

Abstract
Acid-sensitive, two-pore domain potassium channels, K2P3.1 and K2P9.1, are implicated in cardiac and nervous tissue responses to hormones, neurotransmitters and drugs. K2P3.1 and K2P9.1 leak potassium from the cell at rest and directly impact membrane potential. Hence altering channel number on the cell surface drives changes in cellular electrical properties. The rate of K2P3.1 and K2P9.1 delivery to and recovery from the plasma membrane determines both channel number at the cell surface and potassium leak from cells. This study examines the endocytosis of K2P3.1 and K2P9.1. Plasma membrane biotinylation was used to follow the fate of internalized GFP-tagged rat K2P3.1 and K2P9.1 transiently expressed in HeLa cells. Confocal fluorescence images were analyzed using Imaris software, which revealed that both channels are endocytosed by a dynamin-dependent mechanism and over the course of 60 min, move progressively toward the nucleus. Endogenous endocytosis of human K2P3.1 and K2P9.1 was examined in the lung carcinoma cell line, A549. Endogenous channels are endocytosed over a similar time-scale to the channels expressed transiently in HeLa cells. These findings both validate the use of recombinant systems and identify an endogenous model system in which K2P3.1 and K2P9.1 trafficking can be further studied.
Keywords: K2P channel, TASK, potassium channel, endocytosis, recycling, clathrin, caveolin, dynamin, Imaris
Introduction
Two-pore domain potassium (K2P) channels are the molecular component of potassium leak currents1 and key contributors to the resting membrane potential of both excitable and non-excitable cells. While all 15 members of the K2P channel family play key roles in a range of physiological processes, including neuronal excitability, cardiac contraction and smooth muscle tone, the acid-sensitive K2P (TASK) channels, have received considerable interest due to their association with cerebral ischemia, the effects of anesthetics and malignancy.2
TASK channels [K2P3.1 (TASK-1), K2P9.1 (TASK-3) and the non-functional K2P15.1 (TASK-5)] show widespread tissue distribution and are characterized by their sensitivity to extracellular pH, pO2 and volatile anesthetics.2-4 Slow changes in TASK current magnitude have been implicated in cardiac and nervous tissue responses to hormones, glucose concentration, neurotransmitters and drugs, while more acute responses have been shown to have a putative role in peripheral and central chemoreception, T-cell activation and mediating the effects of anesthetics.5-12 Paradoxically, TASK channels are thought to play a role in granule cell deterioration in cerebellum (during development) while also being implicated in enhancing hippocampal cell survival during cellular stress.13,14 A number of reports have also implicated TASK channels in showing altered expression in carcinomas (breast, colorectal and melanoma) and to potentially provide an oncogenic advantage.15-18 As K2P channels are active at physiological resting membrane potentials, changes in channel number at the plasma membrane drastically alter the electrical properties of the cell. For this reason, control of cell surface expression of TASK channels is of paramount importance to cell function.
Delivery of TASK channels to the cell surface is tightly regulated at the transcriptional and post-transcriptional levels, via regulation of biogenesis, sorting and trafficking.19 The export of newly synthesized K2P3.1 (and K2P9.1) channels from the endoplasmic reticulum to the cell surface is subject to tight quality control mechanisms.20-23 Phosphorylation-dependent interaction with auxiliary proteins (including coatomer proteins, annexin II light chain and cytosolic adaptor protein, 14-3-3) determines the number of functional K2P3.1 channels on the cell surface.22-24 Clearly, retrieval from the cell surface, degradation or recycling of K2P channels will have an equally important role in regulating cell surface expression of these channels. Indeed, Gabriel et al.25 suggest that phosphorylation of K2P3.1 and recruitment of 14-3-3 may have a regulatory role in channel endocytosis, while Matsuoka et al.26 propose that nerve growth factor induces endocytosis of K2P3.1 in adrenal medullary cells. While these reports focus on regulators of stimulated endocytosis of TASK channels, to date, the molecular mechanism of K2P channel retrieval from the cell surface has not been characterized.
Cells use various mechanisms to internalize plasma membrane proteins, including clathrin mediated endocytosis (CME) or clathrin independent endocytosis (CIE).27-29 Less well-defined than CME, mechanisms of CIE include caveolae-associated endocytic pathways as well as Clathrin-Independent Carrier (CLIC) or Arf6- or Flotillin-dependent pathways.29 Each pathway utilizes distinct mediators to enable vesicle formation and to recruit specific cargo. Pathway convergence is seen at the early endosomes (EE) from which proteins can either be recycled to the plasma membrane or be degraded via the late endosomes and lysosomes. Distinct endocytic pathways will have their own functions and different types of specialized cells will therefore rely on different pathways to varying extents to control their complement of cell surface proteins.
Understanding the pathways utilized to recover TASK channels from the cell surface will provide critical understanding of the mechanism by which cells control not only their innate excitability and cellular function but also their response to external stimuli. In this study we identify the endocytic pathways utilized by K2P3.1 and K2P9.1 in a recombinant system, which enables us determine intracellular localization of the internalized channels precisely. We then use similar experimental strategies to examine channel internalization in human lung carcinoma cell line, A549, which shows endogenous expression of both channels.
Results
TASK channels retrieved from the cell surface localize within the early endosome
The EE is a point within the endocytic pathway at which a number of independent endocytic routes converge. We, therefore, asked whether rat (r) K2P3.1 is retrieved from the plasma membrane to the EE. A sensitive cell surface biotinylation assay30 was used to follow the fate of plasma membrane GFPrK2P3.1. HeLa cells transiently expressing GFPrK2P3.1 (green; Fig. 1A) underwent surface biotinylation on ice, then were warmed to 37 °C to facilitate endocytosis. Samples were fixed at pre-defined time intervals. Any remaining external biotin was removed and internal biotin was stained to identify internalized cargo (red; Fig. 1A), while the EE was identified by labeling with an antibody against early endosome antigen 1 (EEA1; blue; Fig. 1A). Confocal fluorescence image z-stacks were analyzed first for colocalized channel and biotin (GFPrK2P3.1 + Biotin), representing internalized channel and next for triple fluorescence overlap (GFPrK2P3.1 + Biotin + EEA1), representing endocytosed channel in EE. At time zero, essentially no internalized GFPrK2P3.1 was detected (Fig. 1A). After 60 min, however, punctate structures containing GFPrK2P3.1 and biotin were distributed throughout the cytoplasm, a proportion of which also colocalized with EEA1.

Figure 1. Endocytosis of GFPrK2P3.1. (A) HeLa cells transiently expressing GFPrK2P3.1 were surface-biotinylated on ice and either immediately stripped to remove the external biotin and fixed (upper images, 0 min) or warmed to 37 °C for 60 min (lower images, 60 min) before stripping and fixing. Cells were stained with streptavidin-Alexa Fluor 546 (red; Biotin) to reveal internalized biotin and with anti-EEA1 (blue; EEA1). GFPrK2P3.1 is shown in green. Images are whole cell projections and the Merge panels represent GFPrK2P3.1, biotin and EEA1 superimposed. GFPrK2P3.1 + Biotin colocalization (grayscale) represents internalized GFPrK2P3.1. GFPrK2P3.1 + Biotin + EEA1 colocalization (grayscale) represents internalized GFPrK2P3.1 present in EEA1-positive compartments. Bar represents 10 μm. (B) Images of HeLa cells treated as in (A) were analyzed by Imaris software to identify 0.6 and 1.2 μm vesicles of GFPrK2P3.1, biotin and EEA1 at 0 min and 60 min. Total vesicles: all the spots detected by Imaris software. Colocalized vesicles: all the colocalized GFPrK2P3.1-biotin spots (green and red) and triple colocalized GFPrK2P3.1, biotin and EEA1 spots (green, red and blue). Bar represents 10 μm. Vesicles outlined by a white square (60 min, colocalized vesicles) are shown in the foreground of the enlarged image, taken along the xy plane of the cell. Small patches of nucleic acid stained outside the nucleus are complexes of DNA and jetPEI taken up by the cells during transfection. (C) HeLa cells from the time-course experiment shown in (A and B) were analyzed after 5, 15, 30 and 60 min incubation at 37 °C to identify internalized GFPrK2P3.1. Upper images show colocalized GFPrK2P3.1-biotin spots (green and red) and triple colocalized GFPrK2P3.1, biotin and EEA1 (green, red and blue). Lower images show the same cells, but with the vesicles color-coded according to distance to the nucleus. Magenta vesicles are nearer than 2.5 μm and cyan vesicles are further than 2.5 μm from the nearest nucleus. Numbers represent the proportion of vesicles in each distance category. Scale bars: 10 μm.
To further analyze K2P3.1 endocytosis and localization within EE at these time points, Imaris 7.6 software (Bitplane AG) was utilized to identify puncta or spots of specific dimensions and defined quality criteria that could be modeled as vesicles. Detailed examination of individual puncta within confocal slices enabled the diameter of the puncta to be determined. 0.6 μm was deemed to be an appropriate diameter for many of the spots. Other, larger spots were approximately 1.2 μm in diameter. Many spots of both diameters were detected at time zero (Fig. 1B; total vesicles), but almost none of these showed positive staining for GFPrK2P3.1 with biotin (green and red), or with biotin plus EEA1 (green, red and blue). This finding was expected as the biotinylation had taken place on ice which restricted endocytosis. After a 60-min incubation at 37 °C, many double-positive (GFPrK2P3.1 plus biotin) and triple-positive (GFPrK2P3.1 plus biotin and EEA1) vesicles were apparent (Fig. 1B; colocalized vesicles at 60 min), suggesting that under unstimulated conditions, GFPrK2P3.1 is internalized and enters early endosomes.
A time course of channel endocytosis with samples fixed at 5, 15, 30 and 60 min displays an increase in the number of vesicles containing internalized GFPrK2P3.1 over time (Fig. 1C; upper images). When the distance of triple-stained vesicles from the nucleus is examined, our analysis reveals progression of stained vesicles from the cell periphery toward the nucleus with time (Fig. 1C; lower images, color-coded by distance from the nucleus). At early time points, a large majority of vesicles are greater than 2.5 μm away from the nucleus, whereas at 60 min, closer to half the vesicles are within 2.5 μm of the nucleus. (84% of vesicles are >2.5 μm away from nucleus after 5 min of endocytosis compared with 58% after 60 min).
Surface biotinylation and time-course experiments as outlined above for rK2P3.1 were performed for rK2P9.1 in HeLa cells transiently expressing GFPrK2P9.1. As for rK2P3.1, an accumulation of vesicles containing GFPrK2P9.1 and biotin, or GFPrK2P9.1, biotin and EEA1 after a 60-min incubation was observed (Fig. 2A). Biotin and channel co-stained vesicles were observed at the 5-min time point, with the number of vesicles increasing over time (Fig. 2B, upper panel). When comparing vesicle proximity to the nucleus for both channels, relatively more vesicles containing internalized GFPrK2P9.1 were closer to the nucleus at the early time points (Fig. 2B, lower panel), suggesting that endocytosis of GFPrK2P9.1 occurs more rapidly than that of GFPrK2P3.1.

Figure 2. Endocytosis of GFPrK2P9.1. (A) HeLa cells transiently expressing GFPrK2P9.1 (green) were surface-biotinylated on ice and either immediately stripped to remove the external biotin and fixed (upper images, 0 min) or warmed to 37 °C for 60 min (lower images, 60 min) before stripping and fixing. Cells were stained with streptavidin-Alexa Fluor 546 (red) to reveal internalized biotin and with anti-EEA1 (blue). Confocal z-stacks were analyzed by Imaris to detect 0.6 μm and 1.2 μm vesicles, as in Figure 1. Total vesicles: all the spots detected by Imaris software. Colocalized vesicles: all the colocalized GFPrK2P9.1-biotin spots (green and red) and triple colocalized GFPrK2P9.1, biotin and EEA1 spots (green, red and blue). Bar represents 10 μm. (B) HeLa cells from the time-course experiment shown in (A) were analyzed after 5-, 15-, 30- and 60-min incubation at 37 °C to identify internalized GFPrK2P9.1. Upper images show colocalized GFPrK2P9.1-biotin spots (green and red) and triple colocalized GFPrK2P9.1, biotin and EEA1 (green, red and blue). Lower images show the same cells, but with the vesicles color-coded according to distance to the nucleus. Magenta vesicles are nearer than 2.5 μm and cyan vesicles are further than 2.5 μm from the nearest nucleus. Numbers represent the proportion of vesicles in each distance category. Scale bars: 10 μm.
rK2P9.1 undergoes more rapid retrieval than rK2P3.1
Analysis of the number of 0.6 μm colocalized vesicles containing GFPrK2P3.1 and biotin or GFPrK2P9.1 and biotin (double-positive) over the course of a 60-min incubation compared with the number of triple-positive vesicles, containing EEA1 provided valuable information of rate of channel retrieval (Fig. 3). At 0 min post removing endocytosis block almost no double- or triple-positive vesicles for either channel were observed (Fig. 1B; Fig. 2A). Following 5 min of unstimulated endocytosis, comparable numbers of double-positive vesicles were observed for both channels (K2P3.1 37 ± 4.8 SEM; K2P9.1 53 ± 7.5 SEM; numbers are per 10000 μm3 cytoplasm). Similarly, fewer but comparable numbers of triple-positive vesicles were detected after 5 min (K2P3.1 3.8 ± 1.9 SEM; K2P9.1 2.6 ± 0.8 SEM). After 15 min of unstimulated endocytosis, more double-positive vesicles were observed for GFPrK2P9.1 than GFPrK2P3.1; this reached significance at 30 min (K2P3.1 96 ± 11.4 SEM; K2P9.1 174 ± 12.0 SEM; p = 0.001; Fig. 3A and B) and this trend was maintained for all time points examined While the number of double-positive vesicles increased in number with time, the number of triple-positive vesicles for each channel did not alter significantly with time after 5 min (K2P3.1: 3.8 ± 1.9 SEM, at 5 min and 10.2 ± 2.2 SEM, at 60 min p ≤ 0.111; K2P9.1: 2.6 ± 0.8 SEM, at 5 min and 22 ± 9.6 SEM, at 60 min p ≤ 0.095). Furthermore, despite increased numbers of double-positive vesicles for K2P9.1 compared with K2P3.1 there was no significant difference in number of triple-positive vesicles at any time point between these two groups (Fig. 3B). Significantly, the number of 0.6 μm triple-positive vesicles is only approximately 10% of the internalized channel. Analysis of individual confocal slices suggests that smaller triple vesicles fuse to form larger and more complex structures that are first apparent at 15 min and become much more prominent at 30 min and 60 min (Fig. 3B). These data indicate that GFPrK2P9.1 may be endocytosed more readily than GFPrK2P3.1 and that the channels do not accumulate in the early endosome.

Figure 3. Rates of endocytosis. (A) HeLa cells transiently expressing GFPrK2P3.1 or GFPrK2P9.1 were surface biotinylated on ice, then warmed to 37 °C for the indicated periods of time, before being stripped to remove external biotin, then fixed and stained to detect internalized biotin and the early endosome marker, EEA1. Images were analyzed using Imaris software to calculate the number of colocalized 0.6 μm vesicles per unit volume of cytoplasm (10000 μm3). Numbers are means from n = 3 to 6 fields of view from two independent experiments. Error bars are standard error of the mean. Closed circles: colocalized GFPrK2P3.1 and biotin (endocytosed GFPrK2P3.1); open circles: triple-colocalized GFPrK2P3.1, biotin and EEA1 (endocytosed GFPrK2P3.1 in early endosomes); closed triangles: colocalized GFPrK2P9.1 and biotin (endocytosed GFPrK2P9.1); open triangles: triple-colocalized GFPrK2P9.1, biotin and EEA1 (endocytosed GFPrK2P9.1 in early endosomes). (B) Images from the 30-min samples described above. Endocytosed GFPrK2P3.1 or GFPrK2P9.1, as indicated, in 0.6 μm and 1.2 μm vesicles [colocalized K2P channel (green) with biotin (red)], and endocytosed GFPrK2P3.1 or GFPrK2P9.1 in early endosomes (colocalized green, red and blue spots). Structures consisting of colocalized 1.2 μm spots became more numerous as time progressed, suggesting fusion of smaller vesicles to form larger entities. White arrows indicate the direction from which the magnified perspective views are taken. Nucleus shown in white. Scale bars: 10 μm.
Dynamin-dependent endocytosis of TASK channels
To determine the endocytic routes utilized by TASK channels, we examined colocalization of endocytosed channels with clathrin and the impact of disrupting dynamin-dependent endocytosis (which includes both clathrin-mediated endocytosis (CME) and caveolin-dependent endocytosis)31,32 on colocalization. Using a method similar to that described in Figures 1 and 2 and Imaris Cell to identify 0.6 μm and 1.2 μm vesicles, internalized GFPrK2P3.1 or GFPrK2P9.1 colocalization with clathrin was examined (Fig. 4). After 30 min of unstimulated endocytosis, vesicles positive for biotin (red) channel (green) and clathrin (blue) were detected (Fig. 4A; vesicles). For GFPrK2P3.1 the number of triple-positive 0.6 μm vesicles (GFPrK2P3.1, biotin and clathrin) was 20.5 per 10000 μm3 cytoplasm (SEM 2.2, n = 3 fields of view) demonstrating GFPrK2P3.1 colocalizes with clathrin and likely utilizes CME as a pathway of entry into the cell (colocalized vesicles; Fig. 4A and C).

Figure 4. Dynamin-dependent endocytosis of GFPrK2P3.1 and GFPrK2P9.1. HeLa cells transiently expressing GFPrK2P3.1 [green, (A)] or GFPrK2P9.1 (green, (B)] were incubated with DMSO vehicle (Control) or the dynamin inhibitor, Dynasore (as indicated) before surface biotinylation on ice and warming to 37 °C for different periods of time. Images show cells stripped to remove surface biotin and fixed after a 30-min incubation period, before staining to detect biotin (red) and clathrin (blue). Merge: whole cell projection. Vesicles: image analyzed using Imaris to detect 0.6 μm and 1.2 μm spots. Colocalized vesicles: only the channel-biotin or channel-biotin-clathrin triple-colocalized 0.6 μm and 1.2 μm spots shown. Scale bars: 10 μm. (C) Images were analyzed by Imaris to calculate the number of triple-colocalized (channel, biotin and clathrin) 0.6 μm vesicles per unit volume of cytoplasm. Numbers are the mean and standard error from n = 3 fields of view. Inhibition by Dynasore is significant for GFPrK2P3.1 at p < 0.01; GFPrK2P9.1 at p < 0.05.
Dynasore is a powerful, cell-permeant inhibitor of dynamin-dependent endocytic pathways and therefore disrupts CME.33 Dynasore treatment resulted in a general reduction in the level of channel endocytosis as determined by fewer double-positive (biotin and channel) vesicles (153 ± 25 SEM in control conditions vs. 45 ± 8 SEM following treatment with 80 μM Dynasore; t = 30 min; n = 3 fields of view). Moreover, a marked reduction in triple-positive (biotin, channel and clathrin) vesicles (7.1 per 10000 μm3 cytoplasm SEM 1.1, n = 3 fields of view; p < 0.01; Fig. 4A and C) was observed at 30 min.
Endocytosed GFPrK2P9.1 also showed localization within clathrin-positive vesicles (Fig. 4B) Dynasore treatment had a similar inhibitory effect on endocytosis of GFPrK2P9.1 (Fig. 4B). Under control conditions 22.8 ± 2.5 (SEM; n = 3 fields of view) triple-positive 0.6 μm vesicles were observed, while significantly fewer (9.6 ± 3.0; n = 3 fields of view; p < 0.05) triple-positive vesicles were observed following a 30-min preincubation with 80 μM Dynasore (Fig. 4C).
Dynamin is also required for the fission of caveolae,32 so we asked whether GFPrK2P3.1 and GFPrK2P9.1 colocalize with caveolin-1 (Fig. 5). Marked overlap of the fluorescence of both channels with caveolin was observed throughout the cell. As a measure of the proportion of overlap between caveolin and each of the channels, mean Mander’s coefficients were calculated. The Mander’s coefficient for the degree of colocalization of GFPrK2P3.1 with caveolin is 0.60, (SEM 0.07, n = 3 fields of view), while caveolin with GFPrK2P3.1 is 0.36, (SEM 0.10). The Mander’s coefficient for GFPrK2P9.1 with caveolin is 0.35 (SEM 0.08, n = 3 fields of view), while that of caveolin with GFPK2P9.1 is 0.29 (SEM 0.09). Numerous 0.6 μm and 1.2 μm vesicles of GFPrK2P3.1 and GFPrK2P9.1 colocalized with caveolin vesicles (Fig. 5; colocalized vesicles), together these data suggest that the channels may enter the cell by more than one pathway.
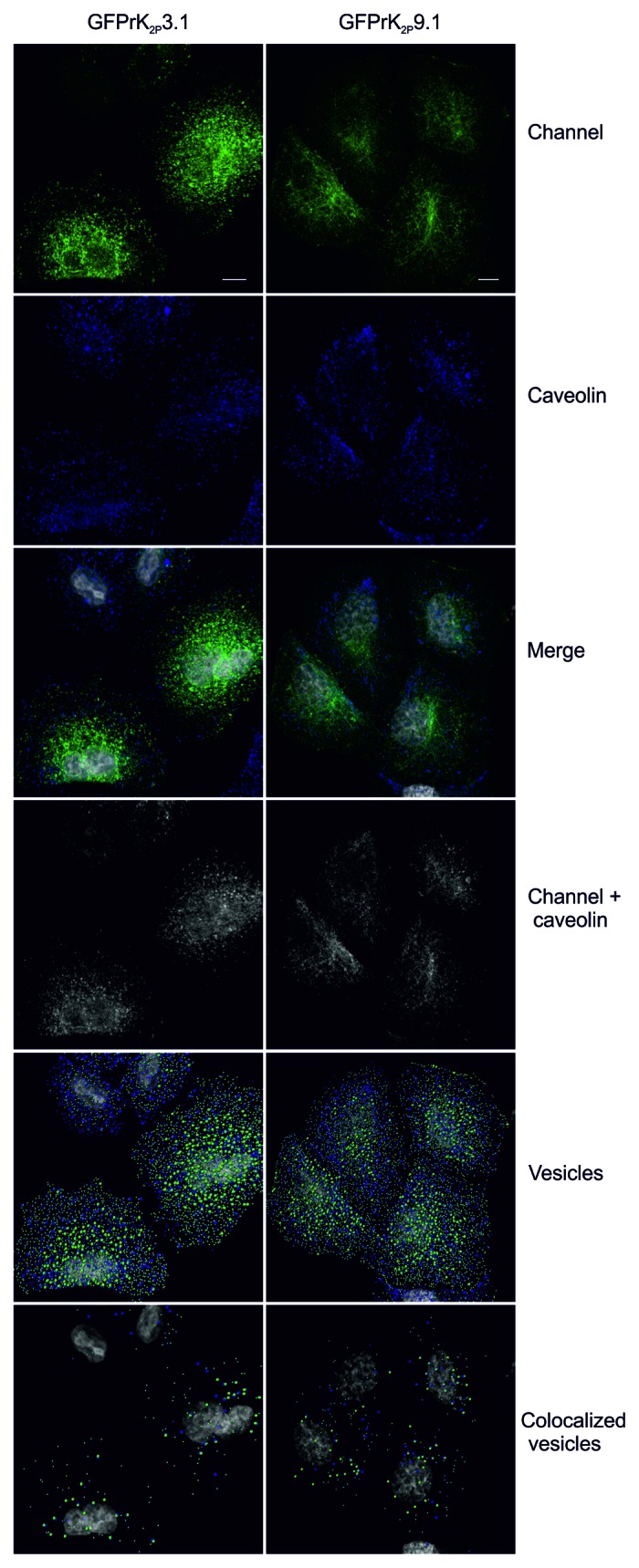
Figure 5. GFPrK2P3.1 and GFPrK2P9.1 colocalize with caveolin. HeLa cells transiently expressing GFPrK2P3.1 and GFPrK2P9.1 (Channel, green) were fixed and stained with anti-caveolin-1 (Caveolin, blue). Images are confocal whole cell projections. Merge: superimposed green and blue channels (nucleus in gray). Channel + caveolin: colocalized K2P channel and caveolin, as determined by the colocalization module of Imaris software, using the automatic thresholding function. Vesicles: 0.6 μm and 1.2 μm spots of GFPrK2P3.1 or GFPrK2P9.1 (green) and caveolin (blue) were detected using Imaris software (nucleus in gray). Colocalized vesicles: colocalized 0.6 μm and 1.2 μm K2P channel and caveolin, detected using Imaris software. Scale bar: 10 μm.
The fate of endocytosed TASK channels
After retrieval from the plasma membrane, proteins may enter rapid or slow recycling pathways, or be targeted for degradation.34,35 To determine if internalized GFPrK2P3.1 or GFPrK2P9.1 were targeted for degradation and hence present in lysosomes we performed endocytosis assays as before and probed for lysosomal localization. After 60-min incubation areas of triple colocalization of GFPrK2P3.1, biotin and RFP-LAMP1 were observed (Fig. 6A; mean Mander’s coefficient for GFPrK2P3.1-biotin with RFP-LAMP1 is 0.55 ± 0.07 SEM, n = 3 fields of view), suggesting that a portion of internalized GFPrK2P3.1 is targeted for degradation. A 3D rendering of colocalized GFPrK2P3.1 and biotin (surface shown in cyan) and its relationship with the lysosome (RFP-LAMP1 surface shown in red) is depicted in large right hand panel in Figure 6A and provides a visual representation of the proportion of channel within lysosomal vesicles and similarly the proportion of lysosomal vesicles containing internalized GFPrK2P3.1.

Figure 6. Fate of endocytosed GFPrK2P3.1 and GFPrK2P9.1. HeLa cells were first transduced with CellLight BacMam 2.0 baculovirus particles to express the lysosomal fusion protein RFP-LAMP1 (shown in red), then transiently transfected with a plasmid encoding either GFPrK2P3.1 [reen, (A)] or GFPrK2P9.1[green, (B)]. After surface biotinylation on ice, cells were warmed to 37 °C for 1 h, then stripped to remove external biotin, fixed and stained to reveal internal biotin (shown in blue). All images are confocal whole cell projections. Merge: superimposed K2P channel, RFP-LAMP1 and biotin. Large images show a 3D reconstruction of colocalized GFPrK2P3.1 and biotin [cyan, (A)] or GFPrK2P9.1 and biotin [cyan, (B)] representing K2P channel endocytosed during the 60 min incubation, together with the distribution of RFP-LAMP1 fusion protein (shown in red), representing the lysosomal compartment. Scale bars: 10 μm. (C) HeLa cells transiently expressing GFPrK2P3.1 (green, upper images) or GFPrK2P9.1 (green, lower images) were surface biotinylated on ice, then warmed to 37 °C for 30 min, before stripping to remove external biotin, fixing and staining with streptavidin-Alexa Fluor 546 to reveal internal biotin (red) and with anti-Rab11 (blue). All images are confocal whole cell projections. Merge: superimposed K2P channel, biotin and Rab11. Vesicles: images were analyzed by Imaris software to detect GFPrK2P3.1 or GFPrK2P9.1 (green), internalized biotin (red) and Rab11 (blue) 0.6 μm diameter spots. Coloc vesicles: colocalized GFPrK2P3.1 or GFPrK2P9.1 and biotin vesicles; triple-colocalized K2P channel, biotin and Rab11 vesicles. Scale bar: 10 μm. (D) Numbers of colocalized 0.6 μm vesicles per unit volume cytoplasm after a 30-min incubation (C). Black bars: GFPrK2P3.1. Gray bars: GFPrK2P9.1. Channel-biotin-Rab11: triple-colocalized 0.6 μm vesicles. Percentages refer to the proportion of endocytosed K2P channel that is in Rab11-positive vesicles. Channel-biotin: endocytosed K2P. Channel-Rab11: number of colocalized K2P and Rab11 0.6 μm vesicles – includes but is not confined to material endocytosed during the 30-min incubation. Numbers are means (n = 2 or 3 fields of view) and error bars are SEM.
Internalized GFPrK2P9.1 was also found to localize within LAMP1 positive vesicles (Fig. 6B). The mean Mander’s coefficient for GFPrK2P9.1-biotin with RFP-LAMP1 is 0.30 (SEM 0.10, n = 3 fields of view).
To determine whether GFPrK2P3.1 or GFPrK2P9.1 might undergo recycling to the cell surface following endocytosis, we examined colocalization of internalized channel with Rab11 as a marker for recycling vesicles. A very small amount of internalized GFPrK2P3.1 was detected in Rab11-containing vesicles after 30-min endocytosis. Figure 6C shows confocal whole cell projections of channel, biotin and Rab11 labeling for both GFPrK2P3.1 and GFPrK2P9.1. The merged image shows each of these images superimposed and enabled detection of small areas of overlap. Imaris Cell was then utilized to identify 0.6 μm vesicles containing channel, biotin or Rab11 (Fig. 6C; vesicles) and the number of triple-positive 0.6 μm vesicles per 10000 μm3 was found to be only 2% of the total internalized GFPrK2P3.1 (coloc vesicles; Fig. 6C and D). For GFPrK2P9.1, the figure was slightly higher with 6% of the internalized channel at 30 min detected in Rab11-positive vesicles. These results suggest that under resting conditions, a modest amount of both channels may enter recycling pathways.
TASK channel endocytosis in an endogenous model system
While recombinant systems are useful in defining the molecular mechanisms in membrane trafficking, a system in which the endogenous distribution, trafficking and expression levels can be examined provides additional valuable information. We therefore sought to identify a cell line that showed endogenous expression of both K2P3.1 and K2P9.1. We screened a range of cell lines by RT-PCR for transcript expression of KCNK3 and KCNK9 and found product of the correct size and sequence for both genes in the human lung carcinoma cell line, A549 (shown in Fig. S1A), as well as in SH-SY5Y (brain) and MCF7 (breast) cell lines. Expression of the channels in A549 cells was confirmed at the protein level by immunofluorescence. Generally, antibodies to acid-sensitive K2P channels do not perform well for immunofluorescence, so we tested a bank of commercially available antibodies and validated their performance in HEK293 cells transiently expressing either GFPhK2P3.1 and GFPhK2P9.1. We found Sigma rabbit anti-K2P3.136 and Santa Cruz goat anti-K2P9.137 to show overlapping signal with GFP and no signal within non-transfected cells (Fig. S1B and C). Following validation, the same conditions were used to probe the presence and sub-cellular localization of both K2P3.1 and K2P9.1 in A549. Both channels were detected on the cell periphery as well as within the endoplasmic reticulum and Golgi apparatus (Fig. S2A and B), as previously observed within recombinant systems.12,20,24
To examine endocytosis of TASK channels in this system, surface biotinylation assays were performed. After 60-min unstimulated endocytosis, channel and biotin signals were found to colocalize (Fig. 7 for hK2P3.1 and Fig. 8 for K2P9.1; endocytosis panels).

Figure 7. Endocytosis of endogenous human K2P3.1. All images are confocal whole cell projections. K2P3.1 is shown in green. Scale bar: 10 μm. Endocytosis panels: A549 cells were surface-biotinylated on ice, warmed to 37 °C for 60 min, then stripped to remove external biotin and fixed. Cells were stained with anti-K2P3.1 and streptavidin-DyLight 650 (blue) to reveal internal biotin. Merge: superimposed K2P3.1 and biotin. Vesicles: 0.6 μm and 1.2 μm spots of K2P3.1 (green) and biotin (blue) detected by Imaris software. Coloc vesicles: colocalized 0.6 μm and 1.2 μm K2P3.1 and biotin spots. EEA1 panels: fixed A549 cells were stained with anti-K2P3.1 and anti-EEA1 (red). Merge: superimposed K2P3.1 and EEA1. Vesicles: 0.6 μm and 1.2 μm spots of K2P3.1 (green) and EEA1 (red) detected by Imaris software. Coloc vesicles: colocalized 0.6 μm and 1.2 μm K2P3.1 and EEA1 spots. Rab7 panels: as for EEA1 panels, except anti-Rab7 (red) as the counterstain. Rab11 panels: fixed A549 cells were stained with anti-K2P3.1 and anti-Rab11 (red). Merge: superimposed K2P3.1 and Rab11. Vesicles: 0.6 μm spots of K2P3.1 (green) and Rab11 (red) were detected by Imaris software. Coloc vesicles: colocalized 0.6 μm spots of K2P3.1 and Rab11.

Figure 8. Endocytosis of endogenous human K2P9.1. All images are confocal whole cell projections. K2P9.1 is shown in green. Scale bar: 10 μm. Endocytosis panels: A549 cells were surface-biotinylated on ice, warmed to 37 °C for 60 min, then stripped to remove external biotin and fixed. Cells were stained with anti-K2P9.1 and streptavidin-DyLight 650 (blue) to reveal internal biotin. Merge: superimposed K2P9.1 and biotin. Vesicles: 0.6 μm and 1.2 μm spots of K2P9.1 (green) and biotin (blue) detected by Imaris software. Coloc vesicles: colocalized 0.6 μm and 1.2 μm K2P9.1 and biotin spots. EEA1 panels: fixed A549 cells were stained with anti-K2P9.1 and anti-EEA1 (red). Merge: superimposed K2P9.1 and EEA1. Vesicles: 0.6 μm and 1.2 μm spots of K2P9.1 (green) and EEA1 (red) detected by Imaris software. Coloc vesicles: colocalized 0.6 μm and 1.2 μm K2P9.1 and EEA1 spots. Rab7 panels: as for EEA1 panels, except anti-Rab7 (red) as the counterstain. Rab11 panels: fixed A549 cells were stained with anti-K2P9.1 and anti-Rab11 (red). Merge: superimposed K2P9.1 and Rab11. Vesicles: 0.6 μm spots of K2P9.1 (green) and Rab11 (red) were detected by Imaris software. Coloc vesicles: colocalized 0.6 μm spots of K2P9.1 and Rab11.
Channel colocalization with key markers of different points within the endocytic pathway (EEA1, Rab7 and Rab11) was also examined. We allowed Imaris to identify 0.6 μm vesicles of hK2P3.1 and each of the markers using automatic quality thresholds for both channel and marker in question. Approximately 2% of all hK2P3.1 positive vesicles colocalized with EEA1 (Fig. 7; EEA1, coloc vesicles image) while 13% colocalized with Rab7 (Fig. 7; Rab7). K2P3.1 showed a small degree of colocalization with Rab11 (mean Mander’s coefficient 0.16, SEM 0.01, n = 3 fields of view; approximately 6% of 0.6 μm hK2P3.1 vesicles).
K2P9.1 also showed colocalization with each of the endocytic pathway markers. Approximately 4% of hK2P9.1 vesicles colocalized with EEA1, 2% with Rab7 and 4% with Rab11 (Fig. 8).
Discussion
Data presented here provide the first insight into the endocytic pathways utilized by acid-sensitive K2P channels, K2P3.1 and K2P9.1. Antibodies to K2P3.1 and K2P9.1 do not recognize external epitopes, which makes tracking channel internalization from the cell surface via traditional primary antibody capture methods difficult without the introduction of external epitope tags to enable distinction between K2P channels from the plasma membrane and other intracellular K2P channels. We adopted an alternative method to investigate channel endocytosis in cells transiently expressing GFP-tagged channels: a “pulse” biotinylation of the cell surface on ice, followed by a 37 °C “chase.”30 We reasoned that vesicles containing both GFP-tagged protein and biotin must have originated from the plasma membrane; therefore by analyzing only structures that contain both GFP and biotin, we can exclude secretory vesicles from our analyses. Using this approach, we detected internalized K2P3.1 and K2P9.1 channels within the EE. Endocytosed channels were also observed within Rab11 positive vesicles, indicative of channel recycling and within LAMP1-positive vesicles, suggestive of channel degradation.
Our colocalization analysis was performed using Imaris software which utilizes the statistical methods of Costes et al.38 Statistically robust Pearson and Mander’s coefficients were generated by the software, and further insight was obtained with Imaris Cell, which enabled detailed qualitative and quantitative analyses of structures; thus providing an appreciation of how different compartments relate to one another in 3D space. This enabled analysis of channel overlap with distinct compartments as well as in relation to their location within the cell and the movement of endocytosed K2P channels to be tracked. Indeed, K2P channels internalized at the plasma membrane were found to move from the cell periphery toward the perinuclear region with time (Fig. 1; Fig. 2); these observations clearly demonstrate a non-random nature to the observed colocalization39 but also may provide insight into the process of transport of endocytosed TASK channels. For example, the observed accumulation of endocytosed GFPrK2P3.1 and GFPrK2P9.1 within the perinuclear region might suggest interaction with the recycling endosome also found within this locale40 and this together with channel localization within Rab11 vesicles is supportive of the hypothesis that TASK channels undergo recycling following endocytosis.
When exploring the possible endocytic pathways utilized by TASK channels, both GFPrK2P3.1 and GFPrK2P9.1 showed colocalization with vesicles positive for clathrin. Dynasore treatment significantly reduced the number of vesicles containing internalised K2P channel and clathrin (as well as the number of structures containing internalised K2P channels). Dynasore is a small molecule that non-competitively inhibits the GTPase activity of dynamin,33 blocking clathrin-dependent endocytosis. We did not observe a complete block of endocytosis, which is either due to incomplete inhibition of dynamin, or the operation of more than one endocytic pathway for GFPrK2P3.1 and GFPrK2P9.1. Dynamin is also necessary for the scission of caveolae to form transport vesicles. Examination of the expression pattern of the caveolar scaffolding protein, caveolin-1, in HeLa cells transfected with GFPrK2P3.1 and GFPrK2P9.1, revealed considerable colocalization of both channels with caveolin-1. While numerous vesicular structures that contained both caveolin-1 and GFPrK2P3.1 and GFPrK2P9.1, which likely represent endocytic, secretory or recycling vesicles were observed,41 it is possible that some of the overlap may be due to an ER and Golgi pool of caveolin-1,42 Further characterization of these pools will enable the precise role of caveolin-dependent endocytosis in TASK channel internalization to be defined.
The number of particular subsets of vesicles within a defined volume of cytoplasm (in this study, 10000 μm3) can be quantified using Imaris cell. When this was performed for the total number of 0.6 μm vesicles positive for internalized GFPrK2P3.1 or GFPrK2P9.1 we found that following surface labeling and supporting endocytosis the number of vesicles carrying internalized TASK channels increased rapidly and plateaued within ca. 20 min (Fig. 3A). More GFPrK2P9.1-positive vesicles were observed with respect to GFPrK2P3.1. Significantly, the number of vesicles positive for internalized channel and the EE marker did not increase in parallel with the increase in total internalized channel. This finding suggests that while endocytosed GFPrK2P3.1 and GFPrK2P9.1 transit through the early endosome, they are rapidly transferred to other parts of the pathway. Indeed, Rab GTPases were used as markers for identifying different endocytic compartments43 and both channels were found to locate within Rab11 positive vesicles. Membrane protein recycling is important for regulating activity of receptors and channels. While some constitutive return to the plasma membrane likely occurs, recycling is also expected to be an important source of channel regulation with Rab11-dependent cargoes previously shown to undergo recycling to the plasma membrane following stimulation.44-46 Acute stimulation of hK2P3.1 endocytosis by the phorbol ester PMA resulted in the appearance of the channel in a transferrin-positive compartment after a 30-min stimulation, but with no overall protein degradation by 60 min.25 This observation would suggest an internal pool of hK2P3.1 that is available for recycling to the plasma membrane given the appropriate stimulus.
It is imperative for cells to carefully regulate the level of expression of leak K+ channels on their cell surface to achieve optimal expression levels for the distinct functions of individual cell types. This is doubtless achieved through the balance of channel delivery to the membrane, internalization and recycling to the cell surface. While studies in recombinant systems in which the movement of tagged channels can be tracked are necessary to our understanding of the general processes at play within these pathways and provide insight into which pathways may recruit these channels; it is crucial to aspire to study these processes in more physiologically relevant settings. To this end, here we report that the human lung carcinoma cell line, A549,47 expresses hK2P3.1 and hK2P9.1 at both the transcript and protein level and show that it can be used as a model system to study the trafficking of endogenous hK2P3.1 and hK2P9.1 using the same methods as for the recombinant system presented in this study. The use of cell surface biotinylation removes the requirement for an external epitope tag and enables the existing commercially available antibodies to be used to analyze endocytosis of endogenous K2P channels. We show that, after a 60-min incubation, both hK2P3.1 and hK2P9.1 can be found in vesicles containing biotin and that the channels are found in a range of endocytic compartments. We observed marked colocalization of both hK2P3.1 and hK2P9.1 with an ER marker, PDI, which is in line with published observations for both channels in recombinant systems.12,20,24 Both channels were observed within the EE and interestingly, a noticeable pool of hK2P3.1 colocalizes with Rab7 in prominently stained vesicles, suggesting that under unstimulated conditions, endocytosed hK2P3.1 may be targeted on a degradative pathway.
These studies not only support the observations drawn from recombinant studies but moreover provide the tools and technology to further investigate both the endocytic pathways and the regulation of entry and transit of TASK channels within these pathways in these and other physiologically relevant cells.
Materials and Methods
Cell culture
HeLa cells were cultured in RPMI/10% FCS; HEK 293 and A549 human lung carcinoma cells were cultured in DMEM/10% FCS. Culture media were routinely supplemented with 50 U/ml penicillin and 50 μg/ml streptomycin; antibiotics were omitted during transfection and subsequent endocytosis experiments. FCS was replaced by low protein growth medium supplement (Nu Serum, BD Biosciences, 355500) for experiments using the inhibitor Dynasore (Sigma-Aldrich, D7693).
Molecular biology
Plasmids encoding GFPrK2P3.1 and GFPrK2P9.1 fusion proteins have been described elsewhere.12 An N-terminal eGFP tag was introduced to human K2P3.1 (GFPhK2P3.1) by ligating a 5′-HindIII-XbaI-3′fragment encoding hK2P3.1 into the plasmid pEGFP-C1 (Clontech, GenBank accession U55763). The identity of the construct was confirmed by restriction digest and DNA sequencing. The plasmid encoding hK2P9.1 with an N-terminal eGFP tag (GFPhK2P9.1) was a gift from Dr. Iain Rowe, Robert Gordon University, Aberdeen. Fusion proteins were expressed transiently after transfection of HeLa cells using jetPEI (Polyplus, 101-10N), as described previously,12 using 3 μg DNA and 6 μl jetPEI per well of a 6-well culture plate (34.8-mm well diameter).
RNA extraction, cDNA synthesis and RT-PCR
Total RNA was extracted from frozen A549 cell pellets using the TRIZOL method. Briefly, cells were lysed in 1 ml TRIZOL (Life Technologies, 15596-026), 200 μl chloroform added and the lysate shaken vigorously for 15 sec. After 5 min at RT, the lysate was centrifuged for 10 min at 12,000 g, 4 °C to separate the mixture into three phases. The top aqueous phase was removed and RNA precipitated in 500 μl isopropanol and 20 μg glycogen for 30 min at −80 °C. The RNA was pelleted and washed twice in 75% ethanol; then the pellet dried for 10 min at RT, before resuspension in 50 μl RNase-free water. The extracted RNA was purified to remove any remaining genomic DNA using the RNeasy Plus Mini Kit (Qiagen, 74134). The RNA obtained was quantified by UV spectrophotometry and stored at −80 °C, before reverse-transcription to cDNA. cDNA synthesis was performed using SuperScript Reverse Transcriptase III (Life Technologies, 18080-044) according to the manufacturer’s protocol, except each reaction contained 1 μg of extracted mRNA, and the random primers were obtained from Promega (C118).
Expression of KCNK3 (hK2P3.1) and KCNK9 (hK2P9.1) was examined using exon-spanning primers: KCNK3 forward: 5′-CAACCTCAGCCAGGGCGGCTAC-3′, and reverse: 5′- GATGAAGCAGTAGTAGTAGGCCTGG-3′ (product 430 bp); KCNK9 forward: 5′- GAAGTACAACATCAGCAGCGAGGAC-3′ and reverse 5′-GGAAGAAGCTCCACTCCTCACACTG-3′ (product 413 bp). Primers designed against the housekeeping gene, transferrin receptor (TFRC; forward: 5′-GCAGAAGCATTATCTTTGCCAG-3′, reverse: 5′-CCCAGTTGCTGTCCTGATATAG-3′, product 252 bp) were used to check the quality of the synthesized cDNA.
RT-PCR reactions were performed using GoTaq DNA polymerase (M300, Promega) Each reaction contained 5 μl template cDNA, 4 μl 5X GoTaq buffer, 0.25 mM dNTP mix, 0.625 μM each of forward and reverse primers, 1.25 U GoTaq DNA polymerase and water to a final volume of 20 μl. The PCR conditions used were initial denaturation at 93 °C for 3 min; 35–40 cycles at 93 °C for 1 min, annealing temperature for 1 min, 72 °C for 1 min; and a final extension step at 72 °C for 10 min.
PCR products were separated using DNA electrophoresis on 1% agarose gels stained with Nancy-520 stain (Sigma-Aldrich 01494). Product bands were cut from the gel and the product extracted using GenElute gel extraction kit (Sigma-Aldrich NA1111). The extracted DNA was sequenced using the following primers: TFRC, 5′-GCAGAAGCATTATCTTTGCCAG-3′; KCNK3, 5′-CTACTTCGCCATCACCGTCA-3′ and KCNK9, 5′-GGAGCTGGTGATCCTGCAGTC-3′.
Antibodies
Antibodies were obtained from the following suppliers and used at the concentrations indicated: Sigma-Aldrich, rabbit anti-K2P3.1 (P0981, 12 μg/ml) and mouse anti-Rab7 (R8779, 4 μg/ml); Santa Cruz, goat anti-K2P9.1 (sc-11320, 10 μg/ml), goat anti-EEA1 (sc-6414, 2 μg/ml), mouse anti-EEA1 (sc-53939, 2 μg/ml), mouse anti-caveolin-1 (sc53564, 2 μg/ml); Abcam, rabbit anti-EEA1 (ab2900, 5 μg/ml) mouse anti-clathrin (ab2731, 6 μg/ml), mouse anti-PDI (ab2792, 10 μg/ml), mouse anti-58 kDa Golgi protein (ab27043, 1 μg/ml); Merck Millipore, mouse anti-Rab11 (05-853, 2.5 μg/ml). Secondary detection reagents were obtained from Life Technologies: Alexa Fluor 488 goat anti-rabbit F(ab′)2 (A-11070), Alexa Fluor 647 goat anti-mouse F(ab′)2, (A-21237), Alexa Fluor 647 chicken anti-rabbit (A-21443), Alexa Fluor 647 chicken anti-goat (A-21469), Alexa Fluor 546-streptavidin (S-11225). DyLight 650-streptavidin was purchased from Pierce (84547). The lysosomal marker was RFP-LAMP1, expressed in HeLa cells after transduction with CellLight BacMam 2.0 baculovirus particles (Life Technologies, C10597; manufacturer’s protocol).
Endocytosis assay
The protocol for the endocytosis assay, based on the method described by Richardson and Mulligan30 has already been described in detail.12 Briefly, sub-confluent HeLa cells on 22 mm coverslips were transfected with DNA encoding GFPrK2P3.1 or GFPrK2P9.1, cultured overnight, then transferred to serum-free medium for 90 min at 37 °C, 5% CO2. Cycloheximide (Sigma-Aldrich, C7698, final concentration 100 μg/ml in DMSO) was added to prevent protein synthesis and the cells returned to the 37°C incubator for a further 30 min. After biotinylation on ice, washes were replaced with 1 ml RPMI / 10% FCS containing 100 μg/ml cycloheximide and coverslips either warmed to 37 °C for a time-course incubation, or immediately placed on ice and stripped to remove external biotin (zero time samples). After 5, 15, 30 or 60 min at 37 °C, coverslips were placed on ice, stripped to remove external biotin, fixed and stained for immunofluorescence microscopy. In experiments to inhibit clathrin-mediated endocytosis, cells in 2 ml serum-free RPMI were preincubated 30 min at 37 °C with 80 μM Dynasore or an equivalent volume, typically 2 μl, of the vehicle DMSO, applied at the same time as the standard cycloheximide protein synthesis blocking step given to all samples. After biotinylation on ice, RPMI containing 10% FCS and 100 μg/ml cycloheximide, plus 80 μM Dynasore or the equivalent volume of DMSO was added to the cells for the duration of the time-course incubation.
Microscopy
Coverslips were mounted in Mowiol and visualized using either a Zeiss Axio Observer D1 or a Leica TCS SP5 confocal scanning microscope in the University of Southampton Biomedical Imaging Unit. Confocal images were acquired using a X63 HCX Plan Apo Lambda Blue NA 1.4 UV oil immersion objective and a pinhole of 1 Airy Unit, with slices every 0.2 μm. Z-stacks were imported by Imaris 7.6 (Bitplane AG) for image analysis. All images shown are whole cell projections, not individual confocal slices, unless stated otherwise. Weakly fluorescent cells were chosen for analysis, to minimize any possible deleterious effects of GFP fusion protein overexpression on the morphology and function of the transfected cells. The Coloc module of Imaris was used to calculate Mander’s coefficients. Where appropriate, regions of interest were defined as transfected (GFP-positive) cells. Next, automatic thresholding of each channel of interest was performed according to38 to exclude false colocalization due to image noise. The software performed simultaneous analysis of randomized images created from the same pixels as the sample image to determine whether the colocalization was statistically significant at the level p ≤ 0.05. Colocalization coefficients were only recorded if the data passed the significance test. P values were typically ≤ 0.01 or ≤ 0.001. For analysis of triple colocalization, a color channel was first constructed representing colocalization of GFPrK2P3.1 or GFPrK2P9.1 with internalized biotin; we assumed this represented endocytosed K2P. Colocalization of this channel with the third marker (e.g., RFP-LAMP1, EEA1, clathrin) was then calculated. Vesicle detection was performed within the Imaris Cell creation wizard. Transfected cells and their nuclei were first identified to enable the calculation of cytoplasm and total cell volume. The creation wizard identifies vesicles as three-dimensional bright spots of user-defined dimensions. We chose 0.6 μm and 1.2 μm as a reasonable approximation of the spot diameters, based on inspection of a large number of images of transfected cells expressing GFPrK2P3.1 and GFPrK2P9.1. Spots were identified using local background subtraction, were assumed spherical and were filtered for “quality” (a parameter related to fluorescence intensity at the center of the spot) according to the following criteria: for 0.6 μm GFPrK2P3.1, GFPrK2P9.1, biotin, clathrin and EEA1 spots, the quality was set manually to 3.0, based on broadly consistent transfection and immunostaining intensities between experiments. For all 1.2 μm spots and where stated for hK2P3.1 and hK2P9.1 in A549 cells, the quality threshold was set automatically by the software. Once vesicles were identified, a MatLab XTension algorithm linked to Imaris was used to map colocalized structures. Colocalized vesicles were those whose distance apart is equal to or less than the radius of an individual vesicle. Another MatLab XTension, “Find Spots Close to Surface,” calculated the distance of each vesicle to the nearest nuclear surface.
Data analysis
All colocalization and vesicle data were first exported to Microsoft Excel for initial calculations, then the graph and statistical analysis software SigmaPlot 12.0 (Systat Software) was used to plot endocytosis data and evaluate significance. Data were first subjected to the Shapiro–Wilk test for normality and. normal populations were analyzed using Student’s t-test. Where appropriate, the Mann–Whitney rank sum test was used instead of Student’s t-test.
Supplementary Material
Acknowledgments
This work was supported by Biotechnology and Biological Sciences Research Council Grant BB-J008168-1 (to I.O.). S.W. is supported by the Gerald Kerkut Charitable Trust. We are grateful to Kelly Wilkinson for technical support and the University of Southampton Biomedical Imaging Unit for provision of confocal imaging facilities.
Glossary
Abbreviations:
- K2P
two pore-domain K+ channel
- TASK
TWIK-related acid-sensitive K+ channel
- GFPrK2P3.1
rat K2P 3.1 tagged with N-terminal GFP
- GFPrK2P9.1
rat K2P9.1 tagged with N-terminal GFP
- CME
clathrin-mediated endocytosis
- CIE
clathin-independent endocytosis
- EE
early endosome
- EEA1
early endosome antigen 1
- PDI
protein disulfide isomerase
- RFP-LAMP1
lysosome-associated membrane protein 1 fused to tagRFP
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/25120
References
- 1.Goldstein SA, Bockenhauer D, O’Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–84. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- 2.Bayliss DA, Barrett PQ. Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci. 2008;29:566–75. doi: 10.1016/j.tips.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 4.Mathie A, Al-Moubarak E, Veale EL. Gating of two pore domain potassium channels. J Physiol. 2010;588:3149–56. doi: 10.1113/jphysiol.2010.192344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci. 1999;2:422–6. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- 6.Buckler KJ. Background leak K+-currents and oxygen sensing in carotid body type 1 cells. Respir Physiol. 1999;115:179–87. doi: 10.1016/S0034-5687(99)00015-8. [DOI] [PubMed] [Google Scholar]
- 7.Buckler KJ, Williams BA, Honoré E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000;525:135–42. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sirois JE, Lei Q, Talley EM, Lynch C, 3rd, Bayliss DA. The TASK-1 two-pore domain K+ channel is a molecular substrate for neuronal effects of inhalation anesthetics. J Neurosci. 2000;20:6347–54. doi: 10.1523/JNEUROSCI.20-17-06347.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayliss DA, Talley EM, Sirois JE, Lei Q. TASK-1 is a highly modulated pH-sensitive ‘leak’ K(+) channel expressed in brainstem respiratory neurons. Respir Physiol. 2001;129:159–74. doi: 10.1016/S0034-5687(01)00288-2. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto Y, Kummer W, Atoji Y, Suzuki Y. TASK-1, TASK-2, TASK-3 and TRAAK immunoreactivities in the rat carotid body. Brain Res. 2002;950:304–7. doi: 10.1016/S0006-8993(02)03181-5. [DOI] [PubMed] [Google Scholar]
- 11.Linden AM, Sandu C, Aller MI, Vekovischeva OY, Rosenberg PH, Wisden W, et al. TASK-3 knockout mice exhibit exaggerated nocturnal activity, impairments in cognitive functions, and reduced sensitivity to inhalation anesthetics. J Pharmacol Exp Ther. 2007;323:924–34. doi: 10.1124/jpet.107.129544. [DOI] [PubMed] [Google Scholar]
- 12.Mant A, Williams S, Roncoroni L, Lowry E, Johnson D, O’Kelly I. N-glycosylation-dependent control of functional expression of background potassium channels K2P3.1 and K2P9.1. J Biol Chem. 2013;288:3251–64. doi: 10.1074/jbc.M112.405167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Cotten JF, Schuyler JA, Fahlman CS, Au JD, Bickler PE, et al. Protective effects of TASK-3 (KCNK9) and related 2P K channels during cellular stress. Brain Res. 2005;1031:164–73. doi: 10.1016/j.brainres.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Lauritzen I, Zanzouri M, Honoré E, Duprat F, Ehrengruber MU, Lazdunski M, et al. K+-dependent cerebellar granule neuron apoptosis. Role of TASK leak K+ channels. J Biol Chem. 2003;278:32068–76. doi: 10.1074/jbc.M302631200. [DOI] [PubMed] [Google Scholar]
- 15.Mu D, Chen L, Zhang X, See L-H, Koch CM, Yen C, et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell. 2003;3:297–302. doi: 10.1016/S1535-6108(03)00054-0. [DOI] [PubMed] [Google Scholar]
- 16.Pei L, Wiser O, Slavin A, Mu D, Powers S, Jan LY, et al. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc Natl Acad Sci U S A. 2003;100:7803–7. doi: 10.1073/pnas.1232448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim CJ, Cho YG, Jeong SW, Kim YS, Kim SY, Nam SW, et al. Altered expression of KCNK9 in colorectal cancers. APMIS. 2004;112:588–94. doi: 10.1111/j.1600-0463.2004.apm1120905.x. [DOI] [PubMed] [Google Scholar]
- 18.Pocsai K, Kosztka L, Bakondi G, Gönczi M, Fodor J, Dienes B, et al. Melanoma cells exhibit strong intracellular TASK-3-specific immunopositivity in both tissue sections and cell culture. Cell Mol Life Sci. 2006;63:2364–76. doi: 10.1007/s00018-006-6166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathie A, Rees KA, El Hachmane MF, Veale EL. Trafficking of neuronal two pore domain potassium channels. Curr Neuropharmacol. 2010;8:276–86. doi: 10.2174/157015910792246146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Kelly I, Butler MH, Zilberberg N, Goldstein SA. Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell. 2002;111:577–88. doi: 10.1016/s0092-8674(02)01040-1. [DOI] [PubMed] [Google Scholar]
- 21.Rajan S, Preisig-Müller R, Wischmeyer E, Nehring R, Hanley PJ, Renigunta V, et al. Interaction with 14-3-3 proteins promotes functional expression of the potassium channels TASK-1 and TASK-3. J Physiol. 2002;545:13–26. doi: 10.1113/jphysiol.2002.027052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Kelly I, Goldstein SAN. Forward Transport of K2p3.1: mediation by 14-3-3 and COPI, modulation by p11. Traffic. 2008;9:72–8. doi: 10.1111/j.1600-0854.2007.00663.x. [DOI] [PubMed] [Google Scholar]
- 23.Mant A, Elliott D, Eyers PA, O’Kelly IM. Protein kinase A is central for forward transport of two-pore domain potassium channels K2P3.1 and K2P9.1. J Biol Chem. 2011;286:14110–9. doi: 10.1074/jbc.M110.190702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Renigunta V, Yuan H, Zuzarte M, Rinné S, Koch A, Wischmeyer E, et al. The retention factor p11 confers an endoplasmic reticulum-localization signal to the potassium channel TASK-1. Traffic. 2006;7:168–81. doi: 10.1111/j.1600-0854.2005.00375.x. [DOI] [PubMed] [Google Scholar]
- 25.Gabriel L, Lvov A, Orthodoxou D, Rittenhouse AR, Kobertz WR, Melikian HE. The acid-sensitive, anesthetic-activated potassium leak channel, KCNK3, is regulated by 14-3-3β-dependent, protein kinase C (PKC)-mediated endocytic trafficking. J Biol Chem. 2012;287:32354–66. doi: 10.1074/jbc.M112.391458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuoka H, Harada K, Nakamura J, Inoue M. Nerve growth factor-induced endocytosis of TWIK-related acid-sensitive K(+) 1 channels in adrenal medullary cells and PC12 cells. Pflugers Arch. 2013 doi: 10.1007/s00424-013-1222-3. In press. [DOI] [PubMed] [Google Scholar]
- 27.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 28.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–12. doi: 10.1038/nrm2216. [DOI] [PubMed] [Google Scholar]
- 29.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 30.Richardson DS, Mulligan LM. Direct visualization of vesicle maturation and plasma membrane protein trafficking. J Fluoresc. 2010;20:401–5. doi: 10.1007/s10895-009-0548-x. [DOI] [PubMed] [Google Scholar]
- 31.van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–63. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh P, McIntosh DP, Schnitzer JE. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J Cell Biol. 1998;141:101–14. doi: 10.1083/jcb.141.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–50. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 34.Hsu VW, Prekeris R. Transport at the recycling endosome. Curr Opin Cell Biol. 2010;22:528–34. doi: 10.1016/j.ceb.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones SA, Morton MJ, Hunter M, Boyett MR. Expression of TASK-1, a pH-sensitive twin-pore domain K(+) channel, in rat myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H181–5. doi: 10.1152/ajpheart.00963.2001. [DOI] [PubMed] [Google Scholar]
- 37.Kovács I, Pocsai K, Czifra G, Sarkadi L, Szucs G, Nemes Z, et al. TASK-3 immunoreactivity shows differential distribution in the human gastrointestinal tract. Virchows Arch. 2005;446:402–10. doi: 10.1007/s00428-005-1205-7. [DOI] [PubMed] [Google Scholar]
- 38.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collinet C, Stöter M, Bradshaw CR, Samusik N, Rink JC, Kenski D, et al. Systems survey of endocytosis by multiparametric image analysis. Nature. 2010;464:243–9. doi: 10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- 40.Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–24. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol. 2010;191:615–29. doi: 10.1083/jcb.201003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–94. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 43.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–25. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 44.Vossenkämper A, Nedvetsky PI, Wiesner B, Furkert J, Rosenthal W, Klussmann E. Microtubules are needed for the perinuclear positioning of aquaporin-2 after its endocytic retrieval in renal principal cells. Am J Physiol Cell Physiol. 2007;293:C1129–38. doi: 10.1152/ajpcell.00628.2006. [DOI] [PubMed] [Google Scholar]
- 45.Seebohm G, Strutz-Seebohm N, Birkin R, Dell G, Bucci C, Spinosa MR, et al. Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ Res. 2007;100:686–92. doi: 10.1161/01.RES.0000260250.83824.8f. [DOI] [PubMed] [Google Scholar]
- 46.Balse E, El-Haou S, Dillanian G, Dauphin A, Eldstrom J, Fedida D, et al. Cholesterol modulates the recruitment of Kv1.5 channels from Rab11-associated recycling endosome in native atrial myocytes. Proc Natl Acad Sci U S A. 2009;106:14681–6. doi: 10.1073/pnas.0902809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–23. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.