

Abstract
All-trans retinoic acid (RA) and interferons (IFNs) have efficacy in treating certain leukemias and lymphomas, respectively, motivating interest in their mechanism of action to improve therapy. Both RA and IFNs induce interferon regulatory factor-1 (IRF-1). We find that in HL-60 myeloblastic leukemia cells which undergo mitogen activated protien kinase (MAPK)-dependent myeloid differentiation in response to RA, IRF-1 propels differentiation. RA induces MAPK-dependent expression of IRF-1. IRF-1 binds c-Cbl, a MAPK related adaptor. Ectopic IRF-1 expression causes CD38 expression and activation of the Raf/MEK/ERK axis, and enhances RA-induced differentiation by augmenting CD38, CD11b, respiratory burst and G0 arrest. Ectopic IRF-1 expression also decreases the activity of aldehyde dehydrogenase 1, a stem cell marker, and enhances RA-induced ALDH1 down-regulation. Interestingly, expression of aryl hydrocarbon receptor (AhR), which is RA-induced and known to down-regulate Oct4 and drive RA-induced differentiation, also enhances IRF-1 expression. The data are consistent with a model whereby IRF-1 acts downstream of RA and AhR to enhance Raf/MEK/ERK activation and propel differentiation.
Keywords: IRF-1, c-Cbl, RA, differentiation, neutrophil, HL-60
Introduction
Interferon regulatory factor-1 (IRF-1), encoded by chromosome 5, a 10 exon gene, is an important regulator of leukocyte differentiation [1]. Deletions of chromosome 5 or parts thereof that contain the IRF-1 gene are the most frequent cytogenetic markers in preleukemic myelodysplastic syndromes (MDS) and subsequent leukemias [2,3]. In MDS, IRF-1 mRNA frequently lacks exons 2 and 3, resulting in an IRF-1 protein without tumor-suppressor activity [4]. In addition to tumor suppression, IRF-1 is also reported to function in differentiation. IRF-1 is up-regulated during differentiation in a murine embryonal carcinoma cell model [5]. IRF-1 protein levels are low in bone marrow stem cells (CD34+), and IRF-1 knockout mice present a maturation block and accumulation of immature granulocytic precursors [6]. Expression of IRF-1 is induced during normal hematopoiesis and during differentiation induction therapy with retinoids and interferons – two important classes of chemotherapeutic agents for leukemias and lymphomas, respectively. It has been shown that IRF-1 induction by interferon γ (IFN γ) requires Mnk1/2 kinases [7], which are activated by ERK1/2 [8].
The standard therapeutic modality for acute promyelocytic leukemia uses retinoic acid (RA). RA induces differentiation, and this action is also mitogen activated protein kinase (MAPK)-dependent [9]. Crosstalk between RA and interferons/IRF-1 pathways, which have been previously reported, may thus involve MAPK signaling pathways. The mechanistic details, however, remain to be clarified. RA and IFN γ driven pathways may have various points of convergence. It has been shown that RA induces IRF-1 gene expression by binding to its GAS (gamma interferon activation site) sequence in many systems, including the HL-60 cell model [10 – 12]. Multiple downstream molecules have been shown to be involved in both RA and IRF pathways. These include Stat1, Stat2, p48, IFN α [13 – 22], cyclin B1 [23], CDKi (cyclin-dependent kinase inhibitor) p21 [24], PML/ RAR α (promyelocytic leukemia/retinoic acid receptor α; reviewed in [25]), TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) [26,27] and others, as well as MAPK. IFNs can also up-regulate retinoid receptors. RA in turn up-regulates IFN α and JAK – STAT (Janus kinase – signal transducer and activator of transcription) pathway members (reviewed in [25,26,28 – 35]). The RA and IRF-1 pathways may thus have a variety of interdependencies which may have the potential to be exploited for combination therapy.
In this study, RA-induced IRF-1 expression, and augmented IRF-1 expression enhanced RA-induced differentiation. We confirmed that RA-induced IRF-1 expression correlated with myeloid differentiation of HL-60 human myeloblastic leukemia cells, a French – American – British (FAB) M1 derived cell model that is a bipotent stem-like cell. IRF-1 stable transfectants were created to investigate how IRF-1 regulates MAPK signaling proteins and determine the role of IRF-1 in RA-induced differentiation. Overexpression of IRF-1 caused increased CD38 expression, enhanced Raf/MEK/ERK activation and promoted RA-induced differentiation and cell cycle arrest. Interestingly, RA-induced IRF-1 protein expression is itself MAPK-dependent. Ectopic IRF-1 may have enhanced RA-induced differentiation by various means. RA-induced differentiation is propelled by MAPK signaling, and CD38 expression has been previously shown to cause enhanced MAPK signaling. IRF-1-induced CD38 may thus have contributed enhanced MAPK signaling to propel differentiation. IRF-1 also bound to c-Cbl, an adaptor that promotes CD38 MAPK signaling. c-Cbl is a versatile adaptor protein that we previously reported to be essential for RA-induced differentiation of HL-60 cells [36]. This may have facilitated the prolonged MAPK signaling needed to propel differentiation and G0 arrest in RA-treated cells. The data also suggest the possibility that, in addition to its well-studied function as a transcription factor, IRF-1 may regulate MAPK signaling through interaction with a signaling complex containing c-Cbl. Aryl hydrocarbon receptor (AhR) up-regulation, either by overexpression or by valproic acid, caused IRF-1 up-regulation. We have reported that RA induces AhR expression and AhR propels RA-induced differentiation [37]. IRF-1 is thus downstream of AhR, and ectopic IRF-1 could potentially thus short-circuit this AhR function to facilitate differentiation. We also reported that RA down-regulates ALDH1 activity consistent with loss of stemness as the cells differentiate, and we find here that IRF-1 expression causes down-regulation of aldehyde dehydrogenase 1 (ALDH1). This too may promote RA-induced differentiation. In sum we show that RA-induced IRF-1 can be related to c-Cbl and AhR, which both propel differentiation, and that they may be key intersection points of retinoic acid and interferon pathways.
Materials and methods
Chemicals
Retinoic acid (Sigma, St. Louis, MO) was dissolved in 100% ethanol with a stock concentration of 5 mM, and used at a final concentration of 1 μM. 1,25-Dihydroxy vitamin D3 (Cayman, Ann Arbor, MI) was used at a final concentration of 0.5 μM from a stock of 1 mM in ethanol. The oligonucleotide primers were synthesized by Integrated DNA Technologies (San Diego, CA).
Cell culture
Human myeloblastic leukemia cells (HL-60) were grown in a humidified atmosphere of 5% CO2 at 37 ° C and maintained in RPMI 1640 supplemented with 5% fetal bovine serum (Invitrogen, Carlsbad, CA). The cells were cultured in constant exponential growth as previously described [37,38]. The experimental cultures were initiated at a cell density of 0.1 × 106 cells/mL.
Reverse transcriptase-PCR and plasmid construction
Total human IRF-1 RNA was isolated from HL-60 cells using the Qiagen RNeasy minikit (Qiagen, Valencia, CA), and the first-strand cDNA was synthesized according to the protocol of the SuperScript first-strand synthesis system (Invitrogen, Carlsbad, CA). For IRF-1 with FLAG tag plasmid (IRF+) construction, DNA fragments carrying complete human IRF-1 gene were generated by polymerase chain reaction (PCR) amplification using oligonucleotides (forward 5′ GCG AAT TCG GAT CCA TGC CCA TCA CTC GGA TGC 3′, reverse 5′ GCG CGG CCG CCG GTG CAC AGG GAA TGG CCT 3′) and cloned into a pIRES-hrGFP II vector using BamH1 and Not1 sites. All inserts were confirmed by sequencing.
IRF-1 stable transfection
Fifty micrograms of plasmid DNA were transfected into HL-60 cells as previously described [39], and selected with G418 (2 mg/mL) for 2 – 3 weeks. Stable transfectants underwent three cycles of cell sorting and amplification to select cells expressing high enhanced green fluorescent protein (EGFP) using fluorescence activated cell sorting (FACS Aria flow cytometer; BD Biosciences, San Jose, CA). Western blots probed with anti-IRF-1 antibody (BD Biosciences) were used to confirm IRF-1 expression. HL-60 wild-type and vector control cells showed no differences in differentiation and behaved indistinguishably in all experiments reported.
Immunoprecipitation and Western blot
A total of 2.5 × 107 cells were lysed using 200 μL lysis buffer (Pierce, Rockford, IL), and lysates were cleared by centrifugation at 16 950 × g for 20 min at 4 ° C. Equal amounts of protein lysates (25 μg) were resolved by sodium dodecyl sulfate-polyacrylamide gel elctrophoresis (SDS-PAGE), transferred to nitrocellulose membranes and probed with antibodies. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-actin and histone H3 antibodies (Cell Signaling, Beverly, MA) were used to check uniform loading. Immunoprecipitation was done starting with 300 μg protein.
CD11b, CD38 expression studies by flow cytometry
HL-60 cells (0.5 × 106) were harvested by centrifugation at 120 × g for 5 min. Cells were resuspended in 200 μL phosphate buffered saline (PBS) containing 2.5 μL of allophycocyanin (APC) conjugated anti-CD11b antibody or APC conjugated anti-CD38 antibody (BD Biosciences). Following incubation for 1 h at 37 ° C, cells were analyzed by flow cytometry (LSRII flow cytometer; BD Biosciences) using 633 nm red laser excitations. The threshold to determine the increase of percentage positive expression was set to exclude 95% of control cells [37].
Measurement of inducible oxidative metabolism
A total of 0.5 × 106 cells were harvested by centrifugation and resuspended in 200 μL 37 ° C PBS containing 10 μM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate acetyl ester (H2-DCFDA; Molecular Probes, Eugene, OR) and 0.4 μg/mL 12-o-tetradecanoylphorbol-13-acetate (PMA; Sigma) with incubation for 20 min in a humidified atmosphere of 5% CO2 at 37 ° C. Dimethylsulfoxide (DMSO; Sigma) was used as carrier. Flow cytometric analysis was done (BD LSRII flow cytometer) using a 488 nm excitation laser and emission collected through a 505 longpass dichroic and 530/30 nm bandpass filter. The shift in fluorescence intensity in response to PMA was used to determine the percentage of cells with the capability to generate inducible superoxide. Gates to determine percentage positive cells were set to exclude 95% of control cells. Control cells with or without PMA and RA-treated cells without PMA typically showed indistinguishable DCF fluorescence histograms [37].
Cell cycle analysis
A total of 0.5 × 106 cells were collected by centrifugation and resuspended in 200 μL of refrigerated hypotonic staining solution containing 50 μg/mL propidium iodine (PI), 1 μL/mL Triton-X and 1 mg/mL sodium citrate. Cells were stored, protected from light, at room temperature for 1 h and analyzed by flow cytometry (BD LSRII flow cytometer) using 488 nm excitation and collection through a 550 longpass dichroic and a 576/26 bandpass [37].
Aldehyde dehydrogenase enzymatic activity assay
ALDH1 enzymatic activity was measured using the Aldefluor kit (Stem Cell Technologies, Durham, NC) as described [37,40].
Statistics
Three independent repeats were conducted in all experiments. Error bars represent standard error of the mean of three repeats. Two-tailed paired Student’s t-test was used to asses the statistical significance.
Results
RA up-regulated IRF-1 expression
To determine the effect of RA on IRF-1 expression, HL-60 human myeloblastic leukemia cells were treated with RA and harvested after 48 h to measure IRF-1 expression by Western blotting. IRF-1 expression was up-regulated by RA (Figure 1). Expression progressively increased over time (from 24 to 72 h) (data not shown). To investigate the functional significance of IRF-1 up-regulation, in particular how IRF-1 regulates MAPK signaling and differentiation in RA-treated cells, stable transfectants ectopically overexpressing IRF-1 with a FLAG tag (IRF+) were created. Western blotting showed that the stable transfectants (IRF+) expressed IRF-1 (Figure 1). The IRF + pooled transfectants had a much stronger IRF-1 protein band than the minimal signal detectable in control parental cells. RA treatment further increased IRF-1 expression in the IRF + stable transfectants (Figure 1). Thus, while wild-type cells express minimal IRF-1, RA clearly induces expression, and levels in untreated transfectants exceed this, with RA treatment causing a further significant increase.
Figure 1.
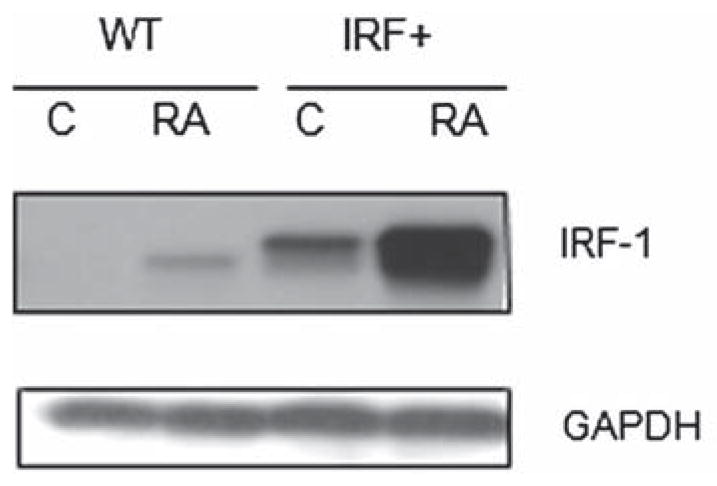
Interferon regulatory factor-1 (IRF-1) expression in wild-type HL-60 and IRF-1 stable transfectant cell lines. Western blot of IRF-1 expression in wild-type and IRF-1 stably transfected (IRF +) HL-60 cells, 48 h. IRF-1 protein expression level was higher in IRF-1 stable transfectants than in parental wild-type cells. RA induced IRF-1 expression in wild-type and even more in IRF-1 overexpressors. RA, C treated or untreated control; RA, 1 μM retinoic acid, 48 h.
IRF-1 enhanced CD38 expression
We could confirm that the transfected IRF-1 was functionally active. The most prominent activity of IRF-1 is its ability to activate transcription from specific promoters. It has been reported that the CD38 promoter contains potential IRF binding sites, and IRF-1 is capable of up-regulating CD38 mRNA and protein levels in leukemic B cells [41,42]. The CD38 ectoenzyme receptor is an early cell surface marker for phenotypic conversion during RA-induced differentiation. We compared CD38 expression in IRF-1 transfectants and parental HL-60. Ectopic IRF-1 expression induced low-level CD38 expression. Flow cytometry showed increased expression from 5% in HL-60 wild-type parental cells to 18% in IRF-1 +cells (p-value <0.05), indicating that the expression of CD38 per cell was significantly higher in IRF-1 transfectants [Figure 2(A)]. IRF-1 expression thus up-regulated CD38 expression. RA treatment further enhanced CD38 expression. In RA-treated cells, CD38 expression was significantly higher in IRF + cells compared to wild-type parental cells [Figure 2(A)]. The stable transfectants thus contain a functionally active IRF-1 that induces expression of an early cell surface differentiation marker, CD38.
Figure 2.

IRF-1 transfectants underwent enhanced cell differentiation. (A) In untreated cells, the expression level of CD38 was significantly higher in IRF-1 stable transfectants (IRF+) than in HL-60 control (WT). In RA-treated cells, IRF-1 overexpressors propelled CD38 expression levels (flow cytometric assay of live cells was carried out setting the logical gate to exclude 95% of untreated cells using APC conjugated anti-CD38 antibody). (B) In RA-treated cells, the expression level of CD11b was higher in IRF-1 stable transfectants than in HL-60 control. (C) In D3-treated cells, the CD11b level was lower in IRF-1 overexpressors compared to wild-type cells (flow cytometric assay of live cells was carried out setting the logical gate to exclude 95% of untreated cells with APC conjugated anti-CD11b antibody). (D) IRF-1 enhanced RA-induced respiratory burst. The percentage of cells capable of PMA inducible oxidative metabolism was analyzed by flow cytometry. The threshold to determine percentage positive cells was set to exclude 95% of control cells. PMA stimulated inducible oxidative metabolism. DMSO, carrier control blank. (E) IRF-1 accelerated G0 arrest induced by RA treatment. Nuclei stained with hypotonic PI staining solution were analyzed by flow cytometry. (F) Growth curves during duration of experiment were constructed by counting cells. Different letters represent significantly different values (p ≤ 0.05). Results are mean ± SEM for at least three repeats.
IRF-1 promoted RA-induced cell differentiation
CD38 expression is known to cause MAPK signaling and propel RA-induced differentiation and G0 cell cycle arrest, motivating the anticipation that IRF-1 expression might facilitate RA-induced differentiation compared to parental cells. The ability of IRF-1 transfectants (IRF+) to differentiate in response to RA was measured using a cell surface marker and a functional differentiation marker. First, CD11b, an α integrin, was used to measure cell differentiation using APC conjugated CD11b antibody by flow cytometry. We compared the percentage of cells expressing CD11b in HL-60 cells and IRF1 + stable transfectants treated with RA for 24, 48 and 72 h. The increase was progressive, and the 72 h (late differentiation) data are presented in Figure 2(B). In RA treatment, IRF-1 transfectants show significant (p ≤ 0.05) enhanced expression of CD11b compared to parental HL-60 cells.
In contrast to RA, vitamin D3 is known to induce monocytic differentiation, and IRF-1 expression impeded D3-induced CD11b expression [Figure 2(C)], although other aspects of differentiation, such as inducible oxidative metabolism and G0 arrest, were not significantly affected (data not shown), indicating potential lineage specificity of the IRF-1 effects seen for RA. The HL-60 cell line is a bipotent leukemic stem cell that undergoes granulocytic differentiation upon RA treatment and monocytic differentiation upon D3 treatment. For THP-1 and U937, IRF-1 was reported to facilitate differentiation toward monocytes [43,44]. However, it is noteworthy that promotion of monocytic differentiation by IRF-1 was found using mediators of inflammation, interleukin-6 (IL-6) and lipopolysaccharide (LPS), whereas in our case, vitamin D3 was used. Moreover, although IRF-1 is a regulator of both neutrophil and monocyte lineage differentiation, the effects involve a tightly correlated balance with other IRF-family members [45,46].
To confirm the role of IRF-1 in propulsion of RA-induced cell differentiation, inducible oxidative metabolism was used as a myeloid functional differentiation marker. The oxidation of the non-fluorescent H2-DCFDA to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) was used to detect the generation of reactive oxygen. Overexpression of IRF-1 accelerated RA-induced functional differentiation compared to parental HL-60 cells [Figure 2(D)]. The difference in DCF-positive percentage between IRF-1 transfectants and parental HL-60 cells was significant after RA treatment for 72 h (31% in HL-60 wild-type cells, 65% in IRF1+ transfectants, p-value < 0.01).
To determine the effects of IRF-1 on RA-induced G1/G0 cell cycle arrest, the percentage of IRF-1 transfectants and parental HL-60 in G1/G0 was measured using flow cytometry. IRF-1 overexpression enhanced G0 arrest upon RA treatment at 72 h [Figure 2(E)], with 75% for parental HL-60 cells and 82% for IRF1+, p-value < 0.001. This was consistent with the differentiation assays, indicating the role of IRF-1 in promoting RA-induced differentiation and cell cycle arrest. To confirm that the enhancement in differentiation and promotion of cell cycle arrest caused by IRF-1 reflected decreased cell growth, we compared IRF transfectants and parental HL-60 growth curves with or without RA [Figure 2(F)]. It is to be noted that the cell proliferation is not totally abrogated for this level of IRF-1 expression, consistent with the fact that not all the cells are in G0/G1 (but rather 82%) and with the fact that this assay cannot discriminate between G0 and G1. IRF-1 transfectants showed curtailed cell growth associated with G0 enrichment compared to parental cells. This indicates that ectopic expression of IRF-1 regulates CD38 expression and promotes RA-induced differentiation and loss of proliferative capability. A summary of the RA-induced differentiation data is presented in Table I.
Table I.
Summary of RA-induced differentiation data (% ± SEM).
| CD38 6 h | CD11b 72 h | Inducible respiratory metabolism 72 h | G0/G1 72 h | |
|---|---|---|---|---|
| C WT | 5.14 ± 0.5 | 5.30 ± 0.15 | 8.86 ± 0.89 | 60.73 ± 1.72 |
| RA WT | 26.14 ± 1.57 | 49.37 ± 3.00 | 31.03 ± 5.67 | 74.93 ± 0.98 |
| C IRF+ | 18.84 ± 6.78 | 5.52 ± 1.07 | 6.49 ± 0.65 | 62.83 ± 1.29 |
| RA IRF+ | 43.42 ± 4.37 | 68.83 ± 3.86 | 64.85 ± 2.48 | 82.31 ± 0.91 |
RA, retinoic acid; C, control; WT, wild-type; IRF+, interferon regulatory factor-1 with FLAG tag plasmid.
Parallel with the observed increase in differentiation we anticipated a decrease in blastic/stemness properties [40,47]. Indeed, ALDH1 enzymatic activity was down-regulated by 1 μM RA, by IRF-1 overexpression and even more by the combination of RA treatment with an IRF-1 elevated background (Figure 3). In sum, IRF-1 expression results in loss of stemness and enhanced RA-induced differentiation and G0 arrest.
Figure 3.
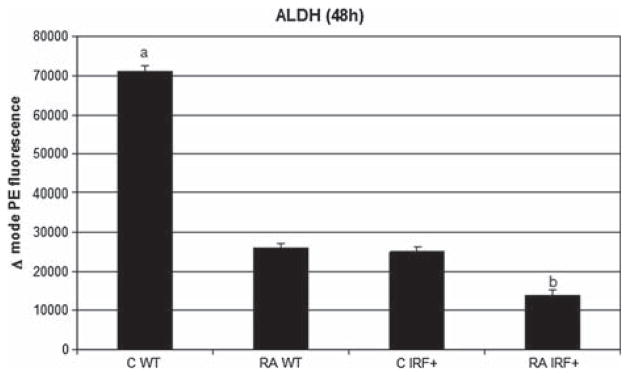
Aldehyde dehydrogenase 1 activity was significantly lower (p = 0.01) in cells treated with RA than in untreated cells. IRF-1 overexpression enhanced this effect. Results of flow cytometric analysis of live cells are expressed as difference in mode phycoerythrin (PE) fluorescence of test sample and control sample (diethylaminobenzaldehyde [DEAB] inhibited). a and b, statistically significant difference between them and compared with rest of samples (p < 0.005 for IFR + RA vs. IRF + C or WT RA and p < 0.001 for WT C vs. WT RA).
IRF-1 caused enhanced Raf/MEK/ERK activation
Raf/MEK/ERK activation is necessary to propel RA-induced differentiation and cell cycle arrest [9]. To investigate whether RA-induced myeloid differentiation and cell cycle arrest enhanced by IRF-1 were related to Raf/MEK/ERK activation, we compared Raf/MEK/ERK expression and phosphorylation in IRF-1 stable transfectants and parental HL-60 cells. IRF1 + transfectants showed increased levels of dual phosphorylated ERK [T(203) and EY(205)] (Figure 4), suggesting that ectopic expression of IRF-1 caused enhanced MAPK signaling and accelerated RA-induced differentiation and cell cycle arrest. RA-treated IRF + transfectants also had more pERK than parental HL-60. Likewise, phosphorylated MEK was enhanced. Raf activation is known to provide critical propulsion for RA-induced differentiation [48]. To investigate how IRF-1 affects Raf, phosphorylation of Raf [S(259) and S(621)] was compared between IRF-1 transfectants and parental HL-60 cells that were untreated or RA-treated. IRF-1 increased Raf kinase activation compared to HL-60 cells, suggesting that overexpression of IRF-1 causes enhanced Raf/MEK/ERK activation, resulting in propulsion of RA-induced cell differentiation and cell cycle arrest.
Figure 4.

IRF-1 caused enhanced Raf/MEK/ERK activation. Western blots of phospho-Raf S259 and -Raf S621, phospho-MEK and phospho-ERK1/2 in WT and IRF+, untreated vs. RA-treated cells for 48 h. GAPDH was used to check protein loading. C, untreated cells; RA, 1 μM RA treated cells for 48 h. Twenty-five micrograms of protein were loaded per well.
RA-induced IRF-1 is associated with c-Cbl and their expression is MEK-dependent
A question emerging from the present data is the link between the observed IRF-1 induction by RA and MAPK activation. c-Cbl is known to be necessary for RA-induced MAPK signaling downstream of CD38, and c-Cbl expression was found to drive RA-induced differentiation as part of a MAPK signaling complex [36,49]. After RA treatment, immunoprecipitation showed that c-Cbl and IRF-1 co-precipitated (Figure 5). This suggests there is a link from IRF-1 to a critical component of the MAPK signaling complex. Furthermore, RA-induced IRF-1 expression itself depends on MAPK signaling. By using PD 98059 pretreatment 4 h before RA treatment, we found that if MEK is inhibited, then IRF-1 is not expressed in RA treated cells. Moreover, the same occurred for c-Cbl, namely, PD 98059 abolishes c-Cbl protein expression. Figure 5 shows IRF-1 and c-Cbl expression in cells that were untreated control or treated with RA or RA plus PD 98059, and the amount of c-Cbl co-immunoprecipitating with IRF-1 under these conditions. Hence, IRF-1 was found to bind a critical adaptor of the MAPK signaling complex driving differentiation, and its RA-induced expression is also MAPK-dependent. The results suggest the possibility that IRF-1 participates in a feedback loop that incorporates CD38 and its downstream MAPK signaling.
Figure 5.

RA caused, in a MEK-dependent manner, enhanced IRF-1 and c-Cbl. IRF-1 and c-Cbl associated Western blots (48 h, first three lanes, and 72 h, last three lanes) of IRF, c-Cbl, c-Cbl immunoprecipitated with IRF-1, actin (lane loading control) and histone 3. Twenty-five micrograms of protein were loaded per well. For IP, 300 μg of protein were used. For treatment, RA final concentration was 1 μM and VPA was 1 mM. C, RA and PD/RA are control, RA-treated and RA plus PD 98059-treated. PD 98059 (1 μM from a 1 mM DMSO stock) treatment was done 4 h prior to RA treatment.
AhR expression induced IRF-1 in presence or absence of RA
While it is known that IRF-1 is downstream of RA as a transcriptionally regulated target, other upstream regulators remain to be elucidated. Of particular interest are relationships of IRF-1 to other regulators that propel RA-induced differentiation. Recently we have shown that AhR is up-regulated by RA and propels RA-induced myeloid differentiation. An emerging question is thus whether AhR up-regulation induces IRF-1 expression. Indeed, as shown in the Western blot in Figure 6, increased AhR expression by ectopic expression in previously described stable transfectants [37] up-regulates IRF-1 expression. Confirming this, up-regulating AhR expression by treating with valproic acid, as we reported previously [37], also up-regulates IRF-1 expression. Hence, IRF-1 expression is downstream of AhR, which is itself up-regulated by RA as well, and propels differentiation [37]. Ectopic IRF-1 overexpression may thus facilitate differentiation by short-circuiting an RA/AhR/ IRF-1 axis. This linkage of AhR and IRF-1 potentially provides a novel basis for possible future combination therapy strategies.
Figure 6.
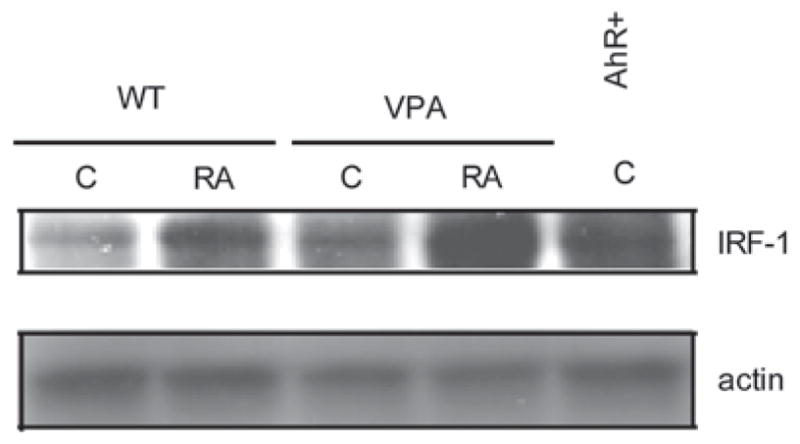
AhR expression induced IRF-1. AhR overexpressors and VPA-treated samples expressed an augmented amount of IRF-1 protein, as determined by Western blotting (shown at 48 h).
Discussion
The present results show that RA induces the expression of IRF-1, and expression of IRF-1 promotes RA-induced differentiation of HL-60 bipotent stem-like leukemic cells. The data implicate four potential mechanisms. (a) IRF-1 expression causes up-regulation of the CD38 receptor. CD38 is an ectoenzyme receptor that is the earliest known cell surface differentiation marker, and CD38 expression propels RAinduced differentiation [50]. IRF-1 may thus accelerate this process. (b) IRF-1 complexes with c-Cbl. RA induces activation of MAPK signaling, which is driven by CD38 using a c-Cbl containing MAPK signaling complex to activate Raf, a kinase known to propel RA-induced differentiation [48]. IRF-1 thus physically interacts with this complex, potentially facilitating the hyperactive MAPK signaling as a possible adaptor. (c) IRF-1 is up-regulated by AhR expression. AhR is up-regulated by RA. AhR expression causes down-regulation of Oct4, a Yamanaka – Thomson factor sustaining stemness, and drives RA-induced differentiation. IRF-1 overexpression may short-circuit this and thereby accelerate induced differentiation. (d) IRF-1 expression causes down-regulation of ALDH1 activity. ALDH1 activity is a trait of stem cells. RA induces ALDH1 activity down-regulation. IRF-1 may thus help propel this to facilitate differentiation.
The present results may have therapeutic implications for differentiation induction therapy by RA that involves IRF-1. RA is an embryonic morphogen specifying the crown to rump axis, and its gradient regulates Hox genes, which are master developmental regulators. It is also a stem cell regulator. In HL-60 leukemic blasts, RA elicits up-regulation of the CD38 membrane receptor. In turn, CD38 recruits a MAPK signaling complex containing c-Cbl and activates Raf to drive the transformation from a bipotent blast to a functional mature myeloid cell [36,48,49]. Downstream of this, IRF-1 is known to regulate myelopoiesis, and its expression is regulated by both RA and MAPK signaling [10 – 12], as well as by AhR. IRF-1 regulates MAPK signaling and binds c-Cbl. c-Cbl is an adaptor that regulates MAPK signaling, which in turn regulates both AhR and RA controlled pathways. A feedback network involving AhR, IRF-1 and MAPK in driving RA-induced myeloid differentiation is thus plausible, and here we present evidence in support of such a hypothesis that c-Cbl, AhR and IRF-1 cooperate in RA-induced myeloid differentiation. These present potential targets of intervention to improve the therapeutic outcome with RA in leukemia therapy. While acute promyelocytic leukemia (APL) has been susceptible to RA, other myeloid leukemias have not, and hence means of enhancing RA efficacy are of interest.
An interesting aspect of the present work is the implication of AhR involvement. Interestingly, two drugs of chemotherapeutic significance, namely arsenic trioxide and valproic acid, are AhR inducers. Arsenic trioxide is in clinical trials for remission maintenance in patients who reached remission by RA; it is a known AhR and IRF-1 inducer able to enhance and maintain RA-induced differentiation [51 – 58]. Arsenic trioxide is an AhR ligand, and hence another means of targeting AhR. It has already been exploited with some degree of success, which supports the idea of targeting AhR. It has been shown that arsenic trioxide induces Mnk [59], and also that IRF-1 induction is Mnk-dependent [7]. The present results contribute to a rationalization of how arsenic trioxide improves or supplants RA. Histone deacetylase inhibitors, such as valproic acid, are also known to modulate AhR expression and activity, and regulate stem cell differentiation [37,60 – 66]. We previously reported that valproic acid up-regulated AhR expression in the present cell system and facilitated RA-induced differentiation [37]. Here we report that valproic acid up-regulates IRF-1 expression, which facilitates differentiation. AhR-targeting agents are thus of potential interest as means of augmenting the IRF-1-dependent RA-induced differentiation.
Acknowledgments
This work was supported by grants from NIH (A.Y.), NYSTEM NY Department of Health (A.Y.) and Cornell Vertebrate Genomics (VERGE) (R.P.B.) NIH/PS-OC (Shuler/AY/RPB).
Footnotes
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article at www.informahealthcare.com/lal.
References
- 1.Yamada G, Ogawa M, Akagi K, et al. Specific depletion of the B-cell population induced by aberrant expression of human interferon regulatory factor 1 gene in transgenic mice. Proc Natl Acad Sci USA. 1991;88:532–536. doi: 10.1073/pnas.88.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tefferi A, Mathew P, Noel P. The 5q-syndrome: a scientific and clinical update. Leuk Lymphoma. 1994;14:375–378. doi: 10.3109/10428199409049692. [DOI] [PubMed] [Google Scholar]
- 3.Willman CL, Sever CE, Pallavicini MG, et al. Deletion of IRF-1, mapping to chromosome 5q31. 1, in human leukemia and preleukemic myelodysplasia. Science. 1993;259:968–971. doi: 10.1126/science.8438156. [DOI] [PubMed] [Google Scholar]
- 4.Harada H, Kondo T, Ogawa S, et al. Accelerated exon skipping of IRF-1 mRNA in human myelodysplasia/leukemia; a possible mechanism of tumor suppressor inactivation. Oncogene. 1994;9:3313–3320. [PubMed] [Google Scholar]
- 5.Harada H, Willison K, Sakakibara J, et al. Absence of the type I IFN system in EC cells: transcriptional activator (IRF-1) and repressor (IRF-2) genes are developmentally regulated. Cell. 1990;63:303–312. doi: 10.1016/0092-8674(90)90163-9. [DOI] [PubMed] [Google Scholar]
- 6.Testa U, Stellacci E, Pelosi E, et al. Impaired myelopoiesis in mice devoid of interferon regulatory factor 1. Leukemia. 2004;18:1864–1871. doi: 10.1038/sj.leu.2403472. [DOI] [PubMed] [Google Scholar]
- 7.Joshi S, Sharma B, Kaur S, et al. Essential role for Mnk kinases in type II interferon (IFNgamma) signaling and its suppressive effects on normal hematopoiesis. J Biol Chem. 2011;286:6017–6026. doi: 10.1074/jbc.M110.197921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waskiewicz AJ, Flynn A, Proud CG, et al. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yen A, Roberson MS, Varvayanis S, et al. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998;58:3163–3172. [PubMed] [Google Scholar]
- 10.Hsu CY, Yung BY. Over-expression of nucleophosmin/B23 decreases the susceptibility of human leukemia HL-60 cells to retinoic acid-induced differentiation and apoptosis. Int J Cancer. 2000;88:392–400. doi: 10.1002/1097-0215(20001101)88:3<392::aid-ijc11>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 11.Pelicano L, Li F, Schindler C, et al. Retinoic acid enhances the expression of interferon-induced proteins: evidence for multiple mechanisms of action. Oncogene. 1997;15:2349–2359. doi: 10.1038/sj.onc.1201410. [DOI] [PubMed] [Google Scholar]
- 12.Matikainen S, Ronni T, Hurme M, et al. Retinoic acid activates interferon regulatory factor-1 gene expression in myeloid cells. Blood. 1996;88:114–123. [PubMed] [Google Scholar]
- 13.Matikainen S, Lehtonen A, Sareneva T, et al. Regulation of IRF and STAT gene expression by retinoic acid. Leuk Lymphoma. 1998;30:63–71. doi: 10.3109/10428199809050930. [DOI] [PubMed] [Google Scholar]
- 14.Alsayed Y, Modi S, Uddin S, et al. All-trans-retinoic acid induces tyrosine phosphorylation of the CrkL adapter in acute promyelocytic leukemia cells. Exp Hematol. 2000;28:826–832. doi: 10.1016/s0301-472x(00)00170-3. [DOI] [PubMed] [Google Scholar]
- 15.Alsayed Y, Uddin S, Mahmud N, et al. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to all-trans-retinoic acid. J Biol Chem. 2001;276:4012–4019. doi: 10.1074/jbc.M007431200. [DOI] [PubMed] [Google Scholar]
- 16.Kambhampati S, Li Y, Verma A, et al. Activation of protein kinase C delta by all-trans-retinoic acid. J Biol Chem. 2003;278:32544–32551. doi: 10.1074/jbc.M301523200. [DOI] [PubMed] [Google Scholar]
- 17.Kambhampati S, Verma A, Li Y, et al. Signalling pathways activated by all-trans-retinoic acid in acute promyelocytic leukemia cells. Leuk Lymphoma. 2004;45:2175–2185. doi: 10.1080/10428190410001722053. [DOI] [PubMed] [Google Scholar]
- 18.Kannan-Thulasiraman P, Dolniak B, Kaur S, et al. Role of the translational repressor 4E-BP1 in the regulation of p21(Waf1/Cip1) expression by retinoids. Biochem Biophys Res Commun. 2008;368:983–989. doi: 10.1016/j.bbrc.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lal L, Li Y, Smith J, et al. Activation of the p70 S6 kinase by all-trans-retinoic acid in acute promyelocytic leukemia cells. Blood. 2005;105:1669–1677. doi: 10.1182/blood-2004-06-2078. [DOI] [PubMed] [Google Scholar]
- 20.Sassano A, Katsoulidis E, Antico G, et al. Suppressive effects of statins on acute promyelocytic leukemia cells. Cancer Res. 2007;67:4524–4532. doi: 10.1158/0008-5472.CAN-06-3686. [DOI] [PubMed] [Google Scholar]
- 21.Sassano A, Lo Iacono M, Antico G, et al. Regulation of leukemic cell differentiation and retinoid-induced gene expression by statins. Mol Cancer Ther. 2009;8:615–625. doi: 10.1158/1535-7163.MCT-08-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schinke C, Goel S, Bhagat TD, et al. Design and synthesis of novel derivatives of all-trans retinoic acid demonstrate the combined importance of acid moiety and conjugated double bonds in its binding to PML-RAR-alpha oncogene in acute promyelocytic leukemia. Leuk Lymphoma. 2010;51:1108–1114. doi: 10.3109/10428191003786766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vuletic A, Konjevic G, Milanovic D, et al. Antiproliferative effect of 13-cis-retinoic acid is associated with granulocyte differentiation and decrease in cyclin B1 and Bcl-2 protein levels in G0/G1 arrested HL-60 cells. Pathol Oncol Res. 2010;16:393–401. doi: 10.1007/s12253-009-9241-2. [DOI] [PubMed] [Google Scholar]
- 24.Giandomenico V, Vaccari G, Fiorucci G, et al. Apoptosis and growth inhibition of squamous carcinoma cells treated with interferon-alpha, IFN-beta and retinoic acid are associated with induction of the cyclin-dependent kinase inhibitor p21. Eur Cytokine Netw. 1998;9:619–631. [PubMed] [Google Scholar]
- 25.Gaboli M, Gandini D, Delva L, et al. Acute promyelocytic leukemia as a model for cross-talk between interferon and retinoic acid pathways: from molecular biology to clinical applications. Leuk Lymphoma. 1998;30:11–22. doi: 10.3109/10428199809050925. [DOI] [PubMed] [Google Scholar]
- 26.Chelbi-Alix MK, Pelicano L. Retinoic acid and interferon signaling cross talk in normal and RA-resistant APL cells. Leukemia. 1999;13:1167–1174. doi: 10.1038/sj.leu.2401469. [DOI] [PubMed] [Google Scholar]
- 27.Clarke N, Jimenez-Lara AM, Voltz E, et al. Tumor suppressor IRF-1 mediates retinoid and interferon anticancer signaling to death ligand TRAIL. EMBO J. 2004;23:3051–3060. doi: 10.1038/sj.emboj.7600302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garattini E, Mologni L, Ponzanelli I, et al. Cross-talk between retinoic acid and interferons: molecular mechanisms of interaction in acute promyelocytic leukemia cells. Leuk Lymphoma. 1998;30:467–475. doi: 10.3109/10428199809057559. [DOI] [PubMed] [Google Scholar]
- 29.Hou X, Zhou R, Wei H, et al. NKG2D-retinoic acid early inducible-1 recognition between natural killer cells and Kupffer cells in a novel murine natural killer cell-dependent fulminant hepatitis. Hepatology. 2009;49:940–949. doi: 10.1002/hep.22725. [DOI] [PubMed] [Google Scholar]
- 30.Kawaguchi S, Ishiguro Y, Imaizumi T, et al. Retinoic acidinducible gene-I is constitutively expressed and involved in IFNgamma-stimulated CXCL9-11 production in intestinal epithelial cells. Immunol Lett. 2009;123:9–13. doi: 10.1016/j.imlet.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Li K, Chen Z, Kato N, et al. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J Biol Chem. 2005;280:16739–16747. doi: 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- 32.Lou YJ, Pan XR, Jia PM, et al. IRF-9/STAT2 [corrected] functional interaction drives retinoic acid-induced gene G expression independently of STAT1. Cancer Res. 2009;69:3673–3680. doi: 10.1158/0008-5472.CAN-08-4922. [DOI] [PubMed] [Google Scholar]
- 33.Percario ZA, Giandomenico V, Fiorucci G, et al. Retinoic acid is able to induce interferon regulatory factor 1 in squamous carcinoma cells via a STAT-1 independent signalling pathway. Cell Growth Differ. 1999;10:263–270. [PubMed] [Google Scholar]
- 34.Vidal M, Ramana CV, Dusso AS. Stat1-vitamin D receptor interactions antagonize 1,25-dihydroxyvitamin D transcriptional activity and enhance stat1-mediated transcription. Mol Cell Biol. 2002;22:2777–2787. doi: 10.1128/MCB.22.8.2777-2787.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao S, Li D, Zhu HQ, et al. RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc Natl Acad Sci USA. 2006;103:16448–16453. doi: 10.1073/pnas.0607830103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen M, Yen A. c-Cbl interacts with CD38 and promotes retinoic acid-induced differentiation and G0 arrest of human myeloblastic leukemia cells. Cancer Res. 2008;68:8761–8769. doi: 10.1158/0008-5472.CAN-08-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bunaciu RP, Yen A. Activation of the aryl hydrocarbon receptor AhR promotes retinoic acid-induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res. 2011;71:2371–2380. doi: 10.1158/0008-5472.CAN-10-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brooks SC, 3rd, Kazmer S, Levin AA, et al. Myeloid differentiation and retinoblastoma phosphorylation changes in HL-60 cells induced by retinoic acid receptor-and retinoid X receptor-selective retinoic acid analogs. Blood. 1996;87:227–237. [PubMed] [Google Scholar]
- 39.Wightman J, Roberson MS, Lamkin TJ, et al. Retinoic acid-induced growth arrest and differentiation: retinoic acid up-regulates CD32 (Fc gammaRII) expression, the ectopic expression of which retards the cell cycle. Mol Cancer Ther. 2002;1:493–506. [PubMed] [Google Scholar]
- 40.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bauvois B, Durant L, Laboureau J, et al. Upregulation of CD38 gene expression in leukemic B cells by interferon types I and II. J Interferon Cytokine Res. 1999;19:1059–1066. doi: 10.1089/107999099313299. [DOI] [PubMed] [Google Scholar]
- 42.Tliba O, Damera G, Banerjee A, et al. Cytokines induce an early steroid resistance in airway smooth muscle cells: novel role of interferon regulatory factor-1. Am J Respir Cell Mol Biol. 2008;38:463–472. doi: 10.1165/rcmb.2007-0226OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manzella L, Conte E, Cocchiaro G, et al. Role of interferon regulatory factor 1 in monocyte/macrophage differentiation. Eur J Immunol. 1999;29:3009–3016. doi: 10.1002/(SICI)1521-4141(199909)29:09<3009::AID-IMMU3009>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 44.Sanceau J, Kaisho T, Hirano T, et al. Triggering of the human interleukin-6 gene by interferon-gamma and tumor necrosis factor-alpha in monocytic cells involves cooperation between interferon regulatory factor-1, NF kappa B, and Sp1 transcription factors. J Biol Chem. 1995;270:27920–27931. doi: 10.1074/jbc.270.46.27920. [DOI] [PubMed] [Google Scholar]
- 45.Tamura T, Yanai H, Savitsky D, et al. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 46.Battistini A. Interferon regulatory factors in hematopoietic cell differentiation and immune regulation. J Interferon Cytokine Res. 2009;29:765–780. doi: 10.1089/jir.2009.0030. [DOI] [PubMed] [Google Scholar]
- 47.Hess DA, Meyerrose TE, Wirthlin L, et al. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood. 2004;104:1648–1655. doi: 10.1182/blood-2004-02-0448. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Yen A. A MAPK-positive feedback mechanism for BLR1 signaling propels retinoic acid-triggered differentiation and cell cycle arrest. J Biol Chem. 2008;283:4375–4386. doi: 10.1074/jbc.M708471200. [DOI] [PubMed] [Google Scholar]
- 49.Shen M, Yen A. c-Cbl tyrosine kinase-binding domain mutant G306E abolishes the interaction of c-Cbl with CD38 and fails to promote retinoic acid-induced cell differentiation and G0 arrest. J Biol Chem. 2009;284:25664–25677. doi: 10.1074/jbc.M109.014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamkin TJ, Chin V, Varvayanis S, et al. Retinoic acid-induced CD38 expression in HL-60 myeloblastic leukemia cells regulates cell differentiation or viability depending on expression levels. J Cell Biochem. 2006;97:1328–1338. doi: 10.1002/jcb.20745. [DOI] [PubMed] [Google Scholar]
- 51.Wu JP, Chang LW, Yao HT, et al. Involvement of oxidative stress and activation of aryl hydrocarbon receptor in elevation of CYP1A1 expression and activity in lung cells and tissues by arsenic: an in vitro and in vivo study. Toxicol Sci. 2009;107:385–393. doi: 10.1093/toxsci/kfn239. [DOI] [PubMed] [Google Scholar]
- 52.Chao HR, Tsou TC, Li LA, et al. Arsenic inhibits induction of cytochrome P450 1A1 by 2,3,7,8 – tetrachlorodibenzo-p-dioxin in human hepatoma cells. J Hazard Mater. 2006;137:716–722. doi: 10.1016/j.jhazmat.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 53.Kann S, Huang MY, Estes C, et al. Arsenite-induced aryl hydrocarbon receptor nuclear translocation results in additive induction of phase I genes and synergistic induction of phase II genes. Mol Pharmacol. 2005;68:336–346. doi: 10.1124/mol.105.011841. [DOI] [PubMed] [Google Scholar]
- 54.Elbekai RH, El-Kadi AO. Modulation of aryl hydrocarbon receptor-regulated gene expression by arsenite, cadmium, and chromium. Toxicology. 2004;202:249–269. doi: 10.1016/j.tox.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 55.Maier A, Dalton TP, Puga A. Disruption of dioxin-inducible phase I and phase II gene expression patterns by cadmium, chromium, and arsenic. Mol Carcinog. 2000;28:225–235. [PubMed] [Google Scholar]
- 56.Hansen LA, Sigman CC, Andreola F, et al. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis. 2000;21:1271–1279. [PubMed] [Google Scholar]
- 57.Chelbi-Alix MK, Bobe P, Benoit G, et al. Arsenic enhances the activation of Stat1 by interferon gamma leading to synergistic expression of IRF-1. Oncogene. 2003;22:9121–9130. doi: 10.1038/sj.onc.1207090. [DOI] [PubMed] [Google Scholar]
- 58.Nayak S, Shen M, Bunaciu RP, et al. Arsenic trioxide cooperates with all trans retinoic acid to enhance mitogen-activated protein kinase activation and differentiation in PML-RARalpha negative human myeloblastic leukemia cells. Leuk Lymphoma. 2010;51:1734–1747. doi: 10.3109/10428194.2010.501535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dolniak B, Katsoulidis E, Carayol N, et al. Regulation of arsenic trioxide-induced cellular responses by Mnk1 and Mnk2. J Biol Chem. 2008;283:12034–12042. doi: 10.1074/jbc.M708816200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bailey DN, Briggs JR. Valproic acid binding to human serum and human placenta in vitro. Ther Drug Monit. 2005;27:375–377. doi: 10.1097/01.ftd.0000158077.07836.9a. [DOI] [PubMed] [Google Scholar]
- 61.Nau H, Zierer R, Spielmann H, et al. A new model for embryotoxicity testing: teratogenicity and pharmacokinetics of valproic acid following constant-rate administration in the mouse using human therapeutic drug and metabolite concentrations. Life Sci. 1981;29:2803–2814. doi: 10.1016/0024-3205(81)90541-5. [DOI] [PubMed] [Google Scholar]
- 62.Jung BH, Kim BJ, Lee MS, et al. Dose-dependent pharmacokinetics of toxic metabolites is not related to increased toxicity follow ing high-dose valproic acid in rats. J Appl Toxicol. 2011;30:775–778. doi: 10.1002/jat.1608. [DOI] [PubMed] [Google Scholar]
- 63.Padmanabhan R, Shafiullah M, Benedict S, et al. Effect of maternal exposure to homocystine on sodium valproate-induced neural tube defects in the mouse embryos. Eur J Nutr. 2006;45:311–319. doi: 10.1007/s00394-006-0600-4. [DOI] [PubMed] [Google Scholar]
- 64.Emmanouil-Nikoloussi EN, Foroglou NG, Kerameos-Foroglou CH, et al. Effect of valproic acid on fetal and maternal organs in the mouse: a morphological study. Morphologie. 2004;88:41–45. doi: 10.1016/s1286-0115(04)97999-4. [DOI] [PubMed] [Google Scholar]
- 65.Koch S, Jager-Roman E, Losche G, et al. Antiepileptic drug treatment in pregnancy: drug side effects in the neonate and neurological outcome. Acta Paediatr. 1996;85:739–746. doi: 10.1111/j.1651-2227.1996.tb14137.x. [DOI] [PubMed] [Google Scholar]
- 66.Nau H, Tzimas G, Mondry M, et al. Antiepileptic drugs alter endogenous retinoid concentrations: a possible mechanism of teratogenesis of anticonvulsant therapy. Life Sci. 1995;57:53–60. doi: 10.1016/0024-3205(95)00242-x. [DOI] [PubMed] [Google Scholar]