyelination in vivo. Subsequently, a recombinant version of sHIgM22, rHIgM22, was engineered by cloning the antibody variable region DNA sequence into an expression vector [59,60] providing the heavy and light chain framework. GMP-grade rHIgM22 antibody was purified in gram quantities for formal toxicology studies and the IND has recently been approved by the FDA for a Phase I, multi-center, double-blind, randomized, placebo-controlled, dose-escalation study designed to evaluate safety, tolerability, pharmacokinetics, and immunogenicity of single intravenous (IV) administrations of rHIgM22 in patients with all clinical presentations of MS (ClinicalTrials.gov Identifier NCT01803867). The Phase I clinical trial involving 60 patients has been in place since March, 2013.
Characterization of the binding properties of rHIgM22 is very attractive; however determination of the antigen(s) for rHIgM22 has been elusive. Therefore mouse IgMs with known receptors that also promote remyelination were investigated using SPR. The antibodies, O1 and O4, are natural autoantibodies [61] that are well-known markers of oligodendrocyte (OL) lineage [62]and have been shown to promote remyelination in mouse models of MS.[63] O4 can initiate Ca2+ signaling in astrocytes and OLs [64] and induce OL differentiation.[65] The primary antigen for O4 is sulfatide (Sulf), a sulfated galactocerebroside that is present from the time OL progenitor cells commit to the OL lineage through terminal differentiation and formation of the myelin membrane. O1, on the other hand, binds to galactocerebroside (GalC), which is inserted in the OL membrane at later stages of development. While O1 and O4 strongly interact with GalC and Sulf respectively, they are polyreactive as is typical of natural autoantibodies. In addition to Sulf, O4 is known to bind to seminolipid and cholesterol, while O1 binds monogalactosyl-diglyceride and psychosine.[66]
Whereas the identity of O1 and O4 antigens is known, little is known about the binding kinetics or the affinities the antibodies have for their antigens. To characterize the O1 and O4 binding properties a variant of SPR that uses nanoholes in a gold film was employed.[67] The SPR sensor was home-built and used a multichannel microfluidic chip for delivery of reagents and analytes to the sensor. Surface functionalization is a key component of a successful SPR sensor. The surface environment must be crafted in a way that resists non-specific adsorption while maintaining selective binding of analytes of interest. In that regard, Sulf and GalC were incorporated into SLBs formed on the surface of the SPR sensor chip. A SLB was formed by first making phosphatidylcholine (PC) vesicles doped with small amounts (2 mole %) of Sulf or GalC. The vesicles were then injected on to the SPR chip, at which point they settled to the surface and spontaneously ruptured due to the presence of a thin (~15 nm) layer of SiO2 on the chip surface. The vesicle rupture process results in a planar SLB covering the sensor surface. It should be noted that there is no commercially available SPR chip that has a SiO2 surface coating.
After formation of the SLB, O1 or O4 were injected into the microfluidic chip. When the SLB contained Sulf, O4 bound to the SLB in a concentration-dependent manner. (Fig. 3a) Likewise, O1 bound to GalC. (Fig. 3b) In the absence of Sulf or GalC, neither O1 nor O4 bound to PC SLBs, (Fig. 3c) proving that the sensor response was due to specific interactions between the antibodies and their antigens. When the antibody injection was completed, PBS was injected into the chip and the antibodies dissociated from their surface-bound state. The kinetics of association and dissociation were analyzed via nonlinear regression (i.e. fits to a biphasic exponential model due to multivalent binding {Kuziemko 1996; Cooper 1999}) to provide association and dissociation rate constants. The ratio of dissociation to association rate constants gives the apparent dissociation constant (KD). The KD for O1 and O4 is strikingly different that for other IgM natural autoantibodies in that it is roughly two orders of magnitude smaller. Most autoantibodies have KD values in the 10−7 to 10−4 M range, [68–70] but O1 bound GalC and O4 bound Sulf with KD values of approximately 10−9 M. Analysis of the binding rate constants showed that the difference in KD compared to other natural autoantibodies was primarily due to the faster association rates observed with O1 and O4. This disparity shows that O1 and O4 have much stronger interactions with their primary binding partners than do other similar antibodies. We also tested O4 binding to a SLB with Sulf and GalC concentrations similar to those found in myelin.[71] With this membrane on the SPR sensor we were able to detect O4 concentrations in the 100 pM range. (Fig. 3d) Possibly these stronger interactions with their corresponding antigens may be one of the reasons that these antibodies are therapeutic to promote remyelination in the CNS.
Figure 3.

SPR characterization of remyelinating IgMs binding to supported lipid bilayers (SLB). (a) SPR kinetics curves of antibody O4 (5 – 50 nM) binding to a SLB containing 2 mole % sulfatide (Sulf). (b) SPR kinetics curves of antibody O1 (5 – 50 nM) binding to a SLB containing 2 mole % galactocerebroside (GalC). (c) Negative controls showing that neither O1 nor O4 bind to SLBs composed solely of egg phosphatidylcholine (egg PC) (solid lines). O1 and O4 did not bind to the sensor protected only by BSA blocking in the absence of a SLB (dashed lines). (d) SPR kinetics curves for 1.25 nM and 310 pM O4 binding to a SLB that mimics the Sulf and GalC content of myelin: 6 mole % Sulf, 16 mole % GalC. In all panels the solid black lines are exponential fits to the data used to extract association and dissociation rate constants. From Wittenberg et al., [67] and used with permission of the American Chemical Society.
Lingo-1 Antagonists
Novel reagents under development for remyelination include high-affinity antibodies and fragments against LINGO-1, which is a component of the Nogo-66 receptor/p75-signaling complex. [72] LINGO-1 antagonists promote OPC differentiation and myelination in vitro and accelerate remyelination in vivo after lysolecithin- or cuprizone-induced demyelination [73] and modulate a rat EAE model. [74] The likely mechanism is the inhibition a natural factor (LINGO-1 or Nogo) that prevents remyelination. To further investigate the structural biology associated with LINGO-1 binding to the Nogo receptor (NgR), Mosyak and coworkers developed a soluble recombinant form of LINGO-1 with a 6-His tag on its C-terminus (LINGO-1-His) and investigated its interactions with NgR using SPR. [75] For NgR, residues 27-451 were fused with a 1D4 epitope tag (NgR 1D4), expressed in CHO cells, purified then immobilized on a carboxymethylated dextran SPR chip using EDC/NHS coupling chemistry. The SPR experiments showed that LINGO-1-His and NgR 1D4 interacted with an equilibrium dissociation constant of 1 μM. The sequence of LINGO-1-His represented the ectodomain of the LINGO-1. Therefore SPR experiments showed that the structural determinants for NgR binding to LINGO-1 are contained within the ectodomain. Characterization of the natural LINGO-1/NgR interaction is crucial in the design of new remyelination therapies and the molecular-level details provided by this work could accelerate the discovery of new LINGO-1 antagonists. Antibodies against LINGO-1 are in clinical trials in MS in an effort to promote CNS remyelination.
Antibody Inhibitors for Neuromyelitis Optica Treatment
NMO is an inflammatory autoimmune disorder of the CNS, almost exclusively relapsing, that causes variable degrees of attack-related disability. Based on recent epidemiologic reports, there are an estimated 4,000 cases in the United States and a half million worldwide. As with many autoimmune conditions, females are more susceptible to the disease than males, at an approximate ratio of 4:1. NMO can affect both children and adults. At this time there is no FDA-approved NMO-specific therapy although immunosuppression with steroids and azathioprine has shown efficacy in clinical practice.[76] Acute attacks respond to plasmapheresis. [77] NMO-IgG directed against aquaporin-4 (AQP4) is believed to play a role in causing NMO disease or relapse. [78,79] AQP4 is the major water-transporting channel in the CNS.[79–81] AQP4 is found primarily on astrocytes and in addition to water transport it facilitates other key cellular functions.[82] The NMO-IgGs bind extracellular domains of AQP4 and induce astrocyte degeneration through complement-dependent cytotoxicity. Interestingly NMO-IgG binding to AQP4 does not alter the water permeability of the channel.[83] It was further demonstrated that AQP4-reactive antibodies appear in the pathologic lesions, [84]and that levels of AQP4 antibody and disease activity appear correlated.[85] NMO is associated with other autoimmune disorders. [86] In addition, auto-antibodies against other common autoimmune diseases, such as Sjögren’s syndrome and systemic lupus erythematosis, appear in the serum of NMO patients [87] but not in the serum of MS patients.[88]
Recently it has been suggested that inhibiting NMO-IgG/AQP4 binding could serve as a therapy for NMO. With this in mind, Verkman and coworkers have developed both small molecules [89] and antibodies to block these interactions. For the antibody-based approach, IgGs (anti-AQP4) were engineered to have high affinity for AQP4. Because the anti-AQP4 antibodies are much larger than AQP4, their binding sterically hinders interactions of pathogenic NMO-IgGs with AQP4. [18](Fig. 4a) SPR was used to characterize binding of the engineered anti-AQP4 antibodies to AQP4. Proteoliposomes containing purified recombinant M1 AQP4 were immobilized on a liposome capture SPR sensor chips. Concentrations of one of the anti-AQP4 antibodies (rAb-53) ranging from 130 nM to 2.2 μM were analyzed with SPR and an equilibrium dissociation constant (KD) of 27 nM was determined. (Fig. 4b) SPR analyses of other engineered anti-AQP4 antibodies were carried out and showed that these antibodies had lower affinities than rAb-53, though one other antibody (rAb-93) appears to have relatively high affinity as well. (Fig. 4c) Fluorescence studies of rAb-53 binding to U87MG cells were also done to assess affinity and show that there is very little washout of antibody over the course of 3 hours unlike other antibodies. rAb-53 is pathogenic in its native state and incubation of cells with rAb-53 resulted in cell death. To eliminate pathogenicity, point mutations were made to the Fc region of rAb-53 to eliminate either complement-dependent cytotoxicity (CDC) (K322A), antibody-dependent cell-mediated cytotoxicity (ADCC) (K326W/E333S) or both types of cytotoxicity (L234A/L235A). Subsequent SPR experiments showed the L234A/L235A mutation of rAb-53 did not significantly alter binding affinity of rAb-53 for AQP4 in proteoliposomes. [18] SPR curves for mutated rAb-53 (L234A/L235A) binding to AQP4 proteoliposomes are shown in Fig. 4d. Most importantly, the mutated forms of rAb-53 greatly reduced cytotoxicity, blocked NMO-IgG binding, prevented cell death due to NMO-IgG, and reduced NMO lesions in a mouse model of NMO.
Figure 4.

High-affinity monoclonal, recombinant anti–aquaporin-4 (AQP4) antibody (rAb) for antibody blocking therapy. (a) Crystal structure of AQP4 tetramer is shown on the same scale with that of an IgG antibody. (b) SPR measurements of recombinant antibody rAb-53 binding to AQP4-reconstituted proteoliposomes. Antibody rAb-53 ranged in concentration from 0.13 to 2.2 μM. (c) SPR measurements of additional recombinant antibodies binding to AQP4 in proteoliposomes. (d) SPR measurements of rAb-53 with the L243A/L235A mutations binding to AQP4 proteoliposomes. These mutations eliminated complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity, but did not significantly alter the binding properties. Adapted from Tradtrantip et al., [18] and used with permission of John Wiley and Sons.
Characterization of β-Amyloid Peptides and Treatments for Alzheimer’s disease
AD is the most prevalent form of dementia, affecting an estimated 5.2 million people in the United States of America in 2013, according to the Alzheimer’s Association. The initial symptom is short-term memory loss, followed by long-term memory loss and reduction of other important cognitive capabilities, eventually leaving patients bedridden and in need of custodial care, ultimately leading to death on average within 9 years of diagnosis. There is currently no treatment that can reverse the pathology of AD, only temporarily improve symptoms (acetylcholinesterase inhibitors), and detection only occurs after permanent damage has occurred. The two histological markers of AD are proteinaceous deposits of extracellular amyloid plaques, and intraneuronal tau neurofibrillary tangles (NFTs). The amyloid plaques are insoluble fibrils of beta-amyloid peptides (Aβ). Aβ peptides originate from the neuronal transmembrane amyloid precursor protein (APP) upon cleavage by secretase enzymes. Two of the peptide lengths associated with AD are 40 and 42 peptides long, Aβ(1-40) and Aβ(1-42), with Aβ(1-42) being the more toxic form. Under certain conditions these peptides lose their native conformation and begin clumping first as soluble aggregates and then into insoluble beta sheets. A cascade of events follow, including an immune response, an increase in free radicals, oxidative stress, and insoluble clumping of hyperphosphorylated tau protein (NFTs). Eventually synapses and neurotransmitters malfunction, leading to neuronal death. [90,91]
One of the leading hypotheses of the causative agents of AD has been the “Amyloid Cascade” hypothesis, which postulates that the primary cause of AD is accumulation of Aβ peptide. [92] One therapeutic strategy has been to target the insoluble fibrils for removal. For this reason, understanding the aggregation mechanism, such as the rate of fibril formation and environmental conditions affecting this rate, has been investigated, and SPR has been a useful tool for this purpose. Other methods had been used to study Aβ polymerization, however these techniques gave information on the hour to week timescale, whereas SPR allows measurement of the polymerization rate on the seconds to minutes timescale. In addition, SPR is well suited towards these experiments as the solution above the chip is continuously replenished. For these experiments, the receptor and ligand pair are monomers and polymers of the same subunit. Following initial studies by Myszka et al.[93]and Hasegawa et al. [94], Cannon et al. [95]immobilized monomers or fibrils of Aβ (1-40) by EDC/NHS-coupling to CM5 chips, and then exposed these to soluble Aβ (1-40) monomers. They found soluble monomers only bound to the fibrils, and did so in a 3 step “dock-and-lock” growth mechanism – a reversible first step (dock), followed by 2 slower steps (lock). Stravalaci et al.[96]extended this technique to monitor the polymerization rates of Aβ (1-42), a more difficult molecule to immobilize on the surface, due to its clumping behavior. They used a novel chemical alteration that had been demonstrated to resist clumping. Other groups have studied the effect of variations in environmental conditions, such as metal ions in solution, on the rate of fibril growth. Hu et al.[97] found the rate of polymerization of Aβ (1-40) to increase with metal ions, Ca2+, Fe2+, Fe3+, and Zn2+, and detected de-polymerization when flowing in a solution with the metal ion chelator, EDTA. Ryu et al. [98] conducted similar experiments with Aβ (1-42) and found nearly 6 times increase in polymerization in the presence of Fe3+.
SPR has also been useful to measure the binding affinities of small molecules and antibodies to amyloid peptides and fibrils. One drug strategy is to disrupt amyloid plaques by binding a molecule more strongly to an amyloid fibril than its constituent subunits, disrupting its structure and further growth. Cairo et al. used SPR to measure the affinity of several such small molecules which bound to plaques and reduced toxicity in cell toxicity assays. [99] They found that the small molecules with the highest affinities (~40 nM) correlated with the greatest reduction in toxicity. Other potential drugs that have been explored and studied with SPR have been retro-inverso-peptide which inhibits Aβ oligomer formation [100], liposomes with curcumin [101], and sulidac sulfide, a modulator of gamma-secretase one of the enzymes that cleaves APP to make Aβ peptide.[102]
Evidence has increasingly pointed towards soluble Aβ oligomers as the causative agents of the amyloid cascade. Therefore many groups have looked specifically for oligomer-targeting molecules, to avoid affecting monomers and dimers which may have natural functions. One drug approach is to target these oligomers with passive immunization for subsequent clearance by the immune system. Lindhagen-Persson et al.[103] hypothesized that a monoclonal IgM, with 10 identical binding sites targeting a subunit of the oligomers, would bind more strongly to an oligomer, due to avidity, than to monomers and dimers. They generated a monoclonal anti-Aβ IgM, OMAB, and immobilized it on a CM5 chip, and then flowed monomers and oligomers of Aβ(1-40) and Aβ(1-42) over them (Figure 5). They found a much slower dissociation rate of oligomers over monomers and dimers, with a t1/2 of days versus minutes, indicating oligomer specificity.
Figure 5.

SPR characterization of binding of a monoclonal IgM, OMAB, to Aβ peptide monomers and oligomers. OMAB was immobilized on the chip followed by injections of (A) Aβ(1-40) monomers or (B) oligomers, or (C) Aβ(1-42) monomers or (D) oligomers. The dissociation curves show that monomers do not stay bound to the IgM very long (t1/2 = ~1 min), whereas the oligomers stay bound to the IgM for much longer (t1/2 = days, or longer), indicating preference for the toxic oligomers. Thus, these IgMs may help capture oligomers for immune system processing, or be used as a capture molecule in a detection scheme. Figure is from Lindhagen-Persson et al. [103]
In another study, Ramakrishnan et al. used many of the capabilities of SPR to evaluate many different anti-Aβ peptide antibodies that had been developed for staining and clinical purposes. They performed epitope mapping, a common usage of SPR, to determine which regions of the Aβ peptide IgG4.1 bound to. By immobilizing the antibodies and injecting fragments of the Aβ peptide, they determined the binding region to be the N-teminal region from peptides 2 to 10. They also immobilized Aβ(1-40) monomers or fibrils and injected various antibodies (6E10, 11A50, IgG4.1, F(ab′)24.1, pIgG4.1, and pF(ab′)24.1) over them, finding all to have affinities in the nanomolar range.[104]
SPR Detection of Alzheimer’s Disease Biomarkers
SPR devices can be used to detect biomarkers in bodily fluids. Other techniques have been in use to detect AD biomarkers, such as polyacrylamide gel electrophoresis (PAGE), immunoprecipitation, mass spectrometry, fluorescent staining, and enzyme-linked immunosorbent assays (ELISA), with ELISA being the best in terms of sensitivity, selectivity, and versatility. However ELISAs require expensive enzyme kits, the use of a carcinogenic substrate molecule for detection, and 1–2 days of processing time. SPR devices do not have any of these constraints and have additional benefits such as low sample consumption and reusable chips.[105]
No chemical diagnosis for AD is currently available. However, the discovery that small soluble Aβ oligomers known as amyloid-derived diffusible ligands (ADDLs) cause memory deficits, [106,107] are associated with synaptic dysfunction, and are found in cerebrospinal fluid (CSF) suggests that if a sensitive assay for ADDLs was developed it could lead to a diagnostic laboratory test of AD. Haes and coworkers used a sandwich assay based on SPR to detect ADDLs from human brain and CSF with a limit of detection less than 100 fM.[108] To carry out the assay anti-ADDL antibodies were first linked to gold nanostructures [109] using EDC coupling chemistry. Then the antibody-functionalized sensors were incubated with ADDL ranging in concentration from 10 nM to 1 fM. After ADDL bound to the immobilized antibody, the second anti-ADDL antibody was introduced to further enhance the signal. (Fig. 6a) Samples with 100 fM and greater concentrations of ADDL had a SPR signal that was easily distinguishable from baseline noise. In extract from postmortem AD positive brains, ADDL was found to average at 1 pM concentration, whereas ADDL was not detected in samples from control brain extracts. In experiments with CSF they found an obvious difference in SPR signal when CSF from an aging control patient was exposed to the sensor vs. a patient showing clinical signs of AD. Although the samples were only from single patients, the magnitude of the difference between the SPR signals (a nearly 5-fold difference, Fig. 6b, c) suggests that the SPR sandwich assay is promising as a laboratory diagnostic, though reports have found that ADDL-like molecules are absent from the CSF of some AD patients.[110] This type of ADDL assays are currently being commercialized by Acumen Pharmaceuticals, Inc.
Figure 6.
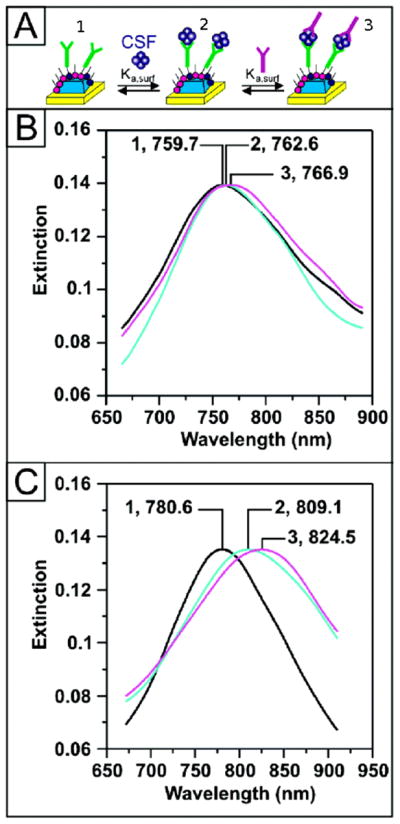
Detection of Alzheimer’s disease biomarker ADDL in human CSF samples using a sandwich assay and SPR. The SPR signal is monitored as peaks in the extinction (i.e. absorption) spectrum of the SPR sensor and molecular binding is indicated by a red-shift in the spectrum. (A) Illustration showing (1) anti-ADDL immobilization on the SPR sensor, (2) capture of ADDL, and (3) addition of second anti-ADDL to amplify changes is SPR signal. (B) Sensor response to CSF of an aging control patient: After functionalization with (B-1) 100 nM anti-ADDL (λmax = 759.7 nm), (B-2) CSF (λmax= 762.6 nm), and (B-3) 100 nM anti-ADDL (λmax = 766.9 nm). (C) Sensor response to CSF from an AD patient: After functionalization with (C-1) 100 nM anti-ADDL (λmax = 780.6 nm), (C-2) CSF (λmax = 809.1 nm), and (C-3) 100 nM anti-ADDL (λmax = 824.5 nm). Figure adapted from Haes et al.[108] and used with permission of the American Chemical Society.
In another approach, Stravalaci et al {Stravalaci 2012} used SPR to elucidate an antibody which could selectively bind toxic Aβ oligomers, to use as an AD biomarker detector. They investigated monoclonal antibody 4G8, which binds to Aβ peptide residues 17-21. Similar to IgM studied by Lindhagen-Persson et al, antibody 4G8 bound to both monomers and oligomers of Aβ peptide, but the monomers dissociated quickly whereas oligomers dissociated very slowly (with an approximate KD of <1 nM for oligomers and 75 nM for monomers). In their SPR experiments they immobilized 4G8 on the surface of a Bio-Rad GLC sensor chip and injected solutions of depsi-Aβ(1-42) which had been allowed to aggregate for different periods of time. The strongest binding aggregates had been incubated for 5 hr, and had a heterogeneous population of monomers, globular proteins, and short protofibrils, with hydrodynamic radii of 10–30 nm and likely masses of 90–400 kDa. For comparison, they demonstrated that anti-Aβ antibody 6E10 bound to Aβ(1-42) monomers and oligomers but could not differentiate the two since both had low dissociation rates. In addition, they demonstrated SPR could monitor the effect of compounds on oligomer formation. They incubated epigallocatechin gallate (EGCG), a compound shown to reduce fibrillogenesis, with Aβ(1-42) peptide for 5 hr, and upon injecting over 4G8, did not detect any of the slow-dissociating species, indicating EGCG had prevented oligomers from forming.
SPR Detection of Multiple Sclerosis Biomarkers (Should this section go after the section on remyelination antibodies?)
Earlier in this review, SPR characterization of remyelinating antibodies was discussed. In addition, SPR is also being applied to the sensitive and selective detection of biomarkers for MS. For example, Real-Fernandez and coworkers used SPR to detect glycopeptide binding autoantibodies in serum of MS patients.[111] This work focused on detection of serum antibodies that bound to CSF114(Glc), a synthetic glycopeptide that was developed by screening peptide libraries for their ability to recognize serum antibodies present in MS patients.[112] The structure of CSF114(Glc) may mimic a type of N-glucosylation that is commonly recognized by autoantibodies present in MS.[113] Previously, CSF114(Glc) had been used in ELISA assays to detect serum antibodies as biomarkers for MS.[112] For the SPR assay, the CSF114(Glc) peptide was linked to the gold surface of a commercially available SPR chip via EDC/NHS coupling chemistry. Serum from MS and healthy control patients was injected onto the SPR chip which was mounted in a Biacore T100 SPR instrument. When diluted serum was analyzed, the SPR sensor gave statistically significantly larger responses for MS patient serum compared to the serum of healthy control patients. This indicates a significantly higher concentration of CSF114(Glc)-binding antibodies in MS patient serum. For MS patients the SPR response averaged 94.6 response units (RU), whereas the healthy controls resulted in an average of 48.9 RU. Using ROC analysis with a 105 RU cutoff, the SPR assay had a sensitivity of 36%, a specificity of 95% and an area under the curve of 0.82, making the SPR assay more sensitive than comparable ELISA assays with CSF114(Glc).[112] Compared to the analogous ELISA experiments, the SPR assay for CSF114(Glc)-binding antibodies offers significant advantages. For example, the sensor coating procedure for SPR requires only 2 hours and only 1 μg of CSF114(Glc) is required to run 100 assays. On the other hand, for the ELISA assay the substrate is incubated overnight to coat with CSF114(Glc) and 1 μg of CSF114(Glc) is required for each assay. After surface coating the SPR assay only requires 10 minutes to run vs. one day for the ELISA assay. Therefore, SPR will be a powerful tool to develop sensitive biomarkers to detect demyelination and possibly to monitor remyelination promoting therapies.
Expert Commentary and Five Year View
Over the past three decades SPR has proven to be a very useful technique when characterizing interactions between potential drugs and their receptors. Over this time SPR evolved through new engineering approaches to increase device sensitivity, throughput and applicability to new problems. As biomedical science becomes increasingly interdisciplinary, collaborations between biological and physical scientists will open up new avenues of biological sensing based on SPR. In particular, there are many opportunities at the intersection of nanoscience and neuroscience. For SPR sensing, there can be certain advantages to using metallic nanostructures. For example, a recent paper by Stevens and coworkers has shown that using the optical properties of metallic nanoparticles and enzyme-mediated metal deposition, it is possible to detect biomolecules in an “inverse” sensing mode where lower concentrations of analytes result in larger changes in signals.[114] This work focused on detection of prostate cancer biomarkers, but it could be applicable to detection of any number of biomarkers for neurodegenerative diseases, or detection of drugs or drug metabolites in biological fluids. Also, with new surface preparation schemes and sensor configurations it possible to better mimic cellular membranes on SPR sensors. This will be crucial moving forward as a number of potential treatments for neurodegenerative disorders target molecules on the surface of CNS cells. Some potential therapeutics target molecules found in lipid rafts, [115] such as the recombinant human antibody rHIgM12 which has shown the ability to promote neurite extension [116] and improve locomotor activity in a mouse model of MS.[117] Other similar human antibodies, such as rHIgM22, are already being tested in MS patients for its ability to promote CNS remyelination (ClinicalTrials.gov Identifier NCT01803867) You do not need to repeat this twice – if put it here then delete the other. Therefore functionalization of SPR sensors with biomembranes that mimic lipid raft compositions will be highly beneficial to determining the affinity and mechanism of action of these antibodies. Beyond cellular membrane mimics, functionalization of SPR sensors with actual cellular membranes will provide a means to characterize binding interactions in the cases where the identity of the specific receptor(s) for drug molecules is elusive. In the neurodegenerative arena there is a great need to develop biomarkers with precise specificity and sensitivity to diagnose and differentiate forms of dementia. SPR maybe a tool to discover these biomarkers since the proteins accumulating in neurons in these disorders are already known. The biomarkers may themselves provide clues to developing novel therapies. In the next five years there should be a major advance in this area similar to the advance that occurred in the last five years in human demyelinating diseases where NMO and MS were distinguished based on antibody reactivity to AQP4. This was not only an academic exercise since the treatments for these two disorders are markedly different such that making the wrong diagnosis and starting the wrong therapy results in deleterious consequences.
Key Issues.
Surface plasmon resonance (SPR) is a label-free optical sensing technique to characterize binding between a soluble ligand and a surface-immobilized receptor.
Commercial SPR instruments employ a gold film illuminated at an angle to generate surface plasmons (SP). The resonance condition for SP excitation and subsequent angle of light reflection is very sensitive to the refractive index at the gold-water interface.
Molecular binding at the gold interface causes in an increase in refractive index, which results in a shift in resonance angle and a change in the SPR signal. Monitoring this change as a function of time allows the calculation of association and dissociation rate constants, which in turn can be used to determine affinity for the binding interaction.
IgM antibodies that bind oligodendrocytes can promote remyelination. Two of these antibodies, mouse IgM O1 and O4, bind lipids that have been incorporated into nanohole array-based SPR sensors. SPR experiments determined that O1 and O4 have much greater affinity for their antigens than do other similar IgMs. Similar human antibodies made in a recombinant form are already in clinical trials for promoting CNS remyelination.
Pathological IgGs against aquaporin-4 cause cytotoxicity associated with NMO. Small molecule and antibody inhibitors are being engineering to prevent pathological IgG binding. Characterization of antibody inhibitors by SPR and complimentary methods has identified an antibody with high-affinity for aquaporin-4 that blocks binding of pathological IgGs.
Amyloid-β fibrilization associated with AD can be characterized by SPR.
SPR can be used for sensitive, specific detection of biomarkers for neurodegenerative diseases like MS and AD.
SPR has the potential to differentiate dementias with specific protein accumulation in neurons (i.e. amyloid-β, tau, alpha–synuclein, TDP-43, FTDP-17 etc).
Acknowledgments
This work was supported by grants from the NIH (R01 GM092993, R01 NS048357 and R21 NS073684), an NSF, (CAREER DBI 1054191) and the National Multiple Sclerosis Society (CA 1060A). This work was also supported by a High-Impact Pilot and Feasibility Award (HIPFA) and Novel Methodology Award (NMDA) from the Mayo Clinic Center for Translational Science Activities (CTSA) and Mayo Clinic CTSA grant number UL1 TR000135 from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH). We also acknowledge with thanks support from the Applebaum, Hilton, Peterson and Sanford Foundations, the Minnesota Partnership Award for Biotechnology and Medical Genomics, the Moon and Marilyn Park Directorship Fund and the McNeilus family.
References
- 1.Arvanitakis Z, Grodstein F, Bienias JL, et al. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology. 2008;70(23):2219–2225. doi: 10.1212/01.wnl.0000313813.48505.86. [DOI] [PubMed] [Google Scholar]
- 2.Driver JA, Logroscino G, Lu L, Gaziano JM, Kurth T. Use of non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease: nested case-control study. BMJ. 2011;342:d198. doi: 10.1136/bmj.d198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin BK, Szekely C, Brandt J, et al. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65(7):896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Consortium WTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 8.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 9.Cooper M. Optical biosensors in drug discovery. Nat Rev Drug Discov. 2002;1(7):515–528. doi: 10.1038/nrd838. [DOI] [PubMed] [Google Scholar]
- 10.Homola J, Yee SS, Gauglitz G. Surface plasmon resonance sensors: review. Sensor Actuat B-Chem. 1999;54(1–2):3–15. [Google Scholar]
- 11.Maynard JA, Lindquist NC, Sutherland JN, et al. Surface plasmon resonance for high-throughput ligand screening of membrane-bound proteins. Biotechnol J. 2009;4(11):1542–1558. doi: 10.1002/biot.200900195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rich RL, Cannon MJ, Jenkins J, et al. Extracting kinetic rate constants from surface plasmon resonance array systems. Anal Biochem. 2008;373(1):112–120. doi: 10.1016/j.ab.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 13.Copeland R, Pompliano D, Meek T. Opinion - Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5(9):730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 14.Nunez S, Venhorst J, Kruse C. Target-drug interactions: first principles and their application to drug discovery. Drug Discov Today. 2012;17(1–2):10–22. doi: 10.1016/j.drudis.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 15.Navratilova I, Dioszegi M, Myszka DG. Analyzing ligand and small molecule binding activity of solubilized GPCRs using biosensor technology. Anal Biochem. 2006;355(1):132–139. doi: 10.1016/j.ab.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 16.Bieri C, Ernst OP, Heyse S, Hofmann KP, Vogel H. Micropatterned immobilization of a G protein-coupled receptor and direct detection of G protein activation. Nat Biotechnol. 1999;17(11):1105–1108. doi: 10.1038/15090. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt EK, Liebermann T, Kreiter M, et al. Incorporation of the acetylcholine receptor dimer from Torpedo californica in a peptide supported lipid membrane investigated by surface plasmon and fluorescence spectroscopy. Biosens Bioelectron. 1998;13(6):585–591. doi: 10.1016/s0956-5663(98)00013-x. [DOI] [PubMed] [Google Scholar]
- 18.Tradtrantip L, Zhang H, Saadoun S, et al. Anti-Aquaporin-4 Monoclonal Antibody Blocker Therapy for Neuromyelitis Optica. Ann Neurol. 2012;71(3):314–322. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rich RL, Errey J, Marshall F, Myszka DG. Biacore analysis with stabilized G-protein-coupled receptors. Anal Biochem. 2011;409(2):267–272. doi: 10.1016/j.ab.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rich RL, Myszka DG. Advances in surface plasmon resonance biosensor analysis. Curr Opin Biotechnol. 2000;11(1):54–61. doi: 10.1016/s0958-1669(99)00054-3. [DOI] [PubMed] [Google Scholar]
- 21.Myszka DG. Analysis of small-molecule interactions using Biacore S51 technology. Anal Biochem. 2004;329(2):316–323. doi: 10.1016/j.ab.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 22.Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr Opin Biotechnol. 1997;8(1):50–57. doi: 10.1016/s0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- 23.Myszka DG. Improving biosensor analysis. J Mol Recognit. 1999;12(5):279–284. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Rich RL, Myszka DG. Survey of the 2009 commercial optical biosensor literature. J Mol Recognit. 2011;24(6):892–914. doi: 10.1002/jmr.1138. [DOI] [PubMed] [Google Scholar]
- 25.Navratilova I, Papalia GA, Rich RL, et al. Thermodynamic benchmark study using Biacore technology. Anal Biochem. 2007;364(1):67–77. doi: 10.1016/j.ab.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 26.Liedberg B, Nylander C, Lundstrom I. Surface-plasmon resonance for gas detection and biosensing. Sensor Actuator. 1983;4(2):299–304. [Google Scholar]
- 27.Samanta D, Sarkar A. Immobilization of bio-macromolecules on self-assembled monolayers: methods and sensor applications. Chem Soc Rev. 2011;40(5):2567–2592. doi: 10.1039/c0cs00056f. [DOI] [PubMed] [Google Scholar]
- 28.Johnsson B, Lofas S, Lindquist G. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal Biochem. 1991;198(2):268–277. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- 29.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem Rev. 2005;105(4):1103–1169. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 30.Im H, Sutherland JN, Maynard JA, Oh SH. Nanohole-based surface plasmon resonance instruments with improved spectral resolution quantify a broad range of antibody-ligand binding kinetics. Anal Chem. 2012;84(4):1941–1947. doi: 10.1021/ac300070t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooper MA. Label-free screening of bio-molecular interactions. Anal Bioanal Chem. 2003;377(5):834–842. doi: 10.1007/s00216-003-2111-y. [DOI] [PubMed] [Google Scholar]
- 32.Yang N, Su X, Tjong V, Knoll W. Evaluation of two- and three-dimensional streptavidin binding platforms for surface plasmon resonance spectroscopy studies of DNA hybridization and protein-DNA binding. Biosens Bioelectron. 2007;22(11):2700–2706. doi: 10.1016/j.bios.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Drews J. Drug discovery: A historical perspective. Science. 2000;287(5460):1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 34.Cooper M, Hansson A, Lofas S, Williams D. A vesicle capture sensor chip for kinetic analysis of interactions with membrane-bound receptors. Anal Biochem. 2000;277(2):196–205. doi: 10.1006/abio.1999.4389. [DOI] [PubMed] [Google Scholar]
- 35.Erb E, Chen X, Allen S, et al. Characterization of the surfaces generated by liposome binding to the modified dextran matrix of a surface plasmon resonance sensor chip. Anal Biochem. 2000;280(1):29–35. doi: 10.1006/abio.1999.4469. [DOI] [PubMed] [Google Scholar]
- 36.Anderluh G, Besenicar M, Kladnik A, Lakey J, Macek P. Properties of nonfused liposomes immobilized on an L1 Biacore chip and their permeabilization by a eukaryotic pore-forming toxin. Anal Biochem. 2005;344(1):43–52. doi: 10.1016/j.ab.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Keller C, Kasemo B. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys J. 1998;75:1397–1402. doi: 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson TH, Min Y, Weirich KL, Zeng H, Fygenson D, Israelachvili JN. Formation of Supported Bilayers on Silica Substrates. Langmuir. 2009;25(12):6997–7005. doi: 10.1021/la900181c. [DOI] [PubMed] [Google Scholar]
- 39.Salafsky J, Groves JT, Boxer SG. Architecture and function of membrane proteins in planar supported bilayers: A study with photosynthetic reaction centers. Biochemistry. 1996;35(47):14773–14781. doi: 10.1021/bi961432i. [DOI] [PubMed] [Google Scholar]
- 40.Im H, Wittenberg N, Lesuffleur A, Lindquist N, Oh S. Membrane protein biosensing with plasmonic nanopore arrays and pore-spanning lipid membranes. Chem Sci. 2010;1:688–696. doi: 10.1039/C0SC00365D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castellana ET, Cremer PS. Solid supported lipid bilayers: From biophysical studies to sensor design. Surf Sci Rep. 2006;61(10):429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurtzke JF, Page WF, Murphy FM, Norman JE. Epidemiology of multiple sclerosis in US veterans 4 Age at onset. Neuroepidemiology. 1992;11(4–6):226–235. doi: 10.1159/000110935. [DOI] [PubMed] [Google Scholar]
- 43.Orton SM, Herrera BM, Yee IM, et al. Sex ratio of multiple sclerosis in Canada: a longitudinal study. Lancet Neurol. 2006;5(11):932–936. doi: 10.1016/S1474-4422(06)70581-6. [DOI] [PubMed] [Google Scholar]
- 44.Ramagopalan SV, Byrnes JK, Orton SM, et al. Sex ratio of multiple sclerosis and clinical phenotype. Eur J Neurol. 2010;17(4):634–637. doi: 10.1111/j.1468-1331.2009.02850.x. [DOI] [PubMed] [Google Scholar]
- 45.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Medical progress: Multiple sclerosis. New Engl J Med. 2000;343(13):938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 46.Bartt RE. Multiple sclerosis, natalizumab therapy, and progressive multifocal leukoencephalopathy. Curr Opin Neurol. 2006;19(4):341–349. doi: 10.1097/01.wco.0000236612.66839.a2. [DOI] [PubMed] [Google Scholar]
- 47.Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 48.Ramos-Cejudo J, Oreja-Guevara C, Stark Aroeira L, Rodriguez de Antonio L, Chamorro B, Diez-Tejedor E. Treatment with natalizumab in relapsing-remitting multiple sclerosis patients induces changes in inflammatory mechanism. J Clin Immunol. 2011;31(4):623–631. doi: 10.1007/s10875-011-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rice GP, Hartung HP, Calabresi PA. Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology. 2005;64(8):1336–1342. doi: 10.1212/01.WNL.0000158329.30470.D0. [DOI] [PubMed] [Google Scholar]
- 50.Blakemore WF, Chari DM, Gilson JM, Crang AJ. Modelling large areas of demyelination in the rat reveals the potential and possible limitations of transplanted glial cells for remyelination in the CNS. Glia. 2002;38(2):155–168. doi: 10.1002/glia.10067. [DOI] [PubMed] [Google Scholar]
- 51.Chari DM, Crang AJ, Blakemore WF. Decline in rate of colonization of oligodendrocyte progenitor cell (OPC)-depleted tissue by adult OPCs with age. J Neuropathol Exp Neurol. 2003;62(9):908–916. doi: 10.1093/jnen/62.9.908. [DOI] [PubMed] [Google Scholar]
- 52.Shields SA, Gilson JM, Blakemore WF, Franklin RJ. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia. 1999;28(1):77–83. doi: 10.1002/(sici)1098-1136(199910)28:1<77::aid-glia9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 53.Wood PM, Bunge RP. Myelination of cultured dorsal root ganglion neurons by oligodendrocytes obtained from adult rats. J Neurol Sci. 1986;74(2–3):153–169. doi: 10.1016/0022-510x(86)90101-2. [DOI] [PubMed] [Google Scholar]
- 54.Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9(11):839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez M, Lennon VA, Benveniste EN, Merrill JE. Remyelination by oligodendrocytes stimulated by antiserum to spinal cord. J Neuropathol Exp Neurol. 1987;46(1):84–95. doi: 10.1097/00005072-198701000-00008. [DOI] [PubMed] [Google Scholar]
- 56.Howe CL, Bieber AJ, Warrington AE, Pease LR, Rodriguez M. Antiapoptotic signaling by a remyelination-promoting human antimyelin antibody. Neurobiol Dis. 2004;15(1):120–131. doi: 10.1016/j.nbd.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 57.Miller DJ, Rodriguez M. A monoclonal autoantibody that promotes central nervous system remyelination in a model of multiple sclerosis is a natural autoantibody encoded by germline immunoglobulin genes. J Immunol. 1995;154(5):2460–2469. [PubMed] [Google Scholar]
- 58.Warrington AE, Asakura K, Bieber AJ, et al. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci USA. 2000;97(12):6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitsunaga Y, Ciric B, Van Keulen V, et al. Direct evidence that a human antibody derived from patient serum can promote myelin repair in a mouse model of chronic-progressive demyelinating disease. FASEB J. 2002;16(10):1325–1327. doi: 10.1096/fj.01-0994fje. [DOI] [PubMed] [Google Scholar]
- 60.Warrington A, Bieber A, Ciric B, Pease L, Van Keulen V, Rodriguez M. A recombinant human IgM promotes myelin repair after a single, very low dose. J Neurosci Res. 2007;85(5):967–976. doi: 10.1002/jnr.21217. [DOI] [PubMed] [Google Scholar]
- 61.Asakura K, Miller D, Pogulis R, Pease L, Rodriguez M. Oligodendrocyte-reactive 01, 04, and HNK-1 monoclonal antibodies are encoded by germline immunoglobulin genes. Mol Brain Res. 1995;34(2):283–293. doi: 10.1016/0169-328x(95)00190-4. [DOI] [PubMed] [Google Scholar]
- 62.Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces - an immunocytological study in the central nervous system. Dev Biol. 1981;83(2):311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- 63.Asakura K, Miller D, Pease L, Rodriguez M. Targeting of IgM kappa antibodies to oligodendrocytes promotes CNS remyelination. J Neurosci. 1998;18(19):7700–7708. doi: 10.1523/JNEUROSCI.18-19-07700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soldan M, Warrington A, Bieber A, et al. Remyelination-promoting antibodies activate distinct Ca2+ influx pathways in astrocytes and oligodendrocytes: redlationship to the mechanism of myelin repair. Mol Cell Neurosci. 2003;22(1):14–24. doi: 10.1016/s1044-7431(02)00018-0. [DOI] [PubMed] [Google Scholar]
- 65.Bansal R, Gard A, Pfeiffer S. Stimulation of oligodendrocyte differentiation in culture by growth in the presence of a monoclonal antibody to sulfated glycolipid. J Neurosci Res. 1988;21(2–4):260–267. doi: 10.1002/jnr.490210218. [DOI] [PubMed] [Google Scholar]
- 66.Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE. Multiple and novel specificities of monoclonal antibodies O1, O4 and R-MAb used in the analysis of oligodendrocyte development. J Neurosci Res. 1989;24(4):548–557. doi: 10.1002/jnr.490240413. [DOI] [PubMed] [Google Scholar]
- 67.Wittenberg NJ, Im H, Xu X, et al. High-Affinity Binding of Remyelinating Natural Autoantibodies to Myelin-Mimicking Lipid Bilayers Revealed by Nanohole Surface Plasmon Resonance. Anal Chem. 2012;84(14):6031–6039. doi: 10.1021/ac300819a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lutz H, Binder C, Kaveri S. Naturally occurring auto-antibodies in homeostasis and disease. Trends Immunol. 2009;30(1):43–51. doi: 10.1016/j.it.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 69.Notkins AL. Polyreactivity of antibody molecules. Trends Immunol. 2004;25(4):174–179. doi: 10.1016/j.it.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 70.Zhou Z, Tzioufas A, Notkins A. Properties and function of polyreactive antibodies and polyreactive antigen-binding B cells. J Autoimmun. 2007;29(4):219–228. doi: 10.1016/j.jaut.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Min Y, Kristiansen K, Boggs JM, Husted C, Zasadzinski JA, Israelachvili J. Interaction forces and adhesion of supported myelin lipid bilayers modulated by myelin basic protein. Proc Natl Acad Sci USA. 2009;106(9):3154–3159. doi: 10.1073/pnas.0813110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mi S, Miller RH, Lee X, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8(6):745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- 73.Mi S, Miller RH, Tang W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65(3):304–315. doi: 10.1002/ana.21581. [DOI] [PubMed] [Google Scholar]
- 74.Mi S, Hu B, Hahm K, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13(10):1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- 75.Mosyak L, Wood A, Dwyer B, et al. The structure of the Lingo-1 ectodomain, a module implicated in central nervous system repair inhibition. J Biol Chem. 2006;281(47):36378–36390. doi: 10.1074/jbc.M607314200. [DOI] [PubMed] [Google Scholar]
- 76.Costanzi C, Matiello M, Lucchinetti CF, et al. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77(7):659–666. doi: 10.1212/WNL.0b013e31822a2780. [DOI] [PubMed] [Google Scholar]
- 77.Magaña SM, Keegan BM, Weinshenker BG, et al. Beneficial plasma exchange response in central nervous system inflammatory demyelination. Arch Neurol. 2011;68(7):870–878. doi: 10.1001/archneurol.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69(24):2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 79.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 81.Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6(7):383–392. doi: 10.1038/nrneurol.2010.72. [DOI] [PubMed] [Google Scholar]
- 82.Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14(4):265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman AS. Neuromyelitis optica IgG does not alter aquaporin-4 water permeability, plasma membrane M1/M23 isoform content, or supramolecular assembly. Glia. 2013;60:2027–2039. doi: 10.1002/glia.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125(Pt 7):1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130(Pt 5):1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- 86.Pittock SJ, Lennon VA, de Seze J, et al. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol. 2008;65(1):78–83. doi: 10.1001/archneurol.2007.17. [DOI] [PubMed] [Google Scholar]
- 87.Wingerchuk DM. Evidence for humoral autoimmunity in neuromyelitis optica. Neurol Res. 2006;28(3):348–353. doi: 10.1179/016164106X98260. [DOI] [PubMed] [Google Scholar]
- 88.Wynn DR, Rodriguez M, O’Fallon WM, Kurland LT. A reappraisal of the epidemiology of multiple sclerosis in Olmsted County, Minnesota. Neurology. 1990;40(5):780–786. doi: 10.1212/wnl.40.5.780. [DOI] [PubMed] [Google Scholar]
- 89.Tradtrantip L, Zhang H, Anderson MO, et al. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J. 2012;26(5):2197–2208. doi: 10.1096/fj.11-201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9(5):387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 91.Tayeb HO, Yang HD, Price BH, Tarazi FI. Pharmacotherapies for Alzheimer’s disease: beyond cholinesterase inhibitors. Pharmacol Ther. 2012;134(1):8–25. doi: 10.1016/j.pharmthera.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 92.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 93.Myszka DG, Wood SJ, Biere AL. Analysis of fibril elongation using surface plasmon resonance biosensors. Meth Enzymol. 1999;309:386–402. doi: 10.1016/s0076-6879(99)09027-8. [DOI] [PubMed] [Google Scholar]
- 94.Hasegawa K, Ono K, Yamada M, Naiki H. Kinetic modeling and determination of reaction constants of Alzheimer’s beta-amyloid fibril extension and dissociation using surface plasmon resonance. Biochemistry. 2002;41(46):13489–13498. doi: 10.1021/bi020369w. [DOI] [PubMed] [Google Scholar]
- 95.Cannon MJ, Williams AD, Wetzel R, Myszka DG. Kinetic analysis of beta-amyloid fibril elongation. Anal Biochem. 2004;328(1):67–75. doi: 10.1016/j.ab.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 96.Stravalaci M, Beeg M, Salmona M, Gobbi M. Use of surface plasmon resonance to study the elongation kinetics and the binding properties of the highly amyloidogenic Aβ(1-42) peptide, synthesized by depsi-peptide technique. Biosens Bioelectron. 2011;26(5):2772–2775. doi: 10.1016/j.bios.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 97.Hu WP, Chang GL, Chen SJ, Kuo YM. Kinetic analysis of beta-amyloid peptide aggregation induced by metal ions based on surface plasmon resonance biosensing. J Neurosci Meth. 2006;154(1–2):190–197. doi: 10.1016/j.jneumeth.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 98.Ryu J, Joung HA, Kim MG, Park CB. Surface plasmon resonance analysis of Alzheimer’s beta-amyloid aggregation on a solid surface: From monomers to fully-grown fibrils. Anal Chem. 2008;80(7):2400–2407. doi: 10.1021/ac7019514. [DOI] [PubMed] [Google Scholar]
- 99.Cairo CW, Strzelec A, Murphy RM, Kiessling LL. Affinity-based inhibition of beta-amyloid toxicity. Biochemistry. 2002;41(27):8620–8629. doi: 10.1021/bi0156254. [DOI] [PubMed] [Google Scholar]
- 100.Taylor M, Moore S, Mayes J, et al. Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochemistry. 2010;49(15):3261–3272. doi: 10.1021/bi100144m. [DOI] [PubMed] [Google Scholar]
- 101.Mourtas S, Canovi M, Zona C, et al. Curcumin-decorated nanoliposomes with very high affinity for amyloid-β1-42 peptide. Biomaterials. 2011;32(6):1635–1645. doi: 10.1016/j.biomaterials.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 102.Richter L, Munter LM, Ness J, et al. Amyloid beta 42 peptide (Abeta42)-lowering compounds directly bind to Abeta and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc Natl Acad Sci USA. 2010;107(33):14597–14602. doi: 10.1073/pnas.1003026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lindhagen-Persson M, Brännström K, Vestling M, Steinitz M, Olofsson A. Amyloid-β oligomer specificity mediated by the IgM isotype--implications for a specific protective mechanism exerted by endogenous auto-antibodies. PLoS One. 2010;5(11):e13928. doi: 10.1371/journal.pone.0013928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ramakrishnan M, Kandimalla KK, Wengenack TM, Howell KG, Poduslo JF. Surface Plasmon Resonance Binding Kinetics of Alzheimer’s Disease Amyloid beta Peptide-Capturing and Plaque-Binding Monoclonal Antibodies. Biochemistry. 2009;48(43):10405–10415. doi: 10.1021/bi900523q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu L, Xia N, Wang JX. Potential applications of SPR in early diagnosis and progression of Alzheimer’s disease. RSC Advances. 2012;2(6):2200–2204. [Google Scholar]
- 106.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from A beta(1-42) are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 108.Haes A, Chang L, Klein W, Van Duyne R. Detection of a biomarker for Alzheimer’s disease from synthetic and clinical samples using a nanoscale optical biosensor. J Am Chem Soc. 2005;127(7):2264–2271. doi: 10.1021/ja044087q. [DOI] [PubMed] [Google Scholar]
- 109.Haes A, Van Duyne R. A nanoscale optical blosensor: Sensitivity and selectivity of an approach based on the localized surface plasmon resonance spectroscopy of triangular silver nanoparticles. J Am Chem Soc. 2002;124(35):10596–10604. doi: 10.1021/ja020393x. [DOI] [PubMed] [Google Scholar]
- 110.Esparza TJ, Zhao H, Cirrito JR, et al. Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73(1):104–119. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Real-Fernandez F, Passalacqua I, Peroni E, et al. Glycopeptide-Based Antibody Detection in Multiple Sclerosis by Surface Plasmon Resonance. Sensors. 2012;12(5):5596–5607. doi: 10.3390/s120505596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lolli F, Mazzanti B, Pazzagli M, et al. The glycopeptide CSF114(Glc) detects serum antibodies in multiple sclerosis. J Neuroimmunol. 2005;167(1–2):131–137. doi: 10.1016/j.jneuroim.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 113.Carotenuto A, Alcaro MC, Saviello MR, et al. Designed glycopeptides with different beta-turn types as synthetic probes for the detection of autoantibodies as biomarkers of multiple sclerosis. J Med Chem. 2008;51(17):5304–5309. doi: 10.1021/jm800391y. [DOI] [PubMed] [Google Scholar]
- 114.Rodriguez-Lorenzo L, de la Rica R, Alvarez-Puebla RA, Liz-Marzan LM, Stevens MM. Plasmonic nanosensors with inverse sensitivity by means of enzyme-guided crystal growth. Nat Mater. 2012;11(7):604–607. doi: 10.1038/nmat3337. [DOI] [PubMed] [Google Scholar]
- 115.Xu X, Warrington AE, Wright BR, et al. A human IgM signals axon outgrowth: coupling lipid raft to microtubules. J Neurochem. 2011;119(1):100–112. doi: 10.1111/j.1471-4159.2011.07416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Warrington AE, Bieber AJ, Van Keulen V, Ciric B, Pease LR, Rodriguez M. Neuron-binding human monoclonal antibodies support central nervous system neurite extension. J Neuropathol Exp Neurol. 2004;63(5):461–473. doi: 10.1093/jnen/63.5.461. [DOI] [PubMed] [Google Scholar]
- 117.Denic A, Macura S, Warrington A, et al. A Single Dose of Neuron–Binding Human Monoclonal Antibody Improves Spontaneous Activity in a Murine Model of Demyelination. Plos One. 2011;6(10) doi: 10.1371/journal.pone.0026001. [DOI] [PMC free article] [PubMed] [Google Scholar]