

Abstract
In recent years, cholesterol has been emerging as a major regulator of ion channel function. We have previously shown that cholesterol suppresses Kir2 channels, a subfamily of constitutively active strongly rectifying K+ channels. Furthermore, our earlier studies have shown that cholesterol sensitivity of Kir2 channels depends on a group of residues that form a belt-like structure around the cytosolic pore of the channel in proximity to the transmembrane domain. In this study, we focus on the contributions of different structural domains of Kir2 channels in the regulation of their cholesterol sensitivity. Focusing on the mildest mutation in the sensitivity belt, L222I, we show that the sensitivity of the channel to cholesterol can be restored by crosstalk between three distinct cytosolic regions: the C-terminal CD loop, the EF and GA loops of the C-terminus, and the βA sheet of the N-terminus. Thus, in addition to the importance of residues that affect the cytosolic G-loop gate in the sensitivity of Kir2 channels to cholesterol, our data suggest an important role to the interactions at the interface between the channel’s N- and C- termini that couple the intracellular domains of its four subunits during gating.
Keywords: inwardly rectifying potassium channels, cholesterol, lipids, Kir2.1, IRK1
Introduction
Cholesterol is one of the major lipid components of the plasma membrane in all mammalian cells, constituting up to 45 mol percent with respect to other membrane phospholipids, and playing critical roles in cell growth and function.1,2 Increased levels of cholesterol, however, have been associated with multiple pathological conditions including the development of cardiovascular disease.3-5 In recent years, it has been demonstrated to regulate the function of almost all known families of ion channels including K+ channels, Ca2+ channels, Na+ channels and Cl- channels, as described in detail in several recent reviews.6-8 For the majority of the channels, increased levels of membrane cholesterol resulted in decreased channel activity due to a decrease in the open probability, in the unitary conductance and/or in the number of active channels in the membrane.8-13 Yet, the mechanisms of cholesterol regulation of ion channel function are only starting to emerge.
Here we focus on the constitutively active inwardly rectifying potassium (Kir) channel Kir2.1 that is critically involved in regulation of the excitability and contraction in a variety of cell types and in maintaining membrane potential in several types of non-excitable cells.14-16 We have previously shown that cholesterol suppressed Kir2.1 whole-cell currents without a significant effect on the channel’s unitary conductance, open probability or on its surface expression suggesting that cholesterol stabilizes Kir2 channels in a closed “silent” state, providing initial mechanistic insights into the regulation of Kir2 channels by cholesterol.9,17
Our earlier studies have shown that mutations of four residues (N216D, K219Q, H221M and L222I) in the cytosolic CD loop that faces the pore of the channel (Fig. S1 for the location of the CD loop) affect the sensitivity of the channel to cholesterol.18-21 Among these, the mutation of L222 to its isomer isoleucine completely abrogated the sensitivity of the channel to cholesterol. Similarly, the reverse I214L mutation at the corresponding position in Kir2.3 resulted in increased sensitivity of Kir2.3 to cholesterol.18 Interestingly, in Kir3.4*, the highly active Kir3.4 pore mutant S143T,22 which is enhanced by cholesterol, the I229L mutation at the equivalent position (see the alignment of representative Kir2 and Kir3 channels in Figure S1A) abrogated the sensitivity of Kir3.4* to cholesterol, suggesting that the importance of this position in cholesterol modulation of channel function extends beyond the Kir2 family.19 Interestingly, the same CD loop residues also play a critical role in regulating the function of different Kir channels by other modulators including the phosphoinositide PI(4,5)P2,23-25 which is required for Kir channel activation,8,23,26-28 and intracellular sodium.29-32
We have previously shown that in addition to Kir2.1, also other members of the Kir2 subfamily were also suppressed by cholesterol.9 However, whereas the sensitivity of Kir2.1 and Kir2.2 to cholesterol was similar, Kir2.3 and Kir2.4 exhibited decreased sensitivity to cholesterol. Using this difference in cholesterol sensitivities as a tool to further study the mechanisms of cholesterol modulation of this subfamily of inwardly rectifying potassium channels, we recently showed that L222 and the three additional CD loop residues listed above were a part of a regulatory site that also included residues in the G-loop (C311), the N-terminus (D51, H53), and the connecting segment between the C-terminus and the inner transmembrane (TM) helix (E191, V194).20,21 Together these residues formed a cytosolic belt that surrounded the pore of the channel close to its interface with the TM domain, and modulated the sensitivity of the channel to cholesterol.20,21 Moreover, our analysis suggested that the underlying reason for the clustering of these residues in proximity to the membrane was the importance of the cholesterol sensitivity belt residues for Kir channel gating. More specifically, within the cytosolic domain, the major gate of Kir2.1 is located at the G-loop,33 which is regarded as the main region of the cytoplasmic pore at which dilation and contraction occurs. Examination of a database of crystallographic structures that include the cytosolic domains of eukaryotic Kir channels suggested that there is correlation between residues of the cholesterol sensitivity belt and residues located in the most flexible region of the G-loop, i.e., its apex.20 In contrast, no significant correlation was found between the G-loop apex residues and residues that were just outside the selectivity belt.20
More recently, using a computational-experimental approach that combines all-atom full-membrane molecular dynamics simulations with site-directed mutagenesis and electrophysiology, we have shown that the N251D mutation restores the sensitivity of the L222I mutant to cholesterol.34 Notably, whereas the CD loop residue, L222, is located close to the interface of the C-terminus with the TM domain, N251 is located further away from the TM domain in the EF loop, ~24Å from L222 (Fig. S1). Furthermore, the same N251D mutation also restored the strength of the interaction of the L222I mutant with PI(4,5)P2.34 These data indicated that a long-distance functional intra-connection within the C-terminus plays an important role in the modulation of Kir2 channels by both cholesterol and PI(4,5)P2.
In the current study, we show that the sensitivity of the channel to cholesterol depends on functional inter-links between the N- and C- termini that couple the motion of the intracellular domains of four subunits of the channel during gating, and that these connections can switch the sensitivity of the channel to cholesterol on or off.
Results
As noted in the Introduction, we have previously used the differential sensitivities of Kir2.1 and Kir2.3 to cholesterol as a tool to identify a group of residues that formed a cholesterol sensitivity belt. Mutations of residues in the sensitivity belt to the corresponding residues in Kir2.3 significantly reduced or abrogated the sensitivity of Kir2.1 to cholesterol.20,21 Surprisingly, however, each of the mutations of the five residues of the cholesterol sensitivity belt that abrogated the sensitivity of Kir2.1 to cholesterol (H53Q, E191Q, V194L, L222I and C311A) also converted these residues to the corresponding residues in Kir2.2. This was unexpected because, as noted above, the sensitivity of Kir2.2 to cholesterol is similar to that of Kir2.1.9 We thus hypothesized that the combination/interaction of each of these positions with other residues that are distinct between Kir2.1 and Kir2.2 underlie the sensitivity of Kir2.2 to cholesterol. And indeed, as described in the Introduction, our recent study has shown that the loss of cholesterol sensitivity of Kir2.1 channel that results from the L222I mutation can be restored by the N251D mutation of a distant C-terminal residue to the equivalent residue in Kir2.2.34 However, comparison between the sequences of the modeled cytosolic domains of Kir2.1 and Kir2.2 revealed over 40 additional differences between the two channels. Thus, in order to uncover further connections between different cytosolic regions in Kir2.1 that control channel modulation by cholesterol, we studied the role of these differences between Kir2.1 and Kir2.2 in the channel’s cholesterol sensitivity.
Cross-talk between two distant cytosolic loops of the C-terminus in cholesterol sensitivity of Kir2.1
We first concentrated on the sequence differences in the modeled C-termini of Kir2.1 and Kir2.2. Focusing on the most conservative mutation that abrogates cholesterol sensitivity, the mutation of the leucine at position 222 to its isomer, isoleucine, we investigated whether in addition to the N251D mutation, mutations of other C-terminal residues in Kir2.1 to the corresponding residues in Kir2.2 could also restore the sensitivity of L222I to cholesterol.
Comparison of the sequences of the modeled C-termini of Kir2.1 and Kir2.2 revealed 32 differences in addition to the differences between the channels in positions 222 and 251 (Fig. 1A and 1B). In order to investigate the role of these differences in determining the sensitivity of the L222I mutant to cholesterol, we divided the C-terminal residues that differ between Kir2.1 and Kir2.2 into groups (Fig. 1A and 1B), and made multiple point mutations of all the residues in Kir2.1 within each group, mutating each of them to the equivalent residues in Kir2.2. As can be seen in Figure 1C, in addition to the N251D mutation (group 3C), also the multiple mutant of group 6C restored the sensitivity of the L222I mutation.

Figure 1. (A) Sequence alignment of the modeled C-terminus of Kir2.1 with the equivalent residues in Kir2.2. Highlighted in black on the sequence alignment are segments that include residues that differ between Kir2.1 and Kir2.2. (B) The C-terminus of one subunit of a model of Kir2.1 based on the cytosolic structure of Kir2.1 (PDB accession number 1U4F) and the TM domain of KirBac3.1 (PDB accession number 1XL4) showing segments 1–8 that correspond to the groups of residues in Figure 1A. The colors correspond to the colors in the alignment in Figure 1A. (C) Whole-cell basal currents at –80mV showing the effect of cholesterol enrichment on Kir2.1, on the L222I mutant and on each of the multiple mutants 1C-8C described in Figure 1A on the background of L222I to the corresponding segments in Kir2.2 (n = 12–43). Significant difference is indicated by an asterisk (*p < 0.05).
Within group 6C, 8 residues in the 280–290 segment of the sequence of Kir2.1 were converted to the equivalent residues in Kir2.2 (Fig. 1A and 1B). In order to identify the mutation(s) responsible for restoring the sensitivity of the L222I mutant to cholesterol, we divided group 6C into two groups, 6Ca (segment 280–283) and 6Cb (segment 284–290) (Fig. 1A). Our data show that mutations of the 3 residues of group 6Ca that differ between Kir2.1 and Kir2.2 restored the cholesterol sensitivity of L222I whereas mutations of the 5 residues of group 6Cb did not (Fig. 2A). We thus tested which of these 3 mutations restored the sensitivity of the L222I mutant to cholesterol. As can be seen in Figure 2B and 2C, mutations of two residues in the GA loop (Fig. S1), Y280F and L282I, reversed the effect of the L222I mutation on the sensitivity of Kir2.1 to cholesterol. Accordingly, both L222I_Y280F and L222I_L282I exhibited a significant decrease in currents following cholesterol enrichment. In contrast, the D281G mutation did not restore the sensitivity of L222I to cholesterol and was insensitive to cholesterol enrichment.

Figure 2. (A and B) Whole-cell basal currents at –80mV showing the effect of cholesterol enrichment on Kir2.1 and on each of the multiple mutants 6Ca and 6Cb on the background of L222I (A) (n = 7–43) and the mutants L222I_Y280F, L222I_D281G and L222I_L282I (B) (n = 7–16). Significant difference is indicated by an asterisk (*p < 0.05). (C) Representative traces of whole-cell basal currents recorded at -80mV/+80mV for L222I_Y280F, L222I_D281G and L222I_L282I mutants in control and in cholesterol-enriched cells. (D) A model of the C-terminus of the cytosolic domain of one subunit of Kir2.1 showing L222 (yellow) in the CD loop, N251 (red) in the EF loop and Y280 (magenta) and L282 (pink) in the GA loop. (E) Surface presentation of the cytosolic domain of Kir2.1 showing the reversal residue chain (dark gray), L222I (yellow), and N251 (red), Y280 (magenta) and L282 (pink).
Recently we analyzed the effect of the L222I and N251D mutations by all-atom full-membrane molecular dynamics (MD) simulations.34 In order to identify structural features that are reversed by the N251D mutation, we compared MD simulations of Kir2.1-WT, L222I, N251D and L222I_N251D. These simulations indicated that the N251D mutation that reversed the effect of L222I on cholesterol sensitivity of the channel, also reversed the effect of the L222I mutation on the distances between the backbone central carbon (Cα) of position 222 and the Cα atoms of a chain of residues in the C-terminus of the channel. This chain of residues linked the residues in the vicinity of L222 to the region proximal to position 251, and from there - continued to surround the cytosolic domain of the channel. As can be seen in Figures 2D and 2E, both Y280 and L282 that restore the sensitivity of the L222I mutant to cholesterol, are located in the vicinity of N251 away from the TM domain. Moreover, L282 is a part of the C-terminal reversal residue-chain.
With the Y280F and L282I mutations each restoring the sensitivity of the L222I mutant to cholesterol, we next examined the effect of each of the mutations on the sensitivity of the WT Kir2.1 protein to cholesterol. As can be seen in Figure 3, both residues abrogate cholesterol sensitivity of Kir2.1 wild type channels when introduced as single mutations further corroborating the importance of these residues in determining the sensitivity of the channel to cholesterol.
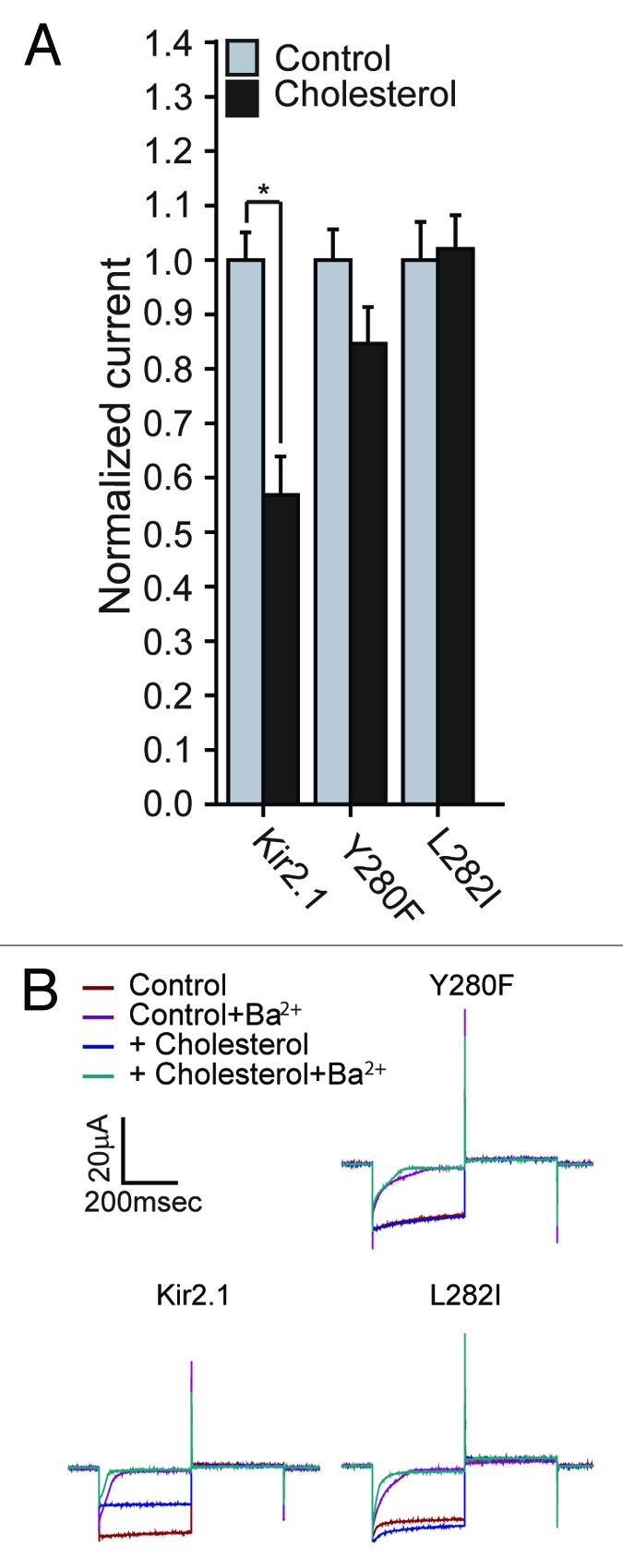
Figure 3. (A) Whole-cell basal currents recorded in Xenopus oocytes at -80mV showing the effect of cholesterol enrichment on Kir2.1 and the mutants Y280F and L282I (n = 11–18). Significant difference is indicated by an asterisk (*p < 0.05). (B) Representative traces of whole-cell basal currents recorded at –80mV/+80mV in Xenopus oocytes showing the effect of cholesterol enrichment on Kir2.1 and the mutants Y280F and L282I. The waveform and the coloring scheme are the same as in Figure 2C.
Cross-talk between C- and N-termini in cholesterol sensitivity of Kir2.1
In addition to the interface between neighboring C-termini of different channel subunits, each C-terminus in Kir channels has an interface with the N-terminus of the adjacent subunit. This interface has been shown to mediate a process of conformational change that extends through the subunits of the channel.35
As noted above, our recent computational analysis of the effect of the L222I and N251D mutations indicated that the N251D mutation reversed the effect of the L222I mutation on the distances between the backbone central carbon (Cα) of position 222 and the Cα atoms of a chain of C-terminal residues.34 Thus, using all-atom molecular dynamics simulations, we tested whether the N251D mutation also reversed the effect of the L222I mutation on the distances between the backbone central carbon (Cα) of position 222 and the Cα atoms of residues in the N-terminus. Our analysis shows that within the modeled N-terminus, the N251D mutation reversed the effect of the L222I mutation on the distance between the backbone central carbon of position 222 and those of 8 residues in the N-terminus of the channel including K49, F58, I59, N60, V61, G62, E63 and R67 (Fig. 4A and 4B). As can be seen in Figure 4B, this N-terminal reversal chain (shown in green sphere presentation) links to the C-terminal reversal chain (shown in dark gray sphere presentation) that we identified earlier.34 Moreover, a major part of the N-terminal reversal chain engulfed R218 at the tip of the CD loop, and which belongs to the C-terminal reversal chain (Fig. 4C). We thus hypothesized that the N-terminus plays an important role in the sensitivity of the channel to cholesterol.

Figure 4. (A) N-terminal residues for which the direction of the changes in the distances relative to position 222 following the L222I mutation is reversed in L222I_N251D. The effect of the L222I mutation on the distances between the Cα atoms of position 222 and the Cα atoms of these residues with respect to the WT is depicted as blue squares whereas the effect of the L222I_N251D mutation compared with the L222I mutation is depicted by the green triangles. The effect of the L222I_N251D mutation compared with the WT channel is also shown as red diamonds. (B) Surface presentation of the cytosolic domain of Kir2.1 showing the N-terminal reversal residue chain (green), Also shown are the C-terminal reversal residue chain (dark gray), L222 (yellow), and N251 (red). (C) Model showing a Close-up of the residue reversal chain residues in the vicinity of L222 (yellow) of the CD loop. The CD loop is shown in light yellow. CD loop residues included in the reversal chain are shown in light yellow. Other C-terminal reversal chain residues in proximity of L222 are shown in dark gray. N-terminal reversal chain residues in the adjacent subunit are shown in green. (D) Sequence alignment of residues 40–68 located in the modeled N-terminus of Kir2.1 with the equivalent residues in Kir2.2 (39–67). Highlighted in black on the sequence alignment are segments that include residues that differ between Kir2.1 and Kir2.2. (E) Whole-cell basal currents recorded in Xenopus oocytes at –80mV showing the effect of cholesterol enrichment on Kir2.1, on the L222I mutant and on each of the multiple mutants 1N-3N that include mutations of the segments shown in Figure 4D on the background of L222I to the corresponding segments in Kir2.2 (n = 12–26). Significant difference is indicated by an asterisk (*p < 0.05).
In order to examine this hypothesis, we investigated the effect of the sequence differences in the modeled N-terminus between Kir2.1 and Kir2.2 on the cholesterol sensitivity of the L222I mutant, employing a similar strategy to the one we used to screen the differences between the C-termini of the two channels. We thus divided the residues in the structure into 3 groups as shown in Figure 4D. As Figure 4E shows, only the multiple mutant of group 3N restored the sensitivity of the L222I mutation. This group converted 6 residues in the 56–65 N-terminal segment of Kir2.1 to the equivalent residues in Kir2.2. Dividing this group into two groups as shown in the alignment in Figure 4D, we show in Figure 5A that mutations of the residues in group 3Na of the 56–59 segment of Kir2.1 restored the sensitivity of the Kir2.1_L222I to cholesterol whereas mutations of the 3 residues of group 3Nb (segment 60–65) did not. Within group 3Na, I59A did not restore the sensitivity of the L222I mutant to cholesterol. On the other hand, both the V56I and the Q57E mutations reversed the effect of the L222I mutation and restored the sensitivity of the channel to cholesterol (Fig. 5B and C). Accordingly, both L222I_V56I and L222I_Q57E exhibited a significant decrease in currents following cholesterol enrichment. V56 and Q57 are located in the N-terminus in proximity to the TM domain (Fig. 5D and 5E). Moreover, this pair of residues is located in the βA sheet of the N-terminus that interfaces the βM sheet of the C-terminus of an adjacent subunit (Fig. 5E and 5F; Fig. S1).
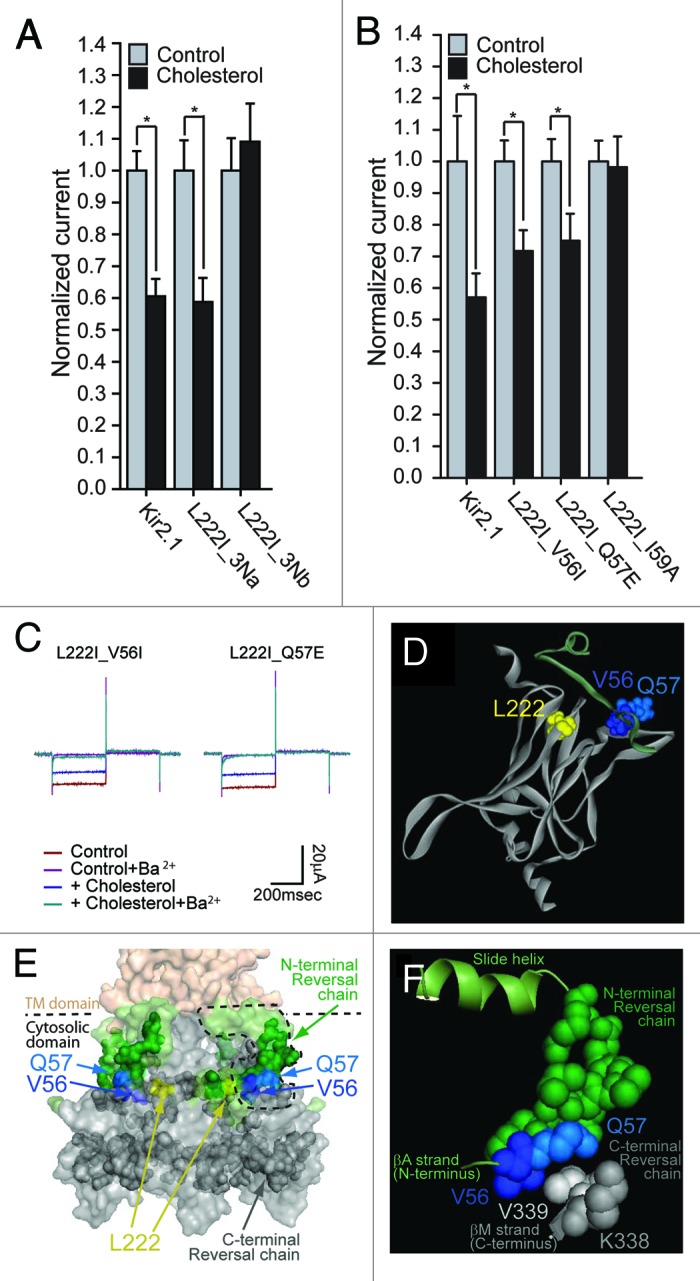
Figure 5. (A) Whole-cell basal currents recorded in Xenopus oocytes at –80mV showing the effect of cholesterol enrichment on Kir2.1 and on each of the multiple mutants 3Na and 3Nb on the background of L222I that include mutations of the segments shown in Figure 4D to the corresponding segments in Kir2.2 (n = 12–43). (B) Whole-cell basal currents recorded in Xenopus oocytes at –80mV showing the effect of cholesterol enrichment on Kir2.1 and the mutants L222I_V56I, L222I_Q57E and L222I_I59A (n = 7–26). Significant difference is indicated by an asterisk (*p < 0.05). (C) Representative traces of whole-cell basal currents recorded at –80mV/+80mV in Xenopus oocytes showing the effect of cholesterol enrichment on the mutants L222I_V56I and L222I_Q57E. The waveform and the coloring scheme is the same as in Figure 2C. (D) A model of the cytosolic domain of one subunit of Kir2.1 showing L222 (yellow) in the CD loop, and the modeled N-terminus of an adjacent subunit showing V56 (blue) and Q57 (light blue) in the βA strand. (E) Surface presentation of the cytosolic domain of Kir2.1 showing the N-terminal reversal chain (green), V56 (blue) and Q57 (light blue). Also shown are the C-terminal reversal residue chain (dark gray) and L222I (yellow). (F) Enlargement of the dashed region in Figure 5E showing V56 (blue) and Q57 (light blue) of the βA strand located in between the N-terminal reversal chain (green) and two of the C-terminal reversal chain residues of the βM strand, K338 (dark gray) and V339 (light gray).
Furthermore, as described above, the C-terminal mutations that restore cholesterol sensitivity of L222I mutant (N251D, Y280F and L282I), also abrogate the sensitivity of the wild type Kir2.1 channel to cholesterol. We thus tested whether this is also the case for the pair of N-terminal mutants that restore the sensitivity of L222I to cholesterol. As Figure 6 shows, also V56I and Q57E abrogate the sensitivity of the wild type Kir2.1 channel to cholesterol.
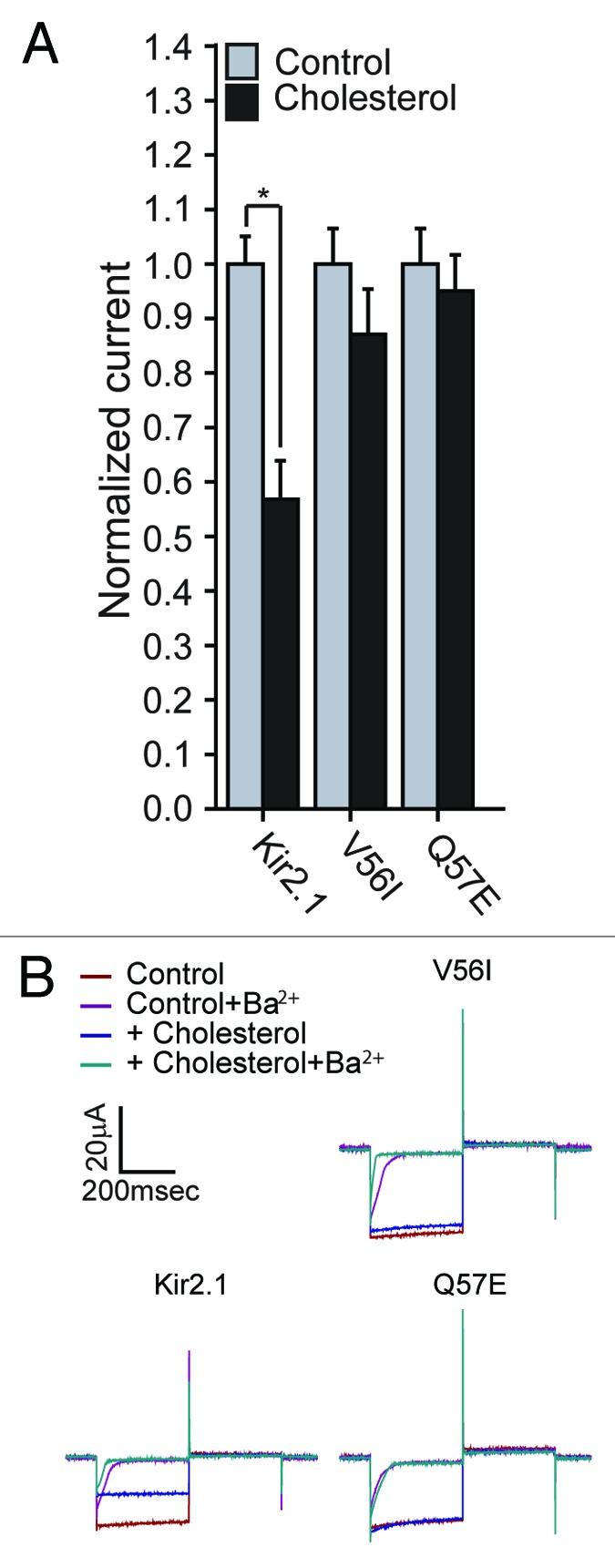
Figure 6. (A) Whole-cell basal currents recorded in Xenopus oocytes at –80mV showing the effect of cholesterol enrichment on Kir2.1 and the mutants V56I and Q57E (n = 7–18). Significant difference is indicated by an asterisk (*p < 0.05). (B) Representative traces of whole-cell basal currents recorded at –80mV/+80mV in Xenopus oocytes showing the effect of cholesterol enrichment on Kir2.1 and the mutants V56I and Q57E. The waveform and the coloring scheme are the same as in Figure 2C.
Discussion
In this study, we identified multiple new functional links between distant non-interacting residues in both the C- and N-termini that control the modulation of Kir2 channels by cholesterol.
Our recent study demonstrated that cholesterol sensitivity of Kir2.1 channels depends on the reciprocal interaction between the L222 residue in the CD-loop of the C-terminus and the N251 residue in the EF-loop even though there is no direct interaction between the two.34 Furthermore, all-atom full-membrane molecular dynamics simulations suggested that these two distant residues are connected by a reversal chain of residues, as defined by their opposite movements as a result of L222I and N251D mutations.34 In this study, we identified additional residues that are coupled to L222 and demonstrate the important role of crosstalk between distinct cytosolic domains in determining the sensitivity of the channel to cholesterol.
Specifically, comparing the differences in the sequences of the modeled C-termini of Kir2.1 and Kir2.2, we identified a pair of C-terminal residues (Y280 and L282) whose mutations both reverse the effect of L222I and abrogate the sensitivity of the WT Kir2.1 channel to cholesterol. Both residues are located in the vicinity of N251 further away from the TM domain in the pore facing GA loop that connects between the G-β-strand and the A-α-helix of the C-terminus. Thus, it is likely that these residues affect the sensitivity of the channel to cholesterol by a similar mechanism to the mechanism that underlies the effect of N251D on cholesterol sensitivity, further reinforcing the importance of this C-terminal region for cholesterol sensitivity.
It is also interesting to note that the same region of the channel has been recently implicated in the regulation of channel’s gating kinetics by PI(4,5)P2.36 Specifically, it has been shown that mutation of E272, which is located between N251 and Y280/L282, affects both the on- and off- gating kinetics of the channel. Moreover, we have recently shown that in addition to reversing the effect of the L222I mutation on the sensitivity of Kir2.1 to cholesterol, N251D also restores the strength of the interaction of the L222I mutant with PI(4,5)P2 to the levels exhibited by the WT Kir2.1 channel.34 It is therefore likely that the flexibility of this region that has been suggested to play a critical role in the regulation of the channel’s gating kinetics by PI(4,5)P2,36 is also important for determining the sensitivity of the channel to cholesterol.
Recently, molecular dynamics simulations have shown that in a Kir3.1 chimera, interaction patterns that involve N-terminal residues are altered when the cytosolic gate of the channel, the G-loop, is dilated in the presence of PI(4,5)P2.37 It was thus suggested that the N-terminus controls the opening of the G-loop gate by coordinating the interactions between the CD loop and the G-loop. One of the specific interactions that differed between the constricted and dilated conformations involved the equivalent residues to R67 and R218 of Kir2.1. Our analysis showed that similarly to R218, which is in the C-terminal reversal chain,34 R67 of the slide helix is included in the N-terminal reversal residue chain (Fig. 4A). This suggests that the N-terminal reversal chain includes key residues for channel gating. As discussed in the introduction, our earlier studies have suggested that cholesterol stabilizes the channel in the closed “silent” state.9,17 We thus hypothesized that the N-terminus plays an important role in the sensitivity of the channel to cholesterol.
And indeed, we identified two N-terminal residues (V56 and Q57) whose mutations both restore the cholesterol sensitivity of the Kir2.1_L222I channel as well as abrogate the cholesterol sensitivity of the WT Kir2.1 channel, suggesting new mechanistic insights into the role of the cytosolic domain in cholesterol sensitivity of the channels. More specifically, this pair of residues is located in the βA sheet of the N-terminus that interfaces the βM sheet of the C-terminus of an adjacent subunit (Fig. 5E and F; Fig. S1). At the residue level, V56 and Q57 are located across from V339 of the βM sheet (Fig. 5F). Recent MD simulations of a Kir3.1 chimera suggested that the interaction pattern between the equivalent residues to Q57 and V339 is altered when the cytosolic domain is dilated in the presence of PI(4,5)P2.37 Notably, V339 of the βM sheet along with its neighboring residue K338 are a part of the C-terminal reversal chain.34 This suggests that the cholesterol sensitivity of Kir2 channels depends not only on the interactions between residues within the same subunit of the channels but also on the interactions between the N- and the C-termini of adjacent subunits. Recent analysis of a series of crystal structures of bacterial KirBac channels has shown that parallel β sheet interactions formed between βA in one subunit and βM of an adjacent subunit couple the motion of each subunit to its neighbor, thus enabling systematic reorientation of the intracellular domain of the channel between its closed (latched) and open (unlatched) conformations.35 Our data demonstrating that the V56I and Q57E mutations that may each affect the hydrogen-bonding network between the βA and βM sheets of adjacent subunits play an important role in the modulation of the channel by cholesterol. This suggests that the interactions between the subunits that couple their coordinated motion are important for this process. This observation is consistent with our previous observation that the L222I mutation has a dominant negative effect on cholesterol sensitivity of the channel,18 and implies that more than one subunit is required for cholesterol to suppress channel activity.
We have previously shown that within the N-terminus, both D51N and H53Q affected the sensitivity of Kir2.1 to cholesterol, and are a part of the cholesterol sensitivity belt of the channel.20 Specifically, D51N decreased the sensitivity of Kir2.1 to cholesterol and H53Q abrogated the channel’s cholesterol sensitivity. However, despite the proximity of positions 51 and 53 to position 56 and 57, these mutations do not restore the sensitivity of the L222I mutant to cholesterol (Fig. 4D and 4E, group 2N). Furthermore, whereas D51 and H53 were both correlated with the C-terminal gate of the channel, the G-loop, Q57 was not.20 These differences between the 51/53 pair and the 56/57 pair indicate that the role of these two pairs in the mechanism underlying channel regulation by cholesterol is different. Thus, the ability of the L222I_V56I and L222I_Q57E double mutants to restore the sensitivity of the corresponding single mutants highlights a unique functional inter-link between the C-terminal L222 and the N-terminus at the neighboring residues V56 and Q57, which is independent of the relationship between the G-loop and the modulation of the channel by cholesterol.
In summary, it is likely that the observations presented here are a result of the intricate relationship between three distinct cytosolic regions and their interrelated roles in the gating mechanism of the channel. Specifically, our study linked the following cytosolic regions of the channel: (a) the CD loop which is in close proximity to the cytosolic G-loop gate, (b) the EF and GA loops of the C-terminus that are distant from the TM domain, and (c) the βA sheet of the N-terminus at the interface with the C-terminus. Accordingly, cholesterol regulation of Kir2 channels depends on two cytosolic constituents: (a) the cytosolic gating machinery that controls the conformational changes at the cytosolic G-loop gate of each subunit, as was shown in our earlier studies,20 and (b) the interactions at the interface between the N- and C- termini of the channel that couple the motion of the intracellular domains of its four subunits during gating.
Materials and Methods
Expression of recombinant channels and electrophysiological recordings in Xenopus oocytes
Point mutations were generated using the Quickchange site-directed mutagenesis kit (Stratagene). cRNAs were transcribed in vitro using the “Message Machine” kit (Ambion). Oocytes were isolated and microinjected as previously described.38 Expression of channel proteins in Xenopus oocytes was accomplished by injection of the desired amount of cRNA. Oocytes were injected with 0.5ng cRNA of the channel. All oocytes were maintained at 17°C. Two-electrode voltage clamp recordings were performed 1 d following injection. Whole-cell currents were measured by conventional two-microelectrode voltage clamp with a GeneClamp 500 amplifier (Axon Instruments), as previously reported.20,38 Basal currents represent the difference of inward currents obtained (at -80 mV) in the presence of 3 mM BaCl2 in HK solution from those in the absence of Ba2+. A minimum of two batches of oocytes were tested for each normalized recording shown. Recordings from different batches of oocytes were normalized to the mean of whole-cell basal currents obtained from control untreated oocytes. The mean of each batch of control untreated oocytes was normalized to one. Statistics (i.e., mean and standard error of the mean) of each construct were calculated from all of the normalized data recorded from different batches of oocytes.
Cholesterol enrichment of Xenopus oocytes
Treatment of Xenopus oocytes with a mixture of cholesterol and lipids has been shown to increase the cholesterol/phospholipid molar ratio of the plasma membrane of the oocytes.39 Thus, in order to enrich the oocytes with cholesterol we used a 1:1:1 (wt:wt:wt) mixture containing cholesterol, porcine brain L-α-phosphatidylethanolamine (PE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (PS) (Avanti Polar Lipids), as described in our earlier studies.20,34 We have shown that the effect of cholesterol on Kir2.1 channels expressed in Xenopus Oocytes is similar to the effect of cholesterol enrichment on Kir2.1 channels transfected into both CHO and HEK cells.18,34 Moreover, these data were in agreement with the effect of cholesterol on native Kir2 channels in aortic endothelial cells40 and in atrial myocytes.41 In addition, we have recently shown that cholesterol treatment of Xenopus oocytes expressing several Kir2.1 mutants including L222I reproduces the effect of cholesterol on the corresponding mutants in HEK cells.34 Therefore, as control and to confirm the quality of each cholesterol preparation for each set of mutants, we tested the effect of cholesterol on Xenopus oocytes that express the wild-type Kir2.1 channel.
Molecular dynamics simulations
The model of the channel used (KDB database ID H011)42 was based on the chimera between the cytosolic domain of Kir3.1 and the TM domain of KirBac1.3 (PDB ID 2QKS; resolution 2.2Å).43 Comparison between the crystal structures33 of the cytosolic domains of Kir2.1 (PDB ID 1U4F, 2.41Å resolution) and Kir3.1 (PDB ID 1U4E, 2.09Å resolution) shows that the structural similarity between the cytosolic domains of the two eukaryotic inwardly rectifying potassium channels is high, with a RMSD of only 1.1Å between the backbone Cα atoms. The simulation system has been constructed using membrane-insertion protocol developed within CHARMM-GUI project,44 as described in our recent study.34 Briefly, the simulation box contains the channel, bound K+ ions in the sites S2:S4 of the selectivity filter and 161 POPC lipid molecules solvated in an explicit 150 mM KCl aqueous solution represented with TIP3 water model45 and optimized CHARMM-27 ion parameters.46 All computations were performed by NAMD version 2.7b147 and analysis was done with CHARMM version c36b2 with the CHARMM-27 force-fields for proteins and lipids.48 The protein structure has been minimized in cycles with gradual decrease of harmonic restraints on heavy atoms. The minimized structures were embedded next into a lipid membrane using a multi-step membrane building procedure used in previous studies.34 MD simulation methods used here are similar to those used in previous studies of membrane systems utilizing NPAT ensemble. Briefly, constant temperature/constant pressure algorithms were applied (with pressure at 1 atm and temperature at 303 K). Electrostatic interactions were treated with the Particle Mesh Ewald (PME) algorithm with a 92 Å/92 Å/144 Å grid for fast Fourier transform. The temperature and pressure in the system were maintained with a combination of Langevin thermostat acting on heavy atoms of the lipid bilayer with Langevin dumping parameter of 5 ps−1 and a Nose-Hoover Langevin piston method. The piston oscillation period was set to 100 fs with a dumping time of 50 fs. The non-bonded interactions were smoothly switched off at 10–13.5 Å. All simulation systems were equilibrated for 1 ns each without any constraints and the production was run for another 10 ns. Figures of models were made using the PyMOL Molecular Graphics System, Version 1.1 (Schrödinger, LLC). Analysis was performed using the Visual Molecular Dynamics package (VMD),49 and the Data Analysis and Graphing Software Origin (OriginLab).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Heikki Vaananen and Sophia Gruszecki (VCU) for oocyte preparation. This work was supported by a Scientist Development Grant (11SDG5190025) from the American Heart Association (to AR-D), and by the US National Institutes of Health (NIH) grants HL-073965, HL-083298 (to IL), and HL-059949 (to DEL). SN was supported by operating funds from the Canadian Institutes of Health Research-CIHR (MOP-186232) and Heart and Stroke Foundation of Alberta and NWT. SN is an Alberta Heritage Foundation for Medical Research (AHFMR) Scholar.
Supplemental Material
Supplemental material may be downloaded here: http://www.landesbioscience.com/journals/channels/article/25437/
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/25437
References
- 1.Yeagle PL. Cholesterol and the cell membrane. Biochim Biophys Acta. 1985;822:267–87. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 2.Yeagle PL. Modulation of membrane function by cholesterol. Biochimie. 1991;73:1303–10. doi: 10.1016/0300-9084(91)90093-G. [DOI] [PubMed] [Google Scholar]
- 3.Kruth HS. Lipoprotein cholesterol and atherosclerosis. Curr Mol Med. 2001;1:633–53. doi: 10.2174/1566524013363212. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 5.Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–7. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]
- 6.Levitan I. Cholesterol and Kir channels. IUBMB Life. 2009;61:781–90. doi: 10.1002/iub.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maguy A, Hebert TE, Nattel S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc Res. 2006;69:798–807. doi: 10.1016/j.cardiores.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Rosenhouse-Dantsker A, Mehta D, Levitan I. Regulation of ion channels by membrane lipids. Compr Physiol. 2012;2:31–68. doi: 10.1002/cphy.c110001. [DOI] [PubMed] [Google Scholar]
- 9.Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH, et al. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–61. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolotina V, Omelyanenko V, Heyes B, Ryan U, Bregestovski P. Variations of membrane cholesterol alter the kinetics of Ca2(+)-dependent K+ channels and membrane fluidity in vascular smooth muscle cells. Pflugers Arch. 1989;415:262–8. doi: 10.1007/BF00370875. [DOI] [PubMed] [Google Scholar]
- 11.Levitan I, Christian AE, Tulenko TN, Rothblat GH. Membrane cholesterol content modulates activation of volume-regulated anion current in bovine endothelial cells. J Gen Physiol. 2000;115:405–16. doi: 10.1085/jgp.115.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toselli M, Biella G, Taglietti V, Cazzaniga E, Parenti M. Caveolin-1 expression and membrane cholesterol content modulate N-type calcium channel activity in NG108-15 cells. Biophys J. 2005;89:2443–57. doi: 10.1529/biophysj.105.065623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu C-C, Su M-J, Chi J-F, Chen W-J, Hsu H-C, Lee YT. The effect of hypercholesterolemia on the sodium inward currents in cardiac myocyte. J Mol Cell Cardiol. 1995;27:1263–9. doi: 10.1016/S0022-2828(05)82388-0. [DOI] [PubMed] [Google Scholar]
- 14.Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K(+) channels. Nat Rev Neurosci. 2003;4:957–67. doi: 10.1038/nrn1244. [DOI] [PubMed] [Google Scholar]
- 15.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 16.Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on I(K1) J Mol Cell Cardiol. 2001;33:625–38. doi: 10.1006/jmcc.2001.1344. [DOI] [PubMed] [Google Scholar]
- 17.Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward-rectifier K+ current by optical isomers of cholesterol. Biophys J. 2002;83:3211–22. doi: 10.1016/S0006-3495(02)75323-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epshtein Y, Chopra AP, Rosenhouse-Dantsker A, Kowalsky GB, Logothetis DE, Levitan I. Identification of a C-terminus domain critical for the sensitivity of Kir2.1 to cholesterol. Proc Natl Acad Sci U S A. 2009;106:8055–60. doi: 10.1073/pnas.0809847106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenhouse-Dantsker A, Leal-Pinto E, Logothetis DE, Levitan I. Comparative analysis of cholesterol sensitivity of Kir channels: role of the CD loop. Channels (Austin) 2010;4:63–6. doi: 10.4161/chan.4.1.10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenhouse-Dantsker A, Logothetis DE, Levitan I. Cholesterol sensitivity of KIR2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J. 2011;100:381–9. doi: 10.1016/j.bpj.2010.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenhouse-Dantsker A, Levitan I. Insights into structural determinants of cholesterol sensitivity of Kir channels. In: Levitan I, Barrantes F, eds. Cholesterol regulation of Ion Channels and Receptors. Hoboken, NJ: Wiley-Blackwell. 2012:47-67. [Google Scholar]
- 22.Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, Logothetis DE. Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J Biol Chem. 1997;272:31553–60. doi: 10.1074/jbc.272.50.31553. [DOI] [PubMed] [Google Scholar]
- 23.Lopes CMB, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 2002;34:933–44. doi: 10.1016/S0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- 24.Rohács T, Lopes CMB, Jin T, Ramdya PP, Molnár Z, Logothetis DE. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci U S A. 2003;100:745–50. doi: 10.1073/pnas.0236364100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garneau L, Klein H, Parent L, Sauvé R. Contribution of cytosolic cysteine residues to the gating properties of the Kir2.1 inward rectifier. Biophys J. 2003;84:3717–29. doi: 10.1016/S0006-3495(03)75100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001;2001:re19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 27.Logothetis DE, Jin T, Lupyan D, Rosenhouse-Dantsker A. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflugers Arch. 2007;455:83–95. doi: 10.1007/s00424-007-0276-5. [DOI] [PubMed] [Google Scholar]
- 28.Rosenhouse-Dantsker A, Logothetis DE. Molecular characteristics of phosphoinositide binding. Pflugers Arch. 2007;455:45–53. doi: 10.1007/s00424-007-0291-6. [DOI] [PubMed] [Google Scholar]
- 29.Ho IH, Murrell-Lagnado RD. Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. J Biol Chem. 1999;274:8639–48. doi: 10.1074/jbc.274.13.8639. [DOI] [PubMed] [Google Scholar]
- 30.Ho IH, Murrell-Lagnado RD. Molecular mechanism for sodium-dependent activation of G protein-gated K+ channels. J Physiol. 1999;520:645–51. doi: 10.1111/j.1469-7793.1999.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, He C, Yan X, Mirshahi T, Logothetis DE. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat Cell Biol. 1999;1:183–8. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]
- 32.Rosenhouse-Dantsker A, Sui JL, Zhao Q, Rusinova R, Rodríguez-Menchaca AA, Zhang Z, et al. A sodium-mediated structural switch that controls the sensitivity of Kir channels to PtdIns(4,5)P(2) Nat Chem Biol. 2008;4:624–31. doi: 10.1038/nchembio.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, et al. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci. 2005;8:279–87. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- 34.Rosenhouse-Dantsker A, Noskov S, Han H, Adney SK, Tang Q-Y, Rodríguez-Menchaca AA, et al. Distant cytosolic residues mediate a two-way molecular switch that controls the modulation of inwardly rectifying potassium (Kir) channels by cholesterol and phosphatidylinositol 4,5-bisphosphate (PI(4,5)P(2)) J Biol Chem. 2012;287:40266–78. doi: 10.1074/jbc.M111.336339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke OB, Caputo AT, Hill AP, Vandenberg JI, Smith BJ, Gulbis JM. Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir potassium channels. Cell. 2010;141:1018–29. doi: 10.1016/j.cell.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 36.An HL, Lü SQ, Li JW, Meng XY, Zhan Y, Cui M, et al. The cytosolic GH loop regulates the phosphatidylinositol 4,5-bisphosphate-induced gating kinetics of Kir2 channels. J Biol Chem. 2012;287:42278–87. doi: 10.1074/jbc.M112.418640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng XY, Zhang HX, Logothetis DE, Cui M. The molecular mechanism by which PIP(2) opens the intracellular G-loop gate of a Kir3.1 channel. Biophys J. 2012;102:2049–59. doi: 10.1016/j.bpj.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He C, Yan X, Zhang H, Mirshahi T, Jin T, Huang A, et al. Identification of critical residues controlling G protein-gated inwardly rectifying K(+) channel activity through interactions with the beta gamma subunits of G proteins. J Biol Chem. 2002;277:6088–96. doi: 10.1074/jbc.M104851200. [DOI] [PubMed] [Google Scholar]
- 39.Santiago J, Guzmàn GR, Rojas LV, Marti R, Asmar-Rovira GA, Santana LF, et al. Probing the effects of membrane cholesterol in the Torpedo californica acetylcholine receptor and the novel lipid-exposed mutation alpha C418W in Xenopus oocytes. J Biol Chem. 2001;276:46523–32. doi: 10.1074/jbc.M104563200. [DOI] [PubMed] [Google Scholar]
- 40.Fang Y, Mohler ER, 3rd, Hsieh E, Osman H, Hashemi SM, Davies PF, et al. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res. 2006;98:1064–71. doi: 10.1161/01.RES.0000218776.87842.43. [DOI] [PubMed] [Google Scholar]
- 41.Deng W, Bukiya AN, Rodríguez-Menchaca AA, Zhang Z, Baumgarten CM, Logothetis DE, et al. Hypercholesterolemia induces up-regulation of KACh cardiac currents via a mechanism independent of phosphatidylinositol 4,5-bisphosphate and Gβγ. J Biol Chem. 2012;287:4925–35. doi: 10.1074/jbc.M111.306134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tai K, Stansfeld PJ, Sansom MS. Ion-blocking sites of the Kir2.1 channel revealed by multiscale modeling. Biochemistry. 2009;48:8758–63. doi: 10.1021/bi9007808. [DOI] [PubMed] [Google Scholar]
- 43.Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–15. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys J. 2009;97:50–8. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–35. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 46.Noskov SY, Bernèche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–4. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 47.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr., Nilsson L, Petrella RJ, Roux B, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8, 27-8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.