

Abstract
Miniature persistent late Na+ currents in cardiomyocytes have been linked to arrhythmias and sudden death. The goals of this study are to establish a stable cell line expressing robust persistent cardiac Na+ currents and to test Class 1 antiarrhythmic drugs for selective action against resting and open states. After transient transfection of an inactivation-deficient human cardiac Na+ channel clone (hNav1.5-CW with L409C/A410W double mutations), transfected mammalian HEK293 cells were treated with 1 mg/ml G-418. Individual G-418-resistant colonies were isolated using glass cylinders. One colony with high expression of persistent Na+ currents was subjected to a second colony selection. Cells from this colony remained stable in expressing robust peak Na+ currents of 996 ± 173 pA/pF at +50 mV (n = 20). Persistent late Na+ currents in these cells were clearly visible during a 4-second depolarizing pulse albeit decayed slowly. This slow decay is likely due to slow inactivation of Na+ channels and could be largely eliminated by 5 μM batrachotoxin. Peak cardiac hNav1.5-CW Na+ currents were blocked by tetrodotoxin with an IC50 value of 2.27 ± 0.08 μM (n = 6). At clinic relevant concentrations, Class 1 antiarrhythmics are much more selective in blocking persistent late Na+ currents than their peak counterparts, with a selectivity ratio ranging from 80.6 (flecainide) to 3 (disopyramide). We conclude that (1) Class 1 antiarrhythmics differ widely in their resting- vs. open-channel selectivity, and (2) stably transfected HEK293 cells expressing large persistent hNav1.5-CW Na+ currents are suitable for studying as well as screening potent open-channel blockers.
Keywords: hNav1.5, cardiac sodium channel, stable cell line, Class 1 antiarrhythmic agents, persistent late sodium current, local anesthetics, inactivation-deficient sodium channel, batrachotoxin, tetrodotoxin
Introduction
Voltage-gated Na+ channels are responsible for the generation of action potentials in excitable membranes. Upon depolarization neuronal Na+ channels open only once with a brief dwell time of ~0.5 ms.1 These open channels are fast inactivated through the action of an inactivation particle, which rapidly plugs the pore.2 However, it has become increasingly evident that various gain-of-function genetic diseases manifest small but persistent late Na+ currents (generally <2%) in cardiac myocytes, skeletal muscle and neurons during prolonged depolarization. Among these diseases are congenital long QT-3 syndromes, paramyotonia congenita, potassium-aggravated myotonia and familial primary erythermalgia.3-5 In addition, small persistent Na+ currents have also been found under pathological conditions such as failing heart6 and hypoxia.7 The discoveries of aberrant persistent late sodium currents have attracted much attention recently as a potential target for drug intervention. Therapeutic drugs that eliminate persistent late Na+ currents preferentially may be beneficial for these life-threatening events.
The human cardiac Na+ channel (hNav1.5) is a transmembrane protein,8 which contains four repeated domains (D1-D4) each with six transmembrane segements (S1-S6). Cardiac Na+ channels show longer open times (~1.0 to 2.0 ms), and have a greater likelihood of multiple openings than neuronal counterparts.9 The hNav1.5-ΔKPQ mutant that manifests the long-QT phenotype generates persistent late Na+ currents during prolonged depolarization.10 The site of ΔKPQ deletion occurs at the D3-D4 intracellular linker. Unfortunately, it was rather difficult to study drug binding with these late currents because of their small amplitude.11,12 Large persistent late Na+ currents could be generated by IFM→QQQ triple mutations at the D3-D4 intracellular linker of the Na+ channel.13 This IFM motif is identified as a crucial part of the inactivation particle.14 A diminished inhibitory effect of antiarrhythmic agent lidocaine on the inactivation-deficient hNav1.5-IFM/QQQ mutant channels have been reported.15 In contrast, flecainide and mexiletine have been found to be potent blockers of small persistent late Na+ currents generated by hNav1.5-ΔKPQ mutant channels.11,12 Both of these drugs have been used to treat LQT-3 syndromes.
Attempts to express hNav1.5-IFM/QQQ mutant channels in human embryonic kidney cells (HEK293) cotransfected with an inward rectifier K+ channel (IRK1)16 yield only partial success. IRK1 channels were introduced to increase the expression of IFM/QQQ mutant Na+ channels by hyperpolarizing the membrane potential. The peak currents in cells expressing the hNav1.5-IFM/QQQ mutant were generally <2 nA, an amplitude too small for detailed drug studies. Previously, we reported a high level of expression of the inactivation-deficient hNav1.5-L409C/A410W mutant in HEK293t cells.17 The position of L409/A410 is located at the C-terminus of D1S6. Since no stable cell line that expresses robust persistent late Nav1.5 Na+ currents exists to date, we asked whether hNav1.5-L409C/A410W mutant clone can be used to generate such a stable cell line. A permanent cell line with robust persistent late cardiac Na+ currents could facilitate detailed studies of drug interactions with the open cardiac Na+ channel.
Results
Robust expression of hNav1.5-CW channels in a permanent HEK293 cell line
We initially isolated a colony, hNav1.5-CW/8, which expressed a high level of Na+ currents under the whole-cell configuration. However, when this cell line was reestablished from a frozen vial, the expression of Na+ currents dropped precipitously within 3–4 passages. We suspected that a revertant might emerge during the initial selection period. To isolate cells with the mutant Na+ channel clone from the total population of cultured hNav1.5-CW/8 cells, we diluted them ~300,000-fold, plated cells into 100-mm cultured dishes, and rescreened for Na+ channel expression as described in the methods section. One rescreened colony was found to remain stable with robust expression of inactivation-deficient hNav1.5-CW Na+ currents for up to 2 mo. This stable cell line (hNav1.5-CW/8–8) was expanded, frozen and stored in a liquid nitrogen tank. Frozen cells were thawed and reestablished in Ti-25 flasks. These cells were kept by continuous passages over a period of 3 mo. G-418 was not needed in the media for the reestablished cell line.
Figure 1A shows superimposed traces of inactivation-deficient hNav1.5-CW Na+ currents. These traces were recorded from a cell reestablished for 2 mo. Under our experimental conditions, inward hNav1.5-CW Na+ currents were activated at the threshold of around −80 mV and outward currents appeared around −10 mV. Large non-inactivated Na+ currents were clearly evident during the 80-ms pulse. The sodium conductance was derived from the peak amplitude, normalized and plotted against membrane potential (Fig. 1B). The graph was fitted using a Boltzmann equation with a V0.5 value of −41.8 ± 1.4 mV (at voltage where 50% channels activated, n = 8) and a slope factor of 10.2 ± 0.6 mV (n = 8). These values are comparable to those found in wild-type hNav1.5 channels.18
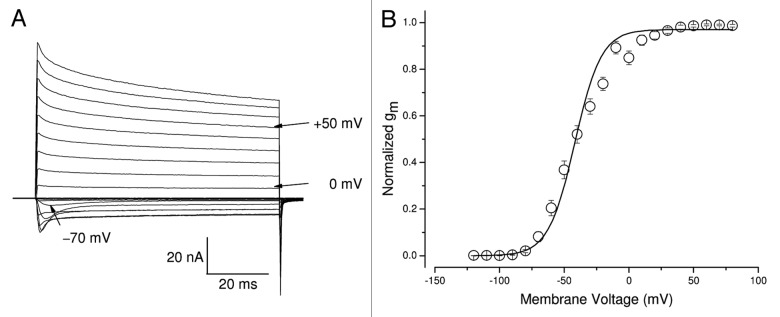
Figure 1. Gating characteristics of hNav1.5-CW mutant channels in stably transfected HEK293 cells. (A) A family of superimposed Na+ current traces was evoked by 80-ms pulses to voltages ranging from −100 to +50 mV in 10-mV increments. The inward current evoked by a pulse to −70 mV and the outward currents evoked by pulses to 0 and +50 mV are labeled. (B) Conductance was calculated by an equation, gm = INa/(Em−ENa), where INa is the peak current, Em is the test voltage and ENa is the estimated reversal potential. Normalized gm values were then plotted against the corresponding voltage (open circles). The plot was fitted with a Boltzmann function. The midpoint voltage (V0.5) and slope (k) of the function were −41.8 ± 1.4 mV and 10.2 ± 0.6 mV, respectively (n = 8). The holding potential was set at −140 mV.
To estimate the level of Na+ channel expression in individual cells we determined its peak amplitude of Na+ current and its capacitance. By and large, these cells expressed 996 ± 173 pA/pF (n = 20) of peak currents at +50 mV. This amount of expression is much higher than that of hNav1.5-IFM/QQQ inactivation-deficient Na+ currents in a permanent HEK293 cell line16 and at least twice as high as the skeletal muscle rNav1.4-WCW stable cell line (~410 pA/pF).19 Evidently, a deficiency in the fast Na+ channel inactivation per se did not impair the stable expression of hNav1.5-CW channels in this permanent HEK293 cell line.
Slow inactivation of hNav1.5-CW mutant cardiac Na+ channels with and without batrachotoxin
Human Nav1.5-CW cardiac Na+ currents were visible during prolonged pulses at voltages between −80 mV to +50 mV (Fig. 2A). These persistent late hNav1.5-CW Na+ currents decreased slowly with two exponential decay constants, likely because of the slow inactivation of the hNav1.5-CW mutant channel.18,20 The majority of current decay followed a slow time constant of 1.64 ± 0.22 sec at +50 mV (68 ± 3%, n = 6). The remaining decreased with a faster time constant of 129 ± 8 ms (25 ± 2%, n = 6) and about 7 ± 1% (n = 6) of currents were non-inactivating during the 4-second pulse. This onset of slow inactivation for inactivation-deficient hNav1.5-CW currents was, however, faster than those found for wild-type hNav1.5 currents. Such phenomenon has been also reported in inactivation-deficient hNav1.5-IFM/QQQ mutant channels20 and demonstrates that slow inactivation is enhanced in fast inactivation-deficient Na+ channels.

Figure 2. BTX prevents the slow decay of hNav1.5-CW mutant Na+ currents. (A) A family of superimposed hNav1.5-CW Na+ current traces was recorded with 4-second test pulses, ranging from −120 to +50 mV. Individual current traces at −80, 0 and +50 mV are labeled. The Na+ reversal potential is near −10 mV. (B) A family of superimposed hNav1.5-CW Na+ current traces were recorded with 4-second test pulses ranging from −140 to +50 mV in the presence of 5 μM BTX within the pipette solution. Notice that BTX-modified Na+ currents were maintained throughout the duration of the pulse. Individual current traces at −130, −100, −50, 0 and +50 mV are labeled. Before this record was taken, 400 repetitive 20-ms pulses at +50 mV were applied in a frequency of 2Hz to facilitate BTX binding with the open channel. The Na+ reversal potential is near −20 mV. Different cells were used in each panel. (C) With 5 μM BTX within the pipette, the Na+ conductance (closed circles) was clearly shifted to the hyperpolarizing direction as compared with that without BTX presence (dashed line as from Fig. 1B). The midpoint voltage (V0.5) and slope (k) of the function were −109.7 ± 1.0 mV and 7.6 ± 0.9 mV, respectively (n = 5). Currents were recorded as shown in Figure 2B and the conductance was calculated as described Figure 1B. Holding potential was set at −140 mV.
Earlier experiments have demonstrated that batrachotoxin (BTX) could eliminate the slow inactivation of voltage-gated Na+ channels.21 BTX also significantly alters the activation process as well as changes the ion selectivity of voltage-gated Na+ channels.22 Our results demonstrated that hNav1.5-L409C/A410W mutant channels could be readily modified by 5 μM BTX. To initiate BTX binding, we first applied repetitive pulses (a total of 400 at +50 mV/24ms) at a frequency of 1 Hz. Figure 2B shows BTX-modified hNav1.5-CW Na+ current traces from −140 to +50 mV at a slow time frame. Inward Na+ currents appeared around −130 mV (Fig. 2B); after activation, these Na+ currents remained during 4-second depolarization with minimal decay. Because of the hyperpolarizing shift in activation by BTX, sizable inward Na+ currents could be observed between −120 to −30 mV (Fig. 2B), whereas only smaller inward Na+ currents were recorded between −70 to −20 mV in the absence of BTX (Fig. 2A). In fact, BTX alters the V0.5 value for activation by −58 mV (from −41.8 mV to −109.7 mV; Fig. 2C, solid vs. dotted line) and eliminates most of the slow inactivation of these inactivation-deficient hNav1.5-L409C/A410W mutant channels (Fig. 2B). The reversal potential for the BTX-modified hNav1.5-CW Na+ channel was leftward shifted, which reversed around −20 mV, probably due to its reduced selectivity for Na+ ions23 and/or Na+ ion loading. This leftward-shift phenomenon of the reversal potential induced by BTX is commonly found in the literature.
Block of hNav1.5-CW mutant Na+ channels by external TTX at the μM range
We have previously shown that TTX blocked skeletal muscle inactivation-deficient hNav1.4-CW Na+ channels potently (IC50 = 15.1 nM).24 In comparison, TTX block the hNav1.5-CW mutant channels at μM range. Figure 3A shows the superimposed current traces before and after the external application of TTX at various concentrations. The dose-response curve Figure 3B (open circle) shows the IC50 for TTX block is estimated 2.32 ± 0.02 μM (Hill coefficient = 0.98 ± 0.01; n = 6) for the peak Na+ current block (Fig. 3B, open circle), demonstrating that cardiac Na+ channel remains relatively resistant to TTX block. This TTX potency of hNav1.5-CW is about 160-fold weaker than its skeletal muscle hNav1.4-CW counterpart. Our estimated IC50 value is nearly identical to that measured in wild-type hNav1.5 expressed in Xenopus oocytes (tonic block; 2.2 μM) but lower than 7.4 μM for hNav1.5-IFM/QQQ inactivation-deficient mutant channels.25 Furthermore, we found that there was no indication of a TTX-induced time-dependent block as shown in Figure 3B (closed circles). Maintained currents were also blocked by TTX during 50-ms depolarization with an IC50 for TTX block of 2.07 ± 0.04 μM (Hill coefficient = 0.94 ± 0.02; n = 6). The difference in IC50 values for peak and persistent late Na+ currents, however, is statistically significant (with a ratio of 1.12; p < 0.05).
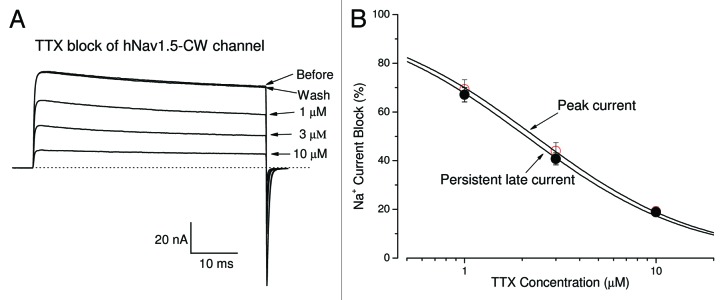
Figure 3. TTX blocks hNav1.5-CW Na+ channels. (A) Superimposed Na+ current traces were recorded in the absence and in the presence of TTX at 1, 3 and 10 μM. Currents were evoked by a 50-ms test pulse to +50 mV every 30 seconds. A steady-state block at each concentration was reached before application of the next solution. The block of Na+ currents by TTX could be completely reversed by the wash of the bath solution. (B) The dose-response curves were constructed using the data set as shown in (A). The peak or late Na+ currents were measured, normalized to the amplitude of the control, and plotted against the TTX concentration. Solid line represents a fit to the data with the Hill equation. The estimated IC50 values ± standard error [Hill coefficient ± S.E.] for peak-current block and the late-current block were 2.32 ± 0.02 μM (0.98 ± 0.01) (n = 6) and 2.07 ± 0.04 μM (0.94 ± 0.02) (n = 6), respectively.
Open-channel block by Class 1c antiarrhythmic drugs flecainide and propafenone during depolarization
Earlier studies indicate that persistent late Nav1.5 Na+ currents could be sensitive to block by Class 1c antiarrhythmic drugs, such as flecainide and propafenone.12,26 We tested these Na+ channel blockers at their therapeutic concentrations to see whether they also effectively blocked hNav1.5-CW Na+ currents in our permanent HEK293 cell line. We found that late hNav1.5-CW currents were far more sensitive to the block of flecainide and propafenone at 0.1 to 10 μM than were peak Na+ currents. Consequently, a strong time-dependent block of hNav1.5-CW Na+ currents by these Class 1c antiarrhythmic drugs appeared during the 100-ms depolarizing pulse. Figure 4A and B show the time-dependent block induced by flecainide and propafenone, respectively. These drugs attained their steady-state block of late hNav1.5-CW Na+ currents at +50 mV within 1.5–5 min. The dose-response curves of the peak and the maintained late hNav1.5-CW Na+ currents for fleicainide and propafernone are shown in Figure 5A and B, respectively. These curves were fitted by a Hill equation, and the estimated 50% inhibitory concentrations (IC50 values) and Hill coefficient for the resting and the open states of Nav1.5-CW Na+ channels are listed in Table 1. We define the resting-channel block as the block of the peak Na+ currents and the open-channel block as the block of the persistent late Na+ currents near the end of the 100-ms pulse at +50 mV. However, the IC50 values for the resting-channel block shown in Figure 5 are likely higher since a fraction of the open-channel block will occur inevitably at the time when the current reaches its peak amplitude, particularly at high drug concentrations. Such contamination of the resting-channel block at the peak amplitude measurement may also contribute to the deviation of the Hill coefficient for flecainide from the unity (0.52 ± 0.04, n = 6; Table 1). Our results nonetheless demonstrate clearly that (1) persistent late hNav1.5-CW Na+ currents in stably transfected HEK293 cells retain their drug sensitivity toward Class 1c antiarrhythmics at clinic relevant concentrations, and (2) the open hNav1.5-CW Na+ channel interacts preferentially with flecainide and propafenone, at least 20-fold more so than does its resting counterpart.
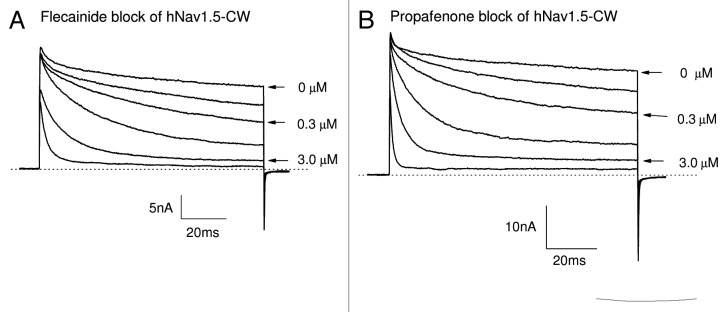
Figure 4. Block of hNav1.5-CW Na+ channels by Class 1c antiarrhythmic drugs. Superimposed hNav1.5-CW Na+ currents were recorded before and after the application of flecainide (A) or propafenone (B) at concentrations of 0.1, 0.3, 1.0, 3.0 and 10 μM. Currents of hNav1.5-CW Na+ channels were evoked every 30 seconds by a 100-ms test pulse at +50 mV. Drugs were applied externally in the external solution, and the currents were recorded after the block reached its steady-state, usually within 5 min. Different cells were used in (A and B). Holding potential was set at −140 mV. Traces of the control (0 μM), 0.3 and 3.0 μM drug concentrations were labeled.
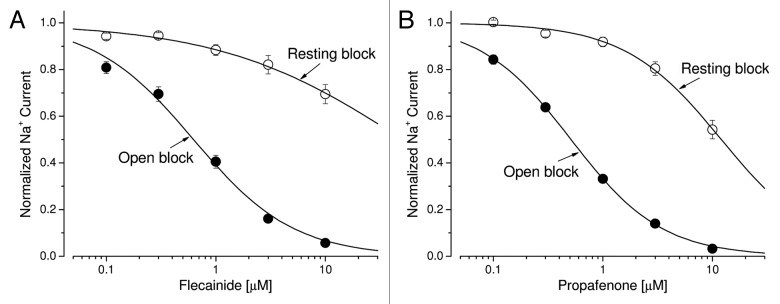
Figure 5. Dose-response curves for Class 1c antiarrhythmic drugs. (A) Dose-response curves for open-channel block and resting-channel block of flecainide were constructed using the data set as described in Figure 4A. For resting-channel block (open circles), we measured the peak amplitude of Na+ currents at various flecainide concentrations, normalized to the peak amplitude of the control, and plotted against drug concentration. Solid lines represent fits to the data with the Hill equation, y = 1 / [1+(x/IC50)n], where IC50 is the 50% inhibitory concentration and n is the Hill coefficient. The IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 50.8 ± 11.5 μM and 0.52 ± 0.04, respectively (n = 8), from the fitted curve. For open-channel block (filled circles), we measured the late current amplitude near the end of the 100-ms pulse at various flecainide concentrations, normalized to the late amplitude of the control, and plotted against drug concentration. The IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 0.63 ± 0.06 μM and 0.95 ± 0.08, respectively (n = 8). (B) Dose-response curves for propafenone were similarly constructed as described above. For resting-channel block (open circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 12.0 ± 0.7 μM and 0.98 ± 0.05, respectively (n = 8). For open-channel block (filled circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 0.51 ± 0.01 μM and 1.04 ± 0.02, respectively (n = 8).
Table 1. Estimates of the 50% inhibitory concentration (IC50) and Hill coefficient of antiarrhythmics for resting and open states of human Nav1.5-CW mutant channels.
| Antiarrhythmic drugs | IC50 value (μM) and Hill coefficient for resting state | IC50 value (μM) and Hill coefficient for open state | Ratio of IC50 (resting/open) |
|---|---|---|---|
| Flecainide (Class 1c) | 50.8 ± 11.5 μM (0.52 ± 0.04) |
0.63 ± 0.06 μM (0.95 ± 0.08) |
80.6 |
| Propafenone (Class 1c) | 12.0 ± 0.7 μM (0.98 ± 0.05) |
0.51 ± 0.01 μM (1.04 ± 0.02) |
23.5 |
| Lidocaine (Class 1b) | 606 ± 15 μM (0.71 ± 0.01) |
35.3 ± 2.7 μM (0.82 ± 0.05) |
17.2 |
| Mexiletine (Class 1b) | 220 ± 54 μM (0.76 ± 0.16) |
14.9 ± 0.9 μM (0.90 ± 0.05) |
14.8 |
| Quinidine (Class 1a) | 72.7 ± 2.9 μM (1.38 ± 0.07) |
8.6 ± 0.4 μM (1.19 ± 0.07) |
8.5 |
| Disopyramide (Class 1a) | 158 ± 11 μM (1.02 ± 0.08) |
52.5 ± 2.8 μM (1.05 ± 0.05) |
3.0 |
IC50 values for resting and open states of hNav1.5-CW channels were estimated as described in Figure 5. Mean and SE of IC50 and Hill coefficient (in parenthesis) are listed (each with n = 6–8).
It is noteworthy that data shown in Figure 4 could be further analyzed to extract the on- and off-rate constants. For example, the time-dependent block at various drug concentrations can be first normalized with the control record in the absence of drug (for removing the slow decay component in the control trace); then the corrected current trace can be fitted with a single exponential fuction to obtain the time constant (τ). The plot of 1/τ vs. drug concentration can be best fitted with a linear function, which yielded a slope and a y intercept. The values of the slope and the y intercept are equal to the on- and off-rate constants, respectively. Details of such analyses have been described previously.26 We were able to obtain the on- and off-rate constant for propafenone of 44.2 ± 2.8 μM−1s−1 and 14.7 ± 1.5 sec−1, respectively (n = 6). The calculated dissociation constant (Kd = off-rate/on-rate or 14.7/44.2) yielded 0.33 μM, a value comparable to the estimated IC50 (Table 1). Since the slow inactivation is eliminated by normalization, the time-dependent block induced by the Class 1 antiarrhythmic is likely to reflect solely the open-channel block. Similar analyses were employed for flecainide, which yielded an on- and off-rate constants of 14.6 ± 1.2 μM−1s−1 and 6.6 ± 0.9 sec−1, respectively (n = 6), and the Kd value of 0.45 μM. Thus, this cell line is well-suited for detailed kinetic analyses of interactions between drug and open Na+ channels.
Open-channel block by Class 1b antiarrhythmic drugs lidocaine and mexiletine during depolarization
Earlier studies have indicated that the block of the inactivated state of cardiac Na+ channels by lidocaine and mexiletine plays a dominant role in their therapeutic action.27,28 However, both of these Class 1b antiarrhythmics also blocked the persistent late hNav1.5-CW Na+ currents effectively. We observed the strong time-dependent block of the late Na+ currents with lidocaine or mexiletine at their clinic relevant concentrations (Fig. 6A and B), similar to those found in Figure 4 for Class 1c antiarrhymthics. This time-dependent block phenotype has previously been well characterized in inactivation-deficient muscle Na+ channels.19,29Figure 7A and B show the dose-response curves of lidocaine and mexiletine for the block of peak and late Na+ currents generated by hNav1.5-CW channels, respectively. Table 1 lists the IC50 of lidocaine and mexiletine for resting state (as measured by the peak current block) and open state (as measured by the late current block). The difference of the IC50 values between the resting and open-channel block is about 15-fold. These results suggest that, in addition to the dominant inactivated-channel block induced by lidocaine and mexiletine at their therapeutic concentrations, the open-channel block of the cardiac Na+ channel could also play a significant role in their antiarrhythmic action.
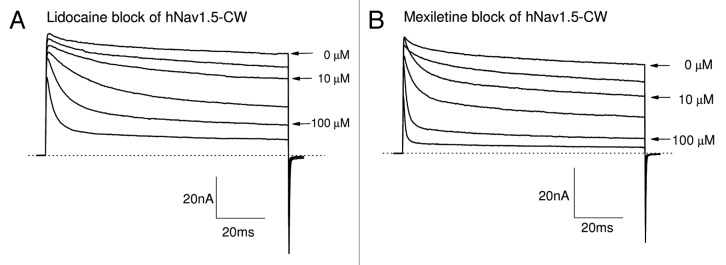
Figure 6. Block of hNav1.5-CW Na+ channels by Class 1b antiarrhythmic drugs. Superimposed Na+ current traces were recorded before and after the application of lidocaine (A) or mexiletine (B) at concentrations ranging from 3.0 to 300 μM. Currents of hNav1.5-CW Na+ channels were evoked every 30 seconds by a 100-ms test pulse at +50 mV. Drugs were applied externally. The currents were recorded after the block reached its steady-state, usually within 3 min. A different cell was used in each panel. Holding potential was set at −140 mV. Traces of the control (0 μM), 10 and 100 μM drug concentrations are labeled.
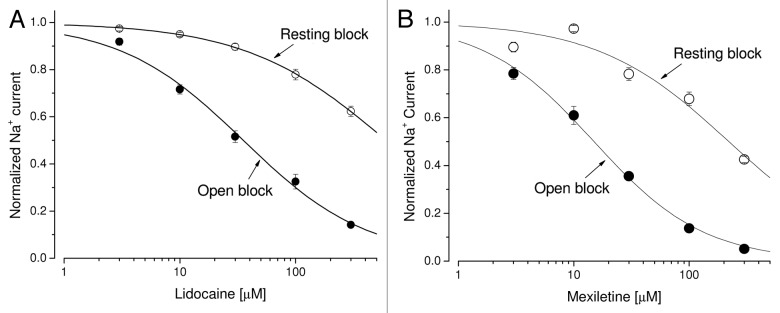
Figure 7. Dose-response curves for Class 1b antiarrhythmic drugs. (A) Dose-response curves for open-channel block and resting-channel block of lidocaine were constructed using the data set as shown in Figure 6A. For resting-channel block (open circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 606 ± 15 μM and 0.71 ± 0.01, respectively (n = 6), from the fitted curve. For open-channel block (filled circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 35.3 ± 2.7 µM and 0.82 ± 0.05, respectively (n = 6). (B) Dose-response curves for mexiletine were constructed as described before. For resting-channel block (open circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 220 ± 54 µM and 0.76 ± 0.16, respectively (n = 6). For open-channel block (filled circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 14.9 ± 0.9 μM and 0.90 ± 0.05, respectively (n = 6).
Block of persistent late hNav1.5-CW Na+ currents by Class 1a antiarrhythmics disopyramide and quinidine
Grant et al. (2000)16 reported a lack of time-dependent block of hNav1.5-IFM/QQQ inactivation-dependent Na+ currents by disopyramide at 200 μM. We thought to corroborate such a finding using the stably transfected cell line as an assay to distinguish the action of disopyramide from other Class 1 antiarrhythmic drugs. Figure 8A shows the superimposed traces of hNav1.5-CW Na+ currents before and after external disopyramide application at various concentrations. We found that a stronger block of the late Na+ currents than that of the peak Na+ currents was still present, albeit less severe than that found for Class 1c and 1b antiarrhythmic drugs. The estimated difference in IC50 values between the block of peak and the late Na+ currents for disopyramide was about 3-fold (Fig. 9A), which is much less than those of the Class 1c and 1b antiarrhythmics (Table 1). Our results thus demonstrate that the phenotype for disopyramide block in hNav1.5-CW and hNav1.5-IFM/QQQ mutant channels is quite different. One (hNav1.5-CW mutant) is with visible time-dependent block, whereas the other (hNav1.5-IFM/QQQ mutant) is without.16
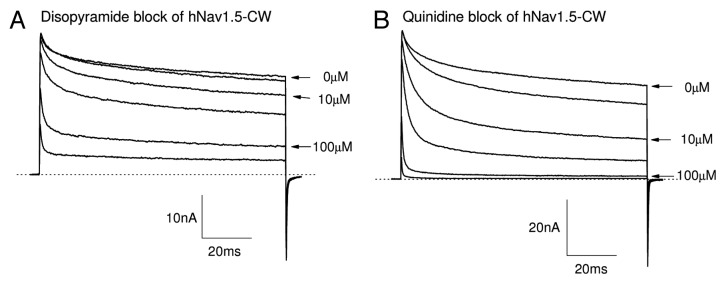
Figure 8. Block of hNav1.5-CW Na+ channels by Class 1a antiarrhythmic drugs. Superimposed Na+ current traces were recorded before and after the application of disopyramide (A) or quinidine (B) at concentrations ranging from 3.0 to 300 μM. Currents of hNav1.5-CW Na+ channels were evoked every 30 seconds by a 100-ms test pulse at +50 mV. Drugs were applied externally. The currents were recorded after the block reached its steady-state, usually within 3 min. A different cell was used in each panel. Holding potential was set at −140 mV. Traces of the control (0 μM), 10 and 100 μM drug concentrations are labeled.
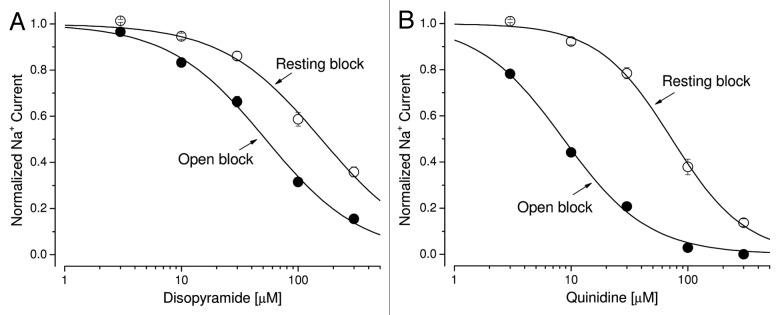
Figure 9. Dose-response curves for Class 1a antiarrhythmic drugs. (A) Dose-response curves for open-channel block and resting-channel block of disopyramide were constructed using the data set as shown in Figure 8A. For resting-channel block (open circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 158 ± 11 μM and 1.02 ± 0.08, respectively (n = 8). For open-channel block (filled circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 52.5 ± 2.8 µM and 1.05 ± 0.05, respectively (n = 8). (B) Dose-response curves for quinidine were constructed as described before. For resting-channel block (open circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 72.7 ± 2.9 μM and 1.38 ± 0.07, respectively (n = 8). For open-channel block (filled circles), the IC50 value (mean ± SEM) and the Hill coefficient (mean ± SEM) were estimated 8.6 ± 0.4 μM and 1.05 ± 0.05, respectively (n = 8).
Within the Class 1a drugs, the time-dependent block for quinidine (Fig. 8B) is more apparent than that of disopyramide. The estimated IC50 values and Hill coefficient of for the resting state and the open state of hNav1.5-CW mutant channels are obtained from the dose-response curve of quinidine (Fig. 9B) and are listed in Table 1. Among Class 1 antiarrhythmic drugs, the ranking order for the selectivity ratio of the IC50 values between resting and open states is as follows: flecainide (80.6) > propafenone (23.5) > lidocaine (17.2) > mexiletine (14.8) > quinidine (8.5) > disopyramide (3.0). The large disparity in this ratio indicates that Class 1c and 1b antiarrhythmics target the persistent late Na+ currents rather selectively whereas the Class 1a drugs show only a modest selectivity toward late Na+ currents.
Discussion
Miniature persistent Na+ currents in cardiomyocytes can cause arrhthymia and sudden death.3,5 Because of their minute size it has been difficult to study these aberrant late currents and to identify therapeutic drugs that may target them selectively. Details of mutant channel behavior, which determine drug interactions and the functional effects of discrete mutations, are likely to depend on both the channel isoform and the species. This report describes for the first time an HEK293 cell line stably transfected with an inactivation-deficient mutant Na+ channel clone that can generate large persistent late human cardiac Na+ currents. Pharmacological profiles and gating properties confirm the identity of the hNav1.5-CW mutant Na+ channel. With such a cell line available we can also address an open question regarding the open-channel vs. resting-channel selectivity among Class 1 antiarrhythmics.
Robust expression of hNav1.5-L409C/A410W mutant Na+ channels in a stable HEK293 cell line
Based on their attempts to establish stably transfected Hek293 cell lines expressing inactivation-deficient cardiac hNav1.5-IFM/QQQ Na+ channels, Grant et al.16 suggested that the overload of Na+ ions from impetuous openings could inhibit hNav1.5-IFM/QQQ mutant channel expression of these cells. After numerous trials we were uncertain whether robust expression of hNav1.5-CW Na+ channels could be attained in a permanent HEK293 cell line. Particularly, we witnessed the rapid loss of inactivation-deficient Na+ currents in cells isolated from a colony with a high level of Na+ channel expression. This observation could be interpreted that the persistent flow of inward Na+ current may be detrimental to the cells. However, with a second colony selection, these isolated cells remain stable in expressing hNav1.5-CW mutant channels. We therefore conclude that robust expression of hNav1.5-CW Na+ channels can be achieved in Hek293 cells despite the reported low expression in the hNav1.5-IFM/QQQ inactivation-deficient mutant.
The availability of a permanent HEK293 cell line expressing robust mutant hNav1.5-CW Na+ channels has a number of advantages. First, the cell line will make it easier for routine exploitations of inactivation-deficient Na+ channels, including biophysical and pharmacological studies of persistent late cardiac hNav1.5-CW Na+ currents. Second, the permanent cell line may be applicable to identify potent open-channel selective Class 1 antiarrhythmic drugs using an automated parallel patch-clamp system.30 There is one disadvantage of using a permanent hNav1.5-CW mutant cell line as the potential drug-binding site may be changed unintentionally by mutations. Such possibility should be taken into consideration when the drug affinity is significantly different from that found in the literature.
TTX and BTX receptors in hNav1.5-L409C/A410W mutant Na+ channels
Both the TTX receptor and the BTX receptor within the Na+ channel have been well characterized, and their locations are mapped along the Na+ permeation pathway.31,32 To check if the binding sites for TTX and BTX within Na+ channels are altered, we studied the effects of these toxins in cells expressing hNav1.5-L409C/A410W cardiac mutant Na+ channels. Our results demonstrated that the binding sites for TTX and BTX in hNav1.5-CW Na+ channels appeared unchanged (Fig. 2; Fig. 3), as their phenotypes induced by these toxins were similar to those found in wild-type counterparts when treated with these ligands. An unchanged binding site for BTX indicates that the BTX receptor within the inner cavity of hNav1.5-CW Na+ channels is comparable to that of the wild-type hNav1.5 channels, whereas an unchanged binding site for TTX suggests that the TTX receptor at the external entryway is also near normal.
It is noteworthy that the cardiac hNav1.5 isoform is relatively TTX insensitive, requiring a high concentration of TTX to block this Na+ channel. We found that TTX blocked both peak and persistent late hNav1.5-CW Na+ currents without a significant time-dependent block during a 50-ms pulse, although it appeared slightly more potent in the block of the late rather than the peak currents (Fig. 3). A clear time-dependent block was found in TTX block of hNav1.5-QQQ mutant during a long 4 sec depolarizing pulse.25 These authors suggested that the extra block of the hNav1.5-QQQ channel found during long depolarization is due to a higher-affinity TTX site, which is modulated by the activation gating of the sodium channel. It remains unknown whether the slow inactivation plays a role in this enhanced block since the slow inactivation is clearly evident in hNav1.5-CW mutant channels during a prolonged pulse (Fig. 2A).
Characterization of open-channel blockers using the stable cell line expressing large inactivation-deficient hNav1.5-CW sodium currents
Class 1 antiarrhythmic drugs target voltage-gated hNav1.5 Na+ channels for their therapeutic action.33,34 These drugs are subgrouped as 1a, 1b and 1c, roughly dependent upon their rates of recovery from drug-induced block under physiological conditions. Unlike the TTX block which shows little time-dependent block of inactivation-deficient Na+ currents during depolarization (Fig. 3), we found that antiarrhythmics, including Class 1c (flecainide, propaferone), Class 1b (mexiletine, lidocaine) and Class 1a (quinidine and disopyramide), blocked persistent late hNav1.5-CW mutant Na+ currents far more effectively than they blocked peak Na+ currents during the depolarizing pulse (Fig. 4; Fig. 6; Fig. 8; Table 1). The Hill coefficient values for the open-channel block are close to the unity ranging from 0.82 for lidocaine to 1.19 for quinidine, suggesting that one drug molecule blocks one open channel. This result is consistent with the finding that there is one local anesthetic/antiarrhythmic receptor within the inner cavity of the Na+ channel with drug-sensing residues situated at multiple S6 segments.35,36 The Hill coefficient values for the resting state, however, deviate from the unity considerably ranging from 0.52 for flecainide to 1.38 for quinidine. One explanation for this deviation is because the resting block is measured as the block of peak Na+ currents. This type of measurement is undoubtedly tainted by the open block of Na+ channels during the channel activation. An additional possibility is that there are multiple resting states and each may have a different affinity toward the antiarrhythmic drug.37
The selectivity ratio of IC50 between the resting-channel block and the open-channel block among Class 1 antiarrhythmic drugs differs significantly, ranging from 80.6 for flecainide to 3.0 for disopyramide (Table 1). Flecainide can therefore be viewed as a genuine open-channel selective blocker whereas disopyramide cannot. For disopyramide, the resting, open and inactivated blocks of Na+ channels all likely play an important role in its efficacy in vivo.37 The reason behind this considerable difference in the selectivity ratio of IC50 between the resting-channel and the open-channel block among Class 1 antiarrhthmic drugs is unclear. It is possible that flecainide and propaferone may have a better induced-fit with their receptor in the open state of the hNav1.5-CW channel than with their counterpart in the resting state because of the dynamic movement of S6 segments during Na+ channel activation.34,36
Physiological significance of the open-channel block by Class 1 antiarrhythmic drugs
Several studies have given two contradictory views concerning the therapeutic role of the open-channel block by lidocaine. First, QX-314 (a quaternary ammonium derivative of lidocaine) fails to inhibit the persistent late Na+ currents in pronase-treated squid axons.38,39 Second, the open-state binding affinity for lidocaine or cocaine is 3- to 10-fold lower than the inactivated-state binding affinity in hNav1.5-IFM/QQQ channels.15,40 These results suggest that the open Na+ channel is not blocked preferentially by lidocaine and therefore the inactivated channel plays the most important role in QX-314/lidocaine/cocaine block of Na+ channels, in agreement with the modulated receptor hypothesis.27,41,42 Additional results of a weak open-channel block by flecainide, RAD-243, and disopyramide16 in hNav1.5-IFM/QQQ mutant channels provide further support for this notion.
In contrast to these findings, various local anesthetics and Class 1 antiarrhythmics preferentially block persistent late Na+ currents created by various conditions. Included are (1) chloramine-T-treated axons,43 (2) gain-of-function genetic diseases that cause LQT-3 symptoms,11,12 (3) hypoxia/failing cardiac myocytes6,44 and (4) inactivation-deficient S6 mutants.19,45 These conflicting results lead to an alternative view that, in addition to the inactivated state of the Na+ channel, the open state also interacts preferentially with lidocaine, mexiletine and flecainide and that such open-channel block is important therapeutically in vivo when persistent late Na+ currents are present.
It is evident that aberrant persistent late Na+ currents generated via chemical/enzymatic modifications, gain-of-function genetic diseases, site-directed mutagenesis or pathological environment are the result of structural alterations of voltage-gated Na+ channels. Theoretically, such alterations could bring about a decrease or an increase in binding affinities for Class 1 antiarrhythmic drugs. In other words, not all persistent late Na+ currents are created equal in their sensitivity toward Class 1 antiarrhythmic drugs. This possibility would account for the two opposing views described above regarding the therapeutic role of the open-channel block. Unfortunately, it is difficult to measure directly the open-channel block in wild-type Na+ channels because these channels showed a brief open time of ~0.5 to ~2.0 ms.1,9 Aside from this uncertainty, the immediate need is to identify drugs that can selectively impede aberrant persistent late Na+ currents in patients, while leaving normal transient Na+ currents intact.46,47 The cell line expressing robust human cardiac hNav1.5-CW channels should be applicable as a tool to identify novel Class 1 antiarrhythmic drugs that preferentially silence persistent late cardiac Na+ currents.
Materials and Methods
Selection of HEK293 cells with stably transfected hNav1.5-L409C/A410W cDNA clone
Full-length wild-type hNav1.5 cDNA in pcDNA3 vector was provided by Dr. Augustus Grant (Duke University, School of Medicine). Mutagenesis of hNav1.5-L409C/A410W cDNA was performed as reported previously.48 The site-directed mutations occurred at position 21–22 of the D1S6 C-terminus. HEK293 cells were purchased from American Type Culture Collection (ATCC) and maintained at 37 °C in a 5% CO2 incubator in DMEM (Life Technologies, Inc.) containing 10% fetal bovine serum (HyClone) and 1% penicillin and streptomycin solution (Sigma). HEK293 cells were transfected with the mutant clone by a calcium phosphate precipitation method.24 One day after transfection, HEK293 cells were treated with 1.0 mg/ml G-418 (Invitrogen, Inc.) in 100-mm culture dishes. About 2 weeks after transfection individual G-418-resistant colonies were isolated using glass cylinders (i.d. = 8 mm). Isolated colonies were trypsinized and replated in 35-mm culture dishes each with a gelatin-treated coverslip. The coverslip was removed from the dish after 2–3 d, and ~5 cells from each coverslip were assayed under the whole-cell configuration. Five positive colonies with inactivation-deficient currents were expanded and frozen individually. One colony initially expressing robust inactivation-deficient sodium currents was later expanded, diluted proportionally to the cell density and subjected to a second colony-selection procedure in 100-mm culture dishes without G-418. The second colony-selection procedure was necessary for this particular colony because the number of cells with the robust expression of inactivation-deficient currents decreased drastically after a few passages. Three out of five colonies were tested positive after the second screening and one remained stable with high expression for up to 2 mo. This colony (identified as hNav1.5-CW/8–8) was expanded and maintained in Ti-25 flasks for studies described here.
Electrophysiology and data acquisition
The whole-cell configuration of a patch-clamp technique49 was used to study Na+ currents in HEK293 cells at room temperature (22 ± 2 °C). Electrode resistance was measured about 0.5 MΩ. Command voltages were elicited with pCLAMP9 software and delivered by Axopatch 200B (Axon Instrument). Cells were held at −140 mV and dialyzed for ~10−15 min before current recording. Most of the capacitance and leak currents were cancelled with a patch-clamp device and by P/−4 subtraction. Liquid junction potential was not corrected. Access resistance was about 1 MΩ; series resistance compensation of ~95% typically resulted in voltage errors of ≤4 mV at +50 mV. Most current measurements were performed at +50 mV for the outward Na+ currents. Such recordings allowed us to avoid the complication of series resistance artifacts and to minimize inward Na+ ion loading.50 Conductance was calculated as gm = INa/(Em−ENa), where INa is the peak current, Em is the test voltage and ENa is the estimated reversal potential. Conductance curves (gm vs Em) were fitted by a Boltzmann equation, gm = gmax/[1+exp((V0.5−Em)/k)], where V0.5 and k are derived as the midpoint voltage and slope factor of the fit, respectively, Em is the test potential and gmax is the maximal Na+ conductance.23 Dose-response curves were fitted with the Hill equation, y = 1/[1+(x/IC50)n], where IC50 is derived as the 50% inhibitory concentration and n as the Hill coefficient.19 Curve fitting was performed by Microcal Origin. A student’s unpaired t-test was used to evaluate estimated parameters (mean ± SE or fitted value ± SE of the fit); P values of <0.05 were considered statistically significant.
Solutions and chemicals
Flecainide-HCl, propafenone-HCl, lidocaine-HCl, mexiletine-HCl, disopyramide sulfate and quinidine HCl were all purchased from Sigma. Tetrodotoxin (TTX) was purchased from Calbiochem Corp., and batrachotoxin (BTX) was a generous gift from Dr. John Daly. TTX at 1 mM was dissolved in H2O and disopyramide sulfate at 100 mM was dissolved in 20 mM HCl. The remaining drugs were dissolved in dimethylsulfoxide (DMSO) as stock solutions and stored at 4 °C. Final drug concentrations were made by serial dilution. The highest DMSO concentration in bath solution was 0.1% except for BTX, which contained 1% DMSO in pipette solution. DMSO at a final concentration of 1% had no effect on Na+ currents. Cells were perfused with an extracellular solution containing (in mM) 65 NaCl, 85 choline-Cl, 2 CaCl2 and 10 HEPES (titrated with tetramethylammonium-OH to pH 7.4). The pipette (intracellular) solution consisted of (in mM) 100 NaF, 30 NaCl, 10 EGTA and 10 HEPES (titrated with cesium-OH to pH 7.2). The rationale of solution choices is given in the last paragraph.
Acknowledgments
This work was previously supported by a grant from National Institutes of Health (HL-66076) and currently by a grant from National Institute of Health (GM-094152). We are grateful to Dr. Augustus Grant (Duke University, School of Medicine) for providing us the hNav1.5-pcDNA3 plasmid and to the late Dr. John Daly (Bethesda, MD) for providing us batrachotoxin.
Glossary
Abbreviations:
- TTX
tetrodotoxin
- BTX
batrachotoxin
- hNav1.5-CW
human cardiac Na+ channel with L409C/A410W mutations
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/25056
References
- 1.Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–41. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–90. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett PB. Long QT syndrome: biophysical and pharmacologic mechanisms in LQT3. J Cardiovasc Electrophysiol. 2000;11:819–22. doi: 10.1111/j.1540-8167.2000.tb00055.x. [DOI] [PubMed] [Google Scholar]
- 4.Cannon SC. An expanding view for the molecular basis of familial periodic paralysis. Neuromuscul Disord. 2002;12:533–43. doi: 10.1016/S0960-8966(02)00007-X. [DOI] [PubMed] [Google Scholar]
- 5.George AL., Jr. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–9. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: studies in failing human myocardium. J Mol Cell Cardiol. 2001;33:923–32. doi: 10.1006/jmcc.2001.1355. [DOI] [PubMed] [Google Scholar]
- 7.Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–47. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gellens ME, George AL, Jr., Chen LQ, Chahine M, Horn R, Barchi RL, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89:554–8. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirsch GE, Brown AM. Kinetic properties of single sodium channels in rat heart and rat brain. J Gen Physiol. 1989;93:85–99. doi: 10.1085/jgp.93.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett PB, Yazawa K, Makita N, George AL., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–5. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 11.Wang DW, Yazawa K, Makita N, George ALJ, Jr., Bennett PB. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest. 1997;99:1714–20. doi: 10.1172/JCI119335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagatomo T, January CT, Makielski JC. Preferential block of late sodium current in the LQT3 DeltaKPQ mutant by the class I(C) antiarrhythmic flecainide. Mol Pharmacol. 2000;57:101–7. [PubMed] [Google Scholar]
- 13.West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–4. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eaholtz G, Colvin A, Leonard D, Taylor C, Catterall WA. Block of brain sodium channels by peptide mimetics of the isoleucine, phenylalanine, and methionine (IFM) motif from the inactivation gate. J Gen Physiol. 1999;113:279–94. doi: 10.1085/jgp.113.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett PB, Valenzuela C, Chen L-Q, Kallen RG. On the molecular nature of the lidocaine receptor of cardiac Na+ channels. Modification of block by alterations in the α-subunit III-IV interdomain. Circ Res. 1995;77:584–92. doi: 10.1161/01.RES.77.3.584. [DOI] [PubMed] [Google Scholar]
- 16.Grant AO, Chandra R, Keller C, Carboni M, Starmer CF. Block of wild-type and inactivation-deficient cardiac sodium channels IFM/QQQ stably expressed in mammalian cells. Biophys J. 2000;79:3019–35. doi: 10.1016/S0006-3495(00)76538-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang GK, Edrich T, Wang SY. Time-dependent block and resurgent tail currents induced by mouse beta4(154-167) peptide in cardiac Na+ channels. J Gen Physiol. 2006;127:277–89. doi: 10.1085/jgp.200509399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Reilly JP, Wang SY, Kallen RG, Wang GK. Comparison of slow inactivation in human heart and rat skeletal muscle Na+ channel chimaeras. J Physiol. 1999;515:61–73. doi: 10.1111/j.1469-7793.1999.061ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S-Y, Mitchell J, Moczydlowski E, Wang GK. Block of inactivation-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway. J Gen Physiol. 2004;124:691–701. doi: 10.1085/jgp.200409128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richmond JE, Featherstone DE, Hartmann HA, Ruben PC. Slow inactivation in human cardiac sodium channels. Biophys J. 1998;74:2945–52. doi: 10.1016/S0006-3495(98)78001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang S-Y, Wang GK. Point mutations in segment I-S6 render voltage-gated Na+ channels resistant to batrachotoxin. Proc Natl Acad Sci U S A. 1998;95:2653–8. doi: 10.1073/pnas.95.5.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khodorov BI. Chemicals as tools to study nerve fiber sodium channels: effect of batrachotoxin and some local anesthetics. Prog Biophys Mol Biol. 1978;45:153–74. [Google Scholar]
- 23.Wasserstrom JA, Liberty K, Kelly J, Santucci P, Myers M. Modification of cardiac Na+ channels by batrachotoxin: effects on gating, kinetics, and local anesthetic binding. Biophys J. 1993;65:386–95. doi: 10.1016/S0006-3495(93)81046-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S-Y, Moczydlowski E, Wang GK. Inactivation-deficient human skeletal muscle Na+ channels (hNav1.4-L443C/A444W) in stably transfected HEK-293 cells. Receptors Channels. 2004;10:131–8. doi: 10.1080/10606820490514914. [DOI] [PubMed] [Google Scholar]
- 25.Dumaine R, Hartmann HA. Two conformational states involved in the use-dependent TTX blockade of human cardiac Na+ channel. Am J Physiol. 1996;270:H2029–37. doi: 10.1152/ajpheart.1996.270.6.H2029. [DOI] [PubMed] [Google Scholar]
- 26.Edrich T, Wang S-Y, Wang GK. State-dependent block of human cardiac hNav1.5 sodium channels by propafenone. J Membr Biol. 2005;207:35–43. doi: 10.1007/s00232-005-0801-4. [DOI] [PubMed] [Google Scholar]
- 27.Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. J Gen Physiol. 1983;81:613–42. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiser T, Qu Y, Catterall WA, Scheuer T. Differential interaction of R-mexiletine with the local anesthetic receptor site on brain and heart sodium channel alpha-subunits. Mol Pharmacol. 1999;56:1238–44. doi: 10.1124/mol.56.6.1238. [DOI] [PubMed] [Google Scholar]
- 29.Wang GK, Russell C, Wang SY. Mexiletine block of wild-type and inactivation-deficient human skeletal muscle hNav1.4 Na+ channels. J Physiol. 2004;554:621–33. doi: 10.1113/jphysiol.2003.054973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanguinetti MC, Bennett PB. Antiarrhythmic drug target choices and screening. Circ Res. 2003;93:491–9. doi: 10.1161/01.RES.0000091829.63501.A8. [DOI] [PubMed] [Google Scholar]
- 31.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/S0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 32.Wang S-Y, Wang GK. Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell Signal. 2003;15:151–9. doi: 10.1016/S0898-6568(02)00085-2. [DOI] [PubMed] [Google Scholar]
- 33.Roden DM. Antiarrhythmic drugs. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Gilman AG, eds. Goodman & Gilman's The Pharmacological Basis of Therapeutics. New York: Macmillan Publishing Company, 2001:933-70. [Google Scholar]
- 34.Sheets MF, Fozzard HA, Lipkind GM, Hanck DA. Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc Med. 2010;20:16–21. doi: 10.1016/j.tcm.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Catterall WA, Mackie K. Local anesthetics. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Gilman AG, eds. Goodman & Gilman's The Pharmacological Basis of Therapeutics. New York: Macmillan Publishing Company, 2001:367-84. [Google Scholar]
- 36.Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- 37.Zilberter YI, Starmer CF, Grant AO. Open Na+ channel blockade: multiple rest states revealed by channel interactions with disopyramide and quinidine. Am J Physiol. 1994;266:H2007–17. doi: 10.1152/ajpheart.1994.266.5.H2007. [DOI] [PubMed] [Google Scholar]
- 38.Cahalan MD. Local anesthetic block of sodium channels in normal and pronase-treated squid giant axons. Biophys J. 1978;23:285–311. doi: 10.1016/S0006-3495(78)85449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeh JZ. Sodium inactivation mechanism modulates QX-314 block of sodium channels in squid axons. Biophys J. 1978;24:569–74. doi: 10.1016/S0006-3495(78)85403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Leary ME, Chahine M. Cocaine binds to a common site on open and inactivated human heart (Na(v)1.5) sodium channels. J Physiol. 2002;541:701–16. doi: 10.1113/jphysiol.2001.016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–98. doi: 10.1016/0304-4157(77)90003-X. [DOI] [PubMed] [Google Scholar]
- 43.Wang GK, Brodwick MS, Eaton DC, Strichartz GR. Inhibition of sodium currents by local anesthetics in chloramine-T-treated squid axons. The role of channel activation. J Gen Physiol. 1987;89:645–67. doi: 10.1085/jgp.89.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ju Y-K, Saint DA, Gage PW. Effects of lignocaine and quinidine on the persistent sodium current in rat ventricular myocytes. Br J Pharmacol. 1992;107:311–6. doi: 10.1111/j.1476-5381.1992.tb12743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang GK, Russell C, Wang SY. State-dependent block of wild-type and inactivation-deficient Na+ channels by flecainide. J Gen Physiol. 2003;122:365–74. doi: 10.1085/jgp.200308857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl 4):iv6–14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eijkelkamp N, Linley JE, Baker MD, Minett MS, Cregg R, Werdehausen R, et al. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135:2585–612. doi: 10.1093/brain/aws225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S-Y, Bonner K, Russell C, Wang GK. Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels. Biophys J. 2003;85:911–20. doi: 10.1016/S0006-3495(03)74530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 50.Cota G, Armstrong CM. Sodium channel gating in clonal pituitary cells. The inactivation step is not voltage dependent. J Gen Physiol. 1989;94:213–32. doi: 10.1085/jgp.94.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]