

Abstract
The use of morpholinos for perturbing gene function in the chick, Gallus gallus, has led to many important discoveries in developmental biology. This technology makes use of in vivo electroporation, which allows gain and loss of function in a temporally, and spatially controlled manner. Using this method, morpholinos can be transfected into embryonic tissues from early to late developmental stages. In this article, we describe the methods currently used in our laboratory to knock down gene function using morpholinos in vivo. We also detail how morpholinos are used to provide consistency of the results, and describe two protocols to visualise the morpholino after electroporation. In addition, we provide guidance on avoiding potential pitfalls, and suggestions for troubleshooting solutions. These revised techniques provide a practical starting point for investigating gene function in the chick.
Keywords: Avian embryo, gene knockdown, in vivo electroporation, loss-of-function
1. Introduction
The chick is a popular model organism to study vertebrate development due to its well-described embryology, rapid development and the relative ease of manipulation at various stages, which has made classical techniques such as grafting and lineage tracing achievable by experimental embryologists for over 100 years [1]. Despite the versatility that the chick has to offer, perturbing gene function, especially generating knock-outs has been a challenge, primarily because of the difficulty in establishing homologous recombination in embryonic stem cells and the long generation time required to produce birds of reproductive age. Recent advances using lentiviral vectors have made it possible to generate germline transgenic chickens expressing GFP at high frequency [2, 3]. However, this method does not allow for reverse genetic approaches to the extent that homologous recombination does in mice. Additionally, maintaining transgenic chicken lines requires a large amount of space and is considerably more expensive than maintaining mouse or zebrafish lines. Over the last decade the chick has overcome this limitation, and become a more powerful model system, primarily because of the introduction of in vivo electroporation, which allows gain- and loss-of-function in a temporally and spatially controlled manner [4–8]. This technology allows introducing siRNA constructs and morpholinos into the embryo to knock down gene function reliably. Although siRNA provides a good strategy [9, 10] unspecific side effects have been reported, especially in young chick embryos [11] and in this review we will not discuss this approach further.
Morpholino phosphorodiamidate oligonucleotides (morpholinos; MOs) are synthetic DNA analogues consisting of about 24 subunits, with a morpholine ring replacing the ribose ring (see [http://www.gene-tools.com/]) (Fig. 1). This adaptation still allows binding of the complementary nucleic sequences by traditional Watson-Crick base pairing. Importantly, it has one major advantage over conventional antisense oligonucleotides: this backbone makes MOs completely resistant to nucleases [12–14] and, unlike other knockdown strategies, MOs do not depend on harnessing the cellular machinery, like the RNA-induced silencing complex and RNAse-H activity. Furthermore, since the backbone does not carry a negative charge, MOs are less likely to interact non-specifically with cellular proteins, and may therefore be less toxic [14].
Fig. 1. Structures of RNA and morpholino oligonucleotides.

(A) RNA oligonucleotide. (B) Morpholino oligonucleotide. Green ring represents the 6-membered morpholino ring (B), which replaces the ribose rings shown in (A). The non-ionic phosphorodiamidate linkages (B, red) replace the phosphodiester linkage in (A). Note the nBase (blue) can be any of the four standard bases (Adenine, Cytosine, Guanine or Thymine).
MOs function by blocking translation [14], or can be designed to prevent normal RNA splicing [15] (Fig. 2). Translation-blocking MOs block initiation of translation by targeting the start codon of the target mRNA or its vicinity, thus preventing protein production completely (Fig. 2C). Additionally, knowledge of the intron-exon structure of the target gene is not required, which is an advantage if the genome is not fully annotated, as is the case with chick. However, efficient protein knockdown can only be tested after the endogenous protein has degraded and requires a specific antibody or, if not available, knockdown needs to be monitored using a tagged version and anti-tag antibodies. Using these approaches, efficiency can be quantified by immunofluorescence or western blotting.
Fig. 2. Effect of splice- and translation-blocking morpholinos.

(A) Splice-blocking MO. Without MO, normal splicing of the mRNA occurs. The MO (purple) targets the splice acceptor site resulting in an abnormally spliced product (exon deletion). (B) Generation of an intron inclusion. (C) Translation-blocking MO. The MO targets a sequence 5' of the translation start site and inhibits the progression of the translation initiation complex.
Splice-inhibiting MOs prevent pre-mRNA processing and can be designed to generate partial or complete exon skipping or intron inclusion [16, 17] (Fig. 2A, B) depending on their exact location this can result in the production of truncated proteins. The main advantage of splice-blocking MOs are that specific effects can be created depending on MO design, that knowledge of the 5'end of the gene is not required and that their efficiency can be tested by RT-PCR. In most cases, this allows for more rapid analysis of the MO effect due to the shorter half-life of RNA as compared to protein. To ensure specificity and control for toxicity, most studies use two different MOs targeting the same gene and a 6bp mismatched control MO (discussed in section 10) (reviewed in reference [18]).
MOs need to be delivered into individual cells, and injection is a feasible method for Xenopus and zebrafish, but not for chick. By the time the egg is laid, the embryo is already multicellular, containing about 20,000 fairly small cells. Thus, to target many cells, MOs (or plasmid DNA) are instead electroporated into the embryo. In contrast to Xenopus and zebrafish, where MOs are mostly injected at very early stages, the chick offers the opportunity for temporally and spatially controlled knockdown, which is an advantage when investigating gene function at different times of development.
In vivo electroporation can introduce MOs into most regions of the chick embryo, and it is especially efficient for younger stages (< HH15). It should be noted however, that electroporation works best for epithelial sheets or cavitated structures (neural tube, otic vesicle). It is more of a challenge for mesenchymal tissues; although various studies have electroporated such tissues successfully [19–21] or alternatively used sonoporation for gene or MO transfer [22, 23]. During electroporation, the application of one or more electric pulses transiently generates pores in the cell membrane. This allows the entry of exogenous substances like DNA and MOs. Upon removal of the electric field the pores close and the cell membrane is re-sealed. After a brief recovery period cells appear to survive without further damage [24].
We currently use electroporation in two different culture systems: in modified New culture [25, 26] and in ovo [8]. The choice of culture depends primarily upon the stage at which the embryo is to be electroporated. We use New culture for young embryos from pre-streak to early somite stages (Eyal-Giladi XI [27] to Hamburger and Hamilton [28] HH8) and in ovo for older embryos. Using both systems, it is possible to examine the effect of MO-mediated knockdown soon after electroporation depending on mRNA and protein turnover. Recently, other ex-ovo culture methods [29, 30] have described successful electroporation, some even at much older stages (> HH22), however we will not discuss these here.
Here, we describe the methods currently used in our laboratory for examining gene function using MOs in the chick. In addition, we will provide guidance on avoiding potential pitfalls, and suggestions for troubleshooting solutions. These revised techniques provide a reliable starting point for investigating the effect of down-regulating gene expression using MOs.
2. Designing and storing morpholinos
2.1. Designing morpholinos
A number of factors must be considered to ensure the successful MO design. The MO should be about 25-bases in length with a GC content of 40–60%, little self-complementarity and little or no secondary structure. This ensures high MO affinity to the target as long oligonucleotides increase binding efficiency and access to the target RNA, and prevents MO-dimer formation and looping. Importantly, to keep the MO water soluble (critical for electroporation), it should contain no more than four contiguous Gs. As a general rule, we check the recommended MO sequence by Basic Local Alignment Search Tool using BLASTn optimised for short, nearly exact matches to confirm that the sequence is specific to the target under investigation. It is also useful to ascertain that the MO sequence does not map to sites of extensive sequence variation such as single nucleotide polymorphisms (SNPs; see [http://genome.ucsu.edu/]. The incomplete annotation of the chick genome can make designing two good MOs for a target gene a challenge. However, the release of sequence information for various strains and of the new genome version (v 4.0, Nov. 2011) has greatly improved annotation and as a consequence MO design.
Translational-blocking MOs require knowledge of the 5' end of the mRNA, and work by blocking translation initiation by targeting the 5' UTR through to the first 25 bases of the coding sequence. MO efficacy decreases as the 3' distance from the translation start site increases. To ensure efficient knockdown of translation-blocking MOs, a good working antibody is needed to visualise the protein either by immunohistochemistry or by western blotting. As discussed, if no antibody is available, a tagged full-length construct of the gene under investigation including the MO target sequence is required to demonstrate efficient knockdown. This can either be co-electroporated with the MO into the embryo [31] or co-transfected into cells [32]; generally a titration curve with different ratios of MO/construct should be performed. Anti-tag antibodies are then used to monitor knockdown by immunofluorescence or western blotting.
MOs designed against any splice junction will generate either a full or partial single exon deletion, or a full or partial single intron insertion depending on the position of the MO. In general, targeting internal intron/exon boundaries results in exon deletion thus requires at least 3 exons. In contrast, targeting the first or last exon/intron boundary generally leads to intron inclusion. Ideally, both strategies should produce a stop codon leading to termination of translation. In some cases, this may generate a truncated protein that could act as dominant negative form or compete with the endogenous protein. RT-PCR is widely used to assess knockdown by splice-inhibiting MO; this requires careful primer design to allow easy distinction of wild type and altered mRNA. Provided that the RNA half-life is relatively short as compared to the protein, splice-blocking MO and RT-PCR allow a rapid evaluation of MO efficiency.
In the chick embryo, electroporation is widely used for MO delivery, although recently a few other methods have been described [33, 34]. For successful electroporation, MOs must be fluorescein-labelled since the negatively charged fluorescein at the 3' end appears to enhance electroporation [4, 13]. The major advantage of fluorescein-labelled MOs is that electroporated cells can be visualised immediately after electroporation and can easily be compared with their wild-type neighbours. However, alternative delivery methods may need to be explored for mesenchymal tissues (see above) and for older embryos due to potential difficulties in accessing the target tissue and the possibility of damaging the heart and blood vessels.
Thus, there are a few limitations for MO-mediated knockdown in chick including design and delivery. Despite this, many studies have successfully used MOs to study gene function [35–40]. Overall, as long as the above recommendations and those provided by GeneTools are followed, MOs provide an efficient and practical technique for gene knockdown in the chick.
2.2. Storage of morpholinos
Generally, we follow instructions provided in the GeneTools data sheet. In brief, lyophilised MOs can be kept on arrival at −20°C. Before use, a MO stock solution of 1–5mM is prepared in ddH2O, heated at 65°C for 10 minutes in a water bath or PCR machine. 5μl aliquots are stored at −20°C until required and diluted to the appropriate concentration. GeneTools recommends storing MOs at RT, but we find evaporation can be a problem, especially for small aliquots.
3. General considerations for efficient electroporation and embryo viability
To obtain reliable and reproducible results several factors need to be considered to maximise efficient MO uptake into the embryo and embryo viability. These factors include quality of electrodes and electroporation chamber, the voltage applied, the correct working concentration of MO and good controls. Here, we explain how to optimise these to ensure consistent results.
The quality of the electrodes and electroporation chamber is critical and both should be made to a good standard. The latter has been described in detail by Voiculescu and colleagues [4]. Briefly, the electroporation chamber is made using acrylic (Perspex) or similar plastic material to construct a rectangular box. A vertical canal (~1.8mm in diameter at 3.0mm depth) is made through the centre of the base of the box, followed by a horizontal canal drilled (~1.6mm in diameter) through the centre of the base to meet the vertical canal. A platinum wire (0.5mm) is inserted into the vertical canal and using water-resistant glue (Epoxy) sealed in place.
Depending on the experiment, different electrodes are required, which are commercially available or can be made in the laboratory. These include fine tungsten electrodes for very focal electroporation [41], silver or platinum electrodes shaped like thin rods and used to target different cell populations, and large, flattened (plate-like) electrodes to electroporate half or the entire embryo. We have successfully used both commercial (Intracel, UK) (Fig 3A, C) and homemade electrodes (Fig 3B). In general, for targeting large areas of the embryo we use homemade silver, flattened electrodes, which are either made by hammering a single piece of silver wire (0.5mm) into a plate-like shape, or by rolling the wire into a spiral of about 2–3mm in diameter. The underside of the electrode (which will face the embryo during electroporation) should be flattened using fine sandpaper. To ensure that the electric field is centred on the target area, it is crucial to insulate the electrodes, except at their tips. This can be achieved using nail varnish.
Fig. 3. Different types of electrodes.

See section 3 for details For electroporation in New culture we use either thin rods (A, a') or flat electrodes (B, b'). (C, c') Double rods used for in ovo electroporation.
The conditions used for electroporation need to be calibrated carefully to ensure efficient MO transfer and maximise embryo viability depending on the age of the embryo and the target tissue. For electroporation in New culture (pre-streak to HH8) we find that five 10V pulses of 50–100ms with an interval of 500–1000ms using a CUY21EX Pulse Generator is sufficient for successful MO uptake without damage to the cells. Older embryos (> HH8) are mostly electroporated in-ovo allowing long-term culture; we generally begin using five 12V pulses of 50–100ms at a 500–1000ms interval using an Intracel TSS20 Ovodyne pulse generator. These conditions are adequate for good electroporation in the neural tube and surface ectoderm and for good viability of embryos over longer culture periods. In both cases, care must be taken to keep sufficient distance between the electrodes and the embryo (no less than 10mm) to avoid tissue damage and cell death. For older embryos in particular, electrodes should not be close to the heart or blood vessels as damage to the circulatory system reduces the survival rate dramatically.
To target different tissues, the electrodes are positioned in different orientations. For example, if targeting the ectoderm in New culture, we simply place the electrode above the target area. Targeting mesoderm and endoderm is straight forward by placing the electrode next to the primitive streak at appropriate positions before cells ingress. In ovo, structures with cavities (neural tube, otic vesicle, epithelial somites) can be filled with MOs; the electrodes are positioned so that different domains are targeted such as the dorsal, lateral or ventral neural tube.
To test successful electroporation, a quick and robust method is to look out for bubbles on the outer edge of the positive electrode. Alternatively, using a volt-ohm meter (VOM), the current or resistance can be measured. To increase viability, cleaning electrodes on a regular basis is important, which is easily achieved by reversing the current a few times.
As described above (section 2.2), we make a MO stock solution of 1–5mM, which is stored at −20°C. However, for each MO the working concentration needs to be tested to ensure efficient transfer and maximise viability. As a general rule we initially start with a working solution of 1mM, but titre each MO to a more suitable concentration depending on specificity, knockdown efficiency, viability and toxicity. Before use, the MO is mixed with carrier DNA (to improve uptake) and fast green (for visualisation) (see section 4.2.3). The amount of MO electroporated per embryo should be kept constant, and we generally use 0.5–1μl of the final MO solution. Successful electroporation is easily detected by fluorescence microscopy. It has been reported that MOs remain in neural tube cells for at least 48 hours after electroporation, with the intensity of the fluorescence being reduced at later time points [13]. We have not systematically tested this, and therefore cannot comment.
To ensure that the phenotype observed is specific to the MO-mediated knockdown, we generally use two different MO targeting the same RNA, standard control MOs and 6bp mismatched control MOs as well as rescue experiments. Each experiment is repeated at least three times with 10–15 embryos per experiment and appropriate controls.
These general `rules' should help to establish successful MO delivery while maintaining good viability of the embryo. At the end of this article, we provide a troubleshooting guide that covers some of these points.
4. Electroporation in New culture
4.2. Preparation of chick eggs
In all experiments, embryos are staged according to morphology by the system of Hamburger and Hamilton [28]. To obtain embryos of the desired stage, fertile chick eggs (Winter Farm, UK) are incubated in a humid incubator at 38°C for the appropriate time.
4.2.1. Materials and reagents
The following nomenclature is used for storage, temperature and preparation:
RT = Room temperature
IU = Prepare immediately before use
FS = Frozen stock
AC = Autoclave
Dissecting microscope with transmitted light base
One pair of coarse forceps
Two pairs of watchmakers' forceps (number 5)
One pair of scissors
Pyrex baking dish ~ 5 cm deep, about 2 l capacity
Fire polished glass Pasteur pipettes (15 cm long)
Blunt ended, wide mouthed glass pipette
Two 50–100 ml beakers (AC)
Two 10 cm glass petridishes
35 mm plastic petridishes with lids (BD Falcon, 351008)
Watch glasses, ~ 5–7 cm diameter
Glass rings, ~ 24mm inner diameter, 1–2mm thick and 3–4mm high
Aspirator tube (Sigma, A51777)
50μl glass capillaries (Drummond Scientific Company 2000050, USA)
Large container for waste collection
Plastic box with lid for incubating culture dishes
Electrodes
Electroporator (Pulse generator CUY21EX, Japan)
38°C humidified incubator
60% sucrose (RT)
Fast green in ddH2O (0.4%) (RT)
Plasmid DNA (~1.5–2.0 μg/μl) (FS)
MOs (FS/IU)
Pannet-Compton (PC) Saline
Solution A: 121 g NaCl, 15.5 g KCl, 10.42 g CaCl2•2H2O, 12.7 g MgCl2• 6H2O, H2O to 1 l (RT/AC).
Solution B: 2.365 g Na2HPO4• 2H2O, 0.188 g Na2HPO4 • 2H2O, H2O to 1 l (RT/AC). Before use mix in order: 120 ml solution A, 2.700 ml H2O and 180 ml solution B (IU).
Tyrode's saline
A 10x concentrated stock can be made and autoclaved for storage (RT/AU): 80 g NaCl, 2 g KCl, 2.71 g CaCl2• 2H2O, 0.5 g NaH2PO4• 2H2O, 2 g MgCl2• 6H2O, 10 g glucose to 1 l. Before use, add 20 ml of 10x stock to 180 ml ddH2O.
4.2.2. Setting up New cultures and electroporation procedure
To prepare the embryos for in vivo electroporation ex ovo, we use a modified version of New's original method [25, 42] (Fig. 4).
-
1.
Fill a clean Pyrex dish with PC saline; there should be sufficient liquid to submerge the egg yolks. (Fig. 4, A)
-
2.
At the blunt end of the egg remove the top part of the shell using the blunt forceps. (Fig. 4, B–D)
-
3.
Discard thick albumen; collect the thin albumen in a sterile 50 ml beaker. (Fig. 4, E–F)
-
4.
Gently tip the egg shell at 45°, allowing the yolk slip into the Pyrex dish; ensure that any remaining thick albumen surrounding the yolk is removed using blunt forceps and scissors. (Fig. 4, G–H)
-
5.
With the embryo facing upwards, cut the vitelline membrane just below the equator of the yolk. Note: Placing no more than 10–15 yolks in the Pyrex dish at any one time can make it easier to manipulate the membranes. (Fig. 4, H–I)
-
6.
Using fine forceps, peel back the vitelline membrane with the embryo remaining attached and place it ventral side up on to the watch glass. Note: To remove the vitelline membrane including the embryo successfully, it helps to peel the membrane back at almost 180° initially. Once peeled back to the position of the embryo, use an angle of 90°; this ensures that the embryo remains attached. Using forceps to push the yolk down while peeling may also help. (Fig. 4, J–M)
-
7.
Position a glass ring over the membrane, with the embryo in the centre. Remove both from the saline, pouring off some of the saline; dry the bottom on a tissue and place under a dissecting microscope. (Fig. 4, M–O)
-
8.
Carefully pull the membrane over the glass ring using fine forceps. It is important to pull it tight, without creases, to keep its tension and to allow the embryo to grow perfectly. Finally, trim the membrane edges inside the ring with scissors. (Fig 4, P)
-
9.
Clean of any remaining yolk in- and outside of the ring by rinsing with PC saline. (Fig. 4, Q)
-
10.
Place the embryos submerged in and surrounded by PC saline under a cover, keep humid. Embryos can be kept at this stage for a few hours at RT. (Fig. 4, A)
Fig. 4. Electroporating MOs in New culture.

See sections 4.2.2 – 4.2.5 for details.
4.2.3. Preparation of MOs and pulling needles
MOs should be prepared once sufficient New cultures have been collected. It is important to control the amount of MO electroporated per embryo. Like others [13], we aim to electroporate a reasonable large volume of MO per embryo. Our guide is to make a final volume of 10 μl; included in this is the MO, plasmid DNA (~1.5μg/μl), fast green (0.4%), and 60% sucrose. From this we add 0.5–1μl per embryo. We find using plasmid DNA (either PCAB:GFP or PC108:GFP) can enhance fluorescence detection, and since the backbone on the DNA has a charge, it can help to take up the MO with electroporation. Note: It has been reported that fast green can inhibit uptake of the MO [13].
-
11.
Thaw MO stock (see section 2.2) at RT and add ddH20 to a suitable working concentration. Heat at 65°C for 10 minutes in a water bath or PCR machine. For a 10 μl working MO concentration; add 0.3 μl 60% sucrose, 1 μl fast green (0.4%) and 1 μl plasmid DNA (~1.5 μg/μl).
-
12.
Mix by gently pipetting up and down. Note: to protect the fluorescein from potential light beaching, keep the MO in foil until required.
-
13.
While heating the MO (as in step 11), pull 50 μl glass capillaries to generate fine injection needles of approximately 5μm in diameter using a single barrel microelectrode puller (Harvard; heat 7, pull 6) or similar. These can be stored for future use.
4.2.4. Electroporation off the vitelline membrane
Prior to electroporation the embryos can be removed from the vitelline membrane.
-
14.
With the embryo submerged in PC saline use a fire-polished Pasteur pipette to loosen one edge of the area opaca from the vitelline membrane by blowing gently; then use a forceps to peel off the embryo. Note: the area opaca is the peripheral, extraembryonic area that surrounds the embryo proper (see reference [28]). Take care to avoid piercing the vitelline membrane.
-
15.
Using a wide-mouth glass pipette collect the embryos in a petridish containing Tyrode's saline.
-
16.
Set up the electroporation chamber containing Tyrode's saline under the microscope; connect the positive lead to the chamber and the negative lead to the top electrode. Check the set up to make sure all the connections are working by a few test pulses; watch out for bubbles.
-
17.
Place embryos individually into the electroporation chamber, dorsal side up. (Fig. 4, R, and T)
-
18.
Manoeuvre the embryo into a position where the area to be targeted is above the hole/platinum wire in the chamber.
-
19.
Using an aspirator tube and a pulled glass capillary, inject no more than 1 μl MO onto the dorsal surface of the embryo, targeting the desired region. Place the negative electrode about 10mm above the target area and apply five pulses of 10 Volts for 50–100ms, at a distance of 500–1000ms, using a CUY21EX pulse generator. (Fig. 4, R, T and V)
-
20.
Collect electroporated embryos in another petridish with fresh PC saline.
-
21.
Once all embryos are electroporated, place each onto a vitelline membrane, ventral side up. Then carefully remove all liquid from within and outside of the ring and slide the ring off the watch glass holding it with a pair of forceps. Note: To avoid tearing the membrane, do not lift the ring up vertically. (Fig. 4, Q)
-
22.
Place each individual New culture into 35 mm petridish, containing ~1ml thin albumen (see step 3). Push down the ring with both pairs of forceps (closed as not to break the tips) so it firmly attaches to the bottom of the dish. This prevents the culture from floating.
-
23.
Using a fire-polished Pasteur pipette, remove any remaining liquid from inside the ring. Excess liquid prevents normal growth of the embryo.
-
24.
Finally, seal the lid of the dish with a small amount of thin albumen; this keeps the individual New cultures secure, reduces the risk of infection and importantly prevents condensation on the lid during culture. The latter can otherwise drip on and destroy the embryo.
-
25.
Stack cultures into a plastic box together with a small piece of wet tissue to keep it moist. Incubate at 38°C until the desired stage is reached. (Fig. 4, U)
Note
We also use an alternative version of the above method and keep the embryos on the membrane when electroporating. One advantage of this method is that the embryo remains largely attached to the vitelline membrane, and as a consequence moves less as it begins to grow. This greatly improves live imaging after electroporation. (Fig. 4, S)
4.2.5. Electroporation on the vitelline membrane
Follow protocol above with the exception of point 14.
-
14*.
Do not remove the embryo from the vitelline membrane. Instead, place the ring including the membrane and embryo into the electroporation chamber. Note: the embryo now faces ventral side up; therefore the current must be reversed. Carefully fill the chamber and ring with Tyrode's saline. With a glass capillary carefully peel back a small area of the area opaca, and insert the pipette tip between the embryo and the vitelline membrane. Aim the pipette at the target area, inject MO solution, and place the top electrode above the target area and electroporate. Remove all liquid from inside the ring and carefully lift the culture out of the chamber. Then follow the protocol described above (step 22–25). Note: To have as little resistance as possible to pass through the vitelline membrane, it is important to remove as much thick albumin from the yolk as possible. (Fig. 4, S)
4.3. Isolation of embryos after culture
After incubation for the appropriate time period, embryos are removed from the culture and processed for in situ hybridisation or immunohistochemistry.
-
1.
Fill a 15 cm petridish with PBS.
-
2.
Using forceps remove the entire ring (including embryo) from the culture dish and place it into the dish of PBS. Note: This step needs to be performed with care; otherwise the membrane can detach from the ring, and may cause damage to the embryo.
-
3.
Using forceps, remove the embryo from the vitelline membrane, and with a blunt ended glass pipette place the embryo dorsal side up in a dish for fixing.
Alternatively
-
1.
Fill each New culture ring with PBS.
-
2.
Using a fire-polished glass pipette and forceps, gently loosen one corner of the area opaca by gently blowing saline, then peel off the embryo.
-
3.
Upon removal of the embryo from the vitelline membrane, use a wide mouth glass pipette to remove the embryo for fixation.
5. In-ovo electroporation
5.1. Preparation of chick eggs
Fertile hens' eggs are incubated in a humid incubator at 38°C until they have reached the desired stage. They should be placed on their side to allow easy access to the embryo.
5.1.1 Materials and reagents
The following nomenclature is used for storage, temperature and preparation:
RT = Room temperature
IU = Prepare immediately before use
FS = Frozen stock
AC = Autoclave
Dissecting microscope
Cold light source
Two pairs of watchmaker's forceps (number 5)
One pair of coarse forceps
One pair of scissors
Scalpel (number 11)
One metal spoon
5ml syringe
1ml syringe
22 gauge needles
30 gauge needle
Electroporator (Intracel TSS20 Ovodyne)
Electrodes
Aspirator tube (Sigma, A51777)
50 μl glass capillaries (Drummond Scientific Company 2000050, USA)
1 egg rest
Small squares of tissue paper
Opaque adhesive tape
38°C humidified egg incubator
Indian Ink (Pelikan, Germany, 221143) (RT)
60% sucrose (RT)
Fast green in ddH20 (0.4%) (RT)
Plasmid DNA (1.5–2.0 μg/μl) (FS)
MOs (GeneTools) (FS/IU)
Penicillin/Streptomycin Antibiotic (10 μl/ml) (FS)
50–100mlTyrode's saline (as in section 4.2.1) (IU)
5.1.2. In-ovo electroporation
The position of the positive electrode depends on the intended target. Here, we describe the method that we routinely use for electroporating the neural tube of HH 8–10 chick embryos (Fig. 5). There are many versions of this method, albeit with slight modifications to suit the user [13, 32, 43, 44]. We advise to work as quickly and as efficiently as possible to prevent drying out of the embryo, which reduces survival rates considerably. Before starting, prepare MO working solution and pull micro capillaries as described in section 4.2.3.
Fig. 5. Electroporating MOs in-ovo.

See section 5.1.2 for detail.
-
1.
Place the egg on the egg rest in the same orientation as it was incubated. Avoid rotating the egg to ensure that the embryo remains on the top-facing side of the egg to gain easy access. (Fig. 5, A–B)
-
2.
Insert a 5ml syringe with 21-gauge needle into one end of the egg. Holding the syringe at a 45° angle, gently remove 3 ml of albumen to lower the yolk slightly away from the shell. Note: it is important to remove the correct amount of albumen; if the yolk sinks but too much, access to the embryo is difficult. (Fig. 5, C)
-
3.
Place one strip of adhesive tape on top of the egg. With a blade, score a 3×2 cm rectangular `window' over the tape and then remove it using scissors or forceps. Note: placing adhesive tape on top of the egg helps to keep the shell as one piece when removing the `window'. This will also prevent any sharp pieces of shell from touching the yolk and potentially breaking the vitelline membrane. (Fig. 5, B, D–F)
-
4.
Dilute Indian ink 1:25 in Tyrode's saline and take up in a 1ml syringe with a 31-gauge needle. Make sure to bend the tip of the needle to 90°, as this will help the next step. Note: Be careful not to take up any air bubbles; bubbles injected underneath the blastoderm will make the embryo float or damage it.
-
5.
At an angle of 90°, carefully penetrate the outer edge of the area opaca and inject sufficient ink into the cavity under the embryo, that the embryo becomes visible to the naked eye. To prevent any unwanted drops of ink during removal of the needle from the membrane, application of gentle suction may be required. (Fig. 5, G–H)
-
6.
Using a 1ml syringe connected to a 31-gauge needle remove a small area of the vitelline membrane above the neural tube. This area needs to be large enough so that electrodes can be easily placed on either side of the embryo, but if too large will result in head deformations. (Fig. 5, I) An alternative method is to use an insect pin (A1) mounted to a glass-pipette.
-
7.
Add 1–2 drops of Tyrode's saline; rapidly place the electrodes on either side of the embryo, where the vitelline membrane has been removed.
-
8.
Fill the tip of a capillary with MO solution including carrier DNA and fast green. Using an aspirator tube inject the solution using air pressure into the lumen of the neural tube. (Fig. 5, J) Note: Injecting more than 2μl can cause opening of the neural tube.
-
9.
Apply five 50ms pulses of 12 Volts, at an interval of 1000ms using Intracel TSS20 ovodyne electroporator. Bubbling at the tip of the positive electrode indicates that the current has successfully passed. Incorporation of the MO into one side of the neural tube can be observed because the solution is faintly green. Note: Tyrode's saline is needed to transmit the current, if no bubbles are visible, apply more Tyrode's saline, and inject the MO again. (Fig. 5, K–M)
-
10.
Once electroporated, lower the yolk by removing 1–2 ml of albumen from the hole made in the eggshell (in step 2). Seal the `window' with opaque adhesive tape, and place in a humid incubator. If embryos are to be incubated for more than 24 hours we suggest adding Penicillin/Streptomycin antibiotic (10 μl/ml), diluted in Tyrode's saline before sealing the egg (Fig. 5, N). Note: whilst electroporating a fluorescein-tagged MO allows visualisation of the electroporated cells immediately after electroporation. We find that allowing the embryo time to `recover' from the electroporation provides consistency with the results (Fig. 5, O).
5.2. Isolation of embryos after in-ovo culture
After incubation for the appropriate time period, the embryo is removed from the egg.
Phosphate buffered saline (PBS)
A 20x concentrated stock can be made and autoclaved for storage (RT/AU): 8.01 g NaCl, 0.20 g KCL, 1.78g NaH2PO4• 2H2O, 0.27g KH2PO4to 1 I with ddH2O. Before use, make a 1x working solution.
-
1.
Using scissors, cut through the `window' to reveal the embryo, or simply peel off the tape to remove the window.
-
2.
Add about 3ml of PBS or Tyrode's saline into the egg (as in 5.1.2, step 2) or through the window (as in 5.1.2, step 3). Note: This floats the embryo to the surface of the egg, making it easier to remove it.
-
3.
Using sharp scissors, cut a square into the vitelline membrane around the embryo, and gently remove the membrane together with the embryo using a spoon. Note: This part can often be difficult due to the Indian ink, which can obscure the embryo.
7. Visualising MO electroporated cells
7.1. Brightening the fluorescein signal
Once the embryos are removed from New culture or the egg, they are routinely fixed in 4% paraformaldehyde for 60 min at RT. We find that shorter fixation times may lead to loss of MO. To visualise fluorescein labelled MOs we use anti-fluorescein antibodies; a similar procedure can be used for other antigens e.g. to detect efficient knockdown of the desired protein or changes in other proteins due to knockdown. Fixation for longer than 60 min may reduce antigen recognition; depending on the antibodies used, different times of fixation or different fixatives may be required. After fixation, embryos are embedded in 7% low-melting point agarose (Invitrogen, 16520-100) for vibratome sectioning (up to 150μm), or in 20% sucrose/gelatine solution for frozen sections [45] (10–25μm) with equally good results.
7.1.1. Materials and reagents
PBS
PBS containing 0.1% Triton-X100 (PBT)
Blocking buffer (5 % goat serum, 3 % BSA, 0.1 % Triton-X100)
For vibratome sections: 24-well plates
For frozen sections:
chromalaun-gelatine coated slides (Sigma Gelatin G-6144, Chromium Potassium Sulphate 24,336–1)
Coplin jar (frozen sections)
α-fluorescein 488 IgG2a (Oregon)
Secondary antibody-488
Mounting media (Mowiol® 4–88, Sigma, 81381, containing 0.1% p-phenylenediamine (PPD), Sigma P6001)
7.1.2.Visualising MOs using fluorescence
During this procedure, all sections are kept in the dark (covered in foil).
-
1.
Collect individual vibratome sections in a 24-well plate and wash 3 times in PBS at RT. Ensure that sections are completely covered with PBS (500 μl is sufficient). Collect frozen sections onto gelatine-coated slides, place into a coplin jar containing PBS and wash twice in PBS at 42°C in a water bath to remove gelatine. Replace PBS with fresh PBS at RT. From here onwards, carry out the entire procedure in a humid chamber.
-
2.
Non-specific binding sites are blocked in blocking buffer for 60 minutes. For vibratome sections use 500μl and place on a rocking platform at RT. For frozen sections, mark an area around the section using a hydrophobic pen and apply 50μl of blocking buffer to this region; do not shake.
-
3.
Incubate sections with primary antibody (1:500; α-fluorescein) diluted in blocking buffer for 2 days at 4 °C. For vibratome sections we use a total volume of 300 μl per well and incubate on a rocking platform. Seal the 24-well plate with parafilm to prevent evaporation. For frozen sections, use 50 μl antibody solution, coverslip the sections and incubate in a humid chamber for 2 days at 4°C.
-
4.
Remove primary antibody and wash sections in PBT 3 times for 5 minutes each. During each wash, place vibratome sections in 24-well plates on a rocker. For frozen sections, remove coverslips by placing slides into a coplin jar with PBS; coverslips will simply float off. Wash sections in the coplin jar. Note: we do not recycle the antibody.
-
5.
Incubate sections in secondary antibody (1:500) in blocking buffer, ON at 4 °C. For vibratome sections place on a rocking platform. For frozen sections incubate in a humid chamber. If required, DAPI (1:1000 mg/ml) can be added at this stage.
-
6.
Remove the secondary antibody (see step 4), and wash sections 3 times for 15 minutes each in PBS.
-
7.
Mount in Mowiol®.
7.2. Visualising MOs using 3'3'-Diaminobenzidine staining
Another method to visualise MO containing cells uses a secondary antibody conjugated to horseradish peroxidase (HRP) and 3,3'-Diaminobenzidine (DAB) as a substrate. This staining procedure works well in combination with whole-mount in situ hybridisation, and sectioning can be carried out thereafter.
7.2.1. Materials and reagents
PBS
Blocking buffer (1 % goat serum, 1 % Triton-100 in PBS)
α-fluorescein (Oregon)
anti rabbit IgG-HRP, (Jackson)
100mMTris-HCl, pH7.4
3'3'-Diaminobenzidine (DAB; Sigma, D4293); make 50 mg/ml stock solution and freeze in 50 μl aliquots
30 % Hydrogen Peroxide (Sigma, H1009)
ddH2O
4 % formaldehyde
7.2.2. DAB staining procedure
The procedure below describes how to perform detection of MO with HRP-coupled antibodies and DAB substrate after whole-mount in situ hybridisation. This method can also be used before whole-mount in situ hybridisation, but lithium chloride needs to be added to all solutions to preserve the RNA if required [46].
-
1.
Wash embryos 3 times for 30 minutes each in PBS on a rocking platform at RT.
-
2.
Non-specific binding sites are blocked in blocking buffer for 60 minutes on a rocking platform, at RT.
-
3.
Incubate embryos in primary antibody (1:500, α-fluorescein) in blocking buffer for 2 days on a rocking platform at 4°C. About 500 μl volume per vial is sufficient to cover the embryos.
-
4.
Remove antibody and wash embryos 4 times for 30 min each in PBS; fill vials to the top and incubate on a rocking platform.
-
5.
Incubate embryos in secondary antibody (anti rabbit IgG-HRP, 1:1000) in blocking buffer ON on a rocking platform at 4 °C.
-
6.
Remove antibody and wash embryos 5 times for 30 min each in PBS; fill vials to the top and incubate on a rocking platform.
-
7.
After the last wash, wash embryos for 5–15 minutes in 100mM Tris-HCl, pH7.4
-
8.
Prepare solution 1: dilute DAB stock solution in 100mM Tris-HCl, pH7.4 to a final concentration of 0.5 mg/ml. Note: DAB is a harmful substance; wear protective clothing and dispose of any liquid and solid waste according to the local health and safety regulations.
-
9.
Prepare solution 2: dilute H202in 100mM Tris-HCl, pH7.4 to a final concentration of 0.03%.
-
10.
Replace washing solution of each vial with 1 ml solution 1; after 5 minutes add 10 μl solution 2 and incubate for 5 minutes in the dark. Check regularly thereafter to detect any colorimetric change. A brown precipitate should appear in all MO carrying cells. Often, it is easier to place one embryo per vial, as the colour reaction will develop at different speed for each embryo.
-
11.
To stop the reaction, wash a few times in ddH2O. Fix embryos in 4 % formaldehyde for at least 30 min at RT.
-
12.
Wash well in PBS before photography.
9. Troubleshooting solutions
There can be a number of difficulties when using MOs and electroporation. Here, we describe some general problems that we have encountered and provide solutions to resolve these issues.
1. Poor embryo survival
A. Infection
This can occur anytime during manipulation of embryos. Solution: autoclave instruments, clean working area with soap and warm water, followed by 70% ethanol. Note: soap can be a problem, see below.
B. Soap
Embryos can disintegrate if in contact with soap. Solution: wash all equipment free from soap, especially rings and watch glasses. Store rings and watch glasses in 70% ethanol when not in use.
C. Egg quality
Embryos should be stored between 12–15°C when not required. After incubation embryos should be roughly +/− 3 hours to the correct stage of Hamburger and Hamilton. Solution: do not use any embryos that are the wrong age or look abnormal (e.g. branching primitive streak).
D. Incubation
If embryos need to be electroporated and then re-incubated we advise working as quickly as possible and not to leave the embryo `waiting' around a long time (>3 hours) before being re-incubated. Solution: For New culture, we aim to do 12–15 cultures per hour. For in ovo, we electroporate one embryo at a time and place straight in the incubator for the desired length of time (at least 60 minutes to `recover' from electroporation).
2. Embryo burned (section 4.2.2)
A. Electrodes too close to the embryo
Solution: The electrode should be at least 10mm away from the embryo.
B. Voltage too high
Solution: Reduce the voltage.
3. No electroporation (section 4.2.2 and 5.1.2)
A. MO concentration too low
Solution: Titrate MO concentration; try starting at 1.0mM.
B. MO volume too low
Solution: Increase volume to 1–2 μl per embryo. Ensure that 1 μl of plasmid DNA has been added to the MO mixture.
C. Voltage too low
Solution: Increase the voltage, check for bubbles surrounding the electrode.
D. Resistance too high
This can be caused by residual thick albumen left on the vitelline membrane. Solution: Remove the ring from the electroporation chamber, and remove any thick albumen.
E. Electrodes not in the correct orientation
Solution: Check that bubbles form around the positive electrode during electroporation – these should be visible to the naked eye, even using low voltage (5.0V). Clean the electrodes by reversing the current and wash in freshly made Tyrode's solution. Then re-peat the procedure.
F. Nail-varnish insulating the entire electrode(s)
Solution: After examination of the electrode(s), you made need to remove some nail-varnish; this can be easily peeled off.
4. Precipitation of MO (section 4.2.2 and 5.1.2)
A. MOs may vary in their solubility (check the data sheet provided by GeneTools), but do stay soluble at 1.0mM in the electroporation mixture.
Solution 1: We generally prepare our electroporation mixture prior to using it, making enough for one experiment. Any leftover MO is re-frozen at −20 °C. This will not precipitate upon re-thawing.
Solution 2: Re-check the design of the MO. Too many (>4) contiguous G's should not be in the design as this affects solubility [47].
5. DAB stain did not work (section 7.2)
A. Cells need to be electroporated well to see the DAB stain. Solution: Check fluorescence after electroporation to assess successful electroporation. Check pH of 100mM Tris: pH 7.4 is critical.
10. Considerations: morpholinos as tools for knockdown approaches in chick
Over the last decade, MOs have become powerful tools to assess gene function during early development: they are stable and resistant to nucleases, they function independent of the cellular machinery, they can be designed to have specific effects (inhibition of translation or splicing) and their efficiency can be monitored. One of the main disadvantages using morpholinos is that they are rather expensive, unlike siRNA constructs, which once generated are cheap to use. Generally, we use two MOs complementary to different regions of the target gene, as well as various control MOs increasing the costs considerably. Occasionally MOs seem to be less efficient, however there are few studies that have compared both approaches systematically.
In chick, MOs are suitable for transient gene knockdown in a temporally and spatially controlled manner. Using fluorescein-tagged MOs has the additional advantage that electroporated cells can be visualised immediately after targeting. Furthermore, electroporation generally results in mosaic knockdown, such that MO-carrying and wild type cells are intermingled allowing their direct comparison in the same embryo. However, as with all knockdown approaches, the major challenge is to ensure that the phenotype observed is specific to the loss or knockdown of the gene under investigation. Thus, MO experiments require careful controls to exclude off-target effects and non-specific changes [12–14].
The typical MO length is 25 base pairs; although there are no systematic studies the general consensus is that a mismatch of 6 base pairs should not bind to or interfere with the same target sequence. Indeed, MOs that differ in only 4 base pairs can have similar effects as the knockdown MO (see review [12, 16, 18, 48]). Therefore, it is possible that a given MO interacts with other, unintended targets, whose knockdown in turn is responsible for the phenotype observed. Below, we highlight important considerations for the interpretation of MO-mediated knockdown experiments.
10.1. Using different MOs targeting the same gene
Whenever possible, two non-overlapping MO against the gene of interest should be designed. It is unlikely that both MOs show off-target binding and, even if so, that they interfere with the same gene. Thus, two independent MO that show the exact same phenotype, considerably increase the confidence in the results observed.
In addition, both MOs can be tested for synergistic effects when electroporated at sub-optimal doses. In this case, three independent experiments should establish a dose-response curve for each MO to determine the concentration at which each MO has no effect. Then both MOs are co-electroporated at a non-effective dose. Ideally, this should produce the same results as a higher concentration of each MO. To establish a dose response curve it is particularly important to control the MO volume injected. Although we generally use air pressure for injections, in this case the use of a picospritzer is advisable to allow injection of consistent volumes.
10.2. Control MOs
Control MO should be used to assess the effect of electroporation, transfection of large amounts of exogenous oligonucleotides into tissues or cells, and potential off-target effects.
One control is to use MOs that target a gene that is not expressed in the target tissue. GeneTools offers a standard control MO specific for the mutated β-globin sequence of human thalassemia patients; this is unlikely to have an effect in chick embryos. This controls for electroporation artefacts and loading cells with exogenous oligonucleotides, but may not always provide a good control for every experimental MO, e.g. for toxicity. It may therefore be desirable to design a control MO more similar to the experimental MO. Some studies have used inverted [49], scrambled [50] or 5–6 base pair mismatched control MOs [51] with the idea that they contain the same base pair composition and are most similar to the experimental MO. However, with the lack of systematic studies on the effect of these potential controls, it is difficult to provide general rules about the ideal control MO.
10.3. Rescue experiments
Ideally, each knockdown experiment should be coupled to an appropriate rescue experiment that introduces the gene of interest or a downstream target. This requires the availability of a full-length construct that can be co-electroporated with the experimental MO and a proper titration of both to obtain a clear dose-response (see above). Importantly, the expression construct must be immune to MO-mediated knockdown. For translation blocking MOs this generally requires the introduction of silent mutations into the sequence targeted by the MO. In contrast, the effect of splice-blocking MO should easily be rescued by a wild type misexpression construct, as relevant exon/intron boundary sequences are not present. One potential problem is that over- or misexpression of the gene of interest can have a very strong gain-of-function phenotype. In this case, as an alternative approach it may be possible to use a downstream target to rescue the phenotype [31].
10.4. Titration of MOs
We further control toxicity and non-specific effects by careful titration of the MO: initially we inject a concentration of 1mM and then titrate this down to an optimal concentration that shows an effect, but no unspecific side effects. This was recently exemplified nicely by Kos and colleagues [13], where low MO concentration showed no observable difference to controls, while intermediate concentrations had a specific, but minor effect, and high concentrations a strong phenotype. This shows the importance of trying a range of concentrations, as well as being aware of non-specific effects.
10.5. Off-target effects: activation of pro-apoptotic genes
In zebrafish, a major problem for MO-mediated knockdown has been reported to be the activation of p53 and associated apoptosis [52–55]. In fish, this problem is relieved by co-injections of MOs that target p53. In chick there are no systematic studies regarding this issue; in our hands apoptosis does not occur after MO electroporation [11] provided that electroporation conditions are appropriately controlled and MOs are titrated properly.
11. Concluding remarks
Here, we have described our current methods using MOs to examine gene function in the chick. We provide up to date protocols describing different techniques to electroporate MOs ex ovo and in ovo as well as two protocols for to visualise MOs after electroporation. In addition, we give a broad introduction to these methods and details of potential pitfalls, as well as suggesting troubleshooting ideas for the user. These developed and revised techniques provide a starting point for using MOs to study the effects of perturbing gene function in the chick.
Highlights
Morpholinos as successful tools to examine gene function in chick.
Electroporation can be used to introduce morpholinos into the chick embryo as soon as the egg is laid.
We provide detailed guidance on avoiding potential pitfalls, and suggest troubleshooting solutions to ensure efficient uptake of the morpholino and embryo viability.
Fig. 6. DAB staining to visualise MO electroporated cells.
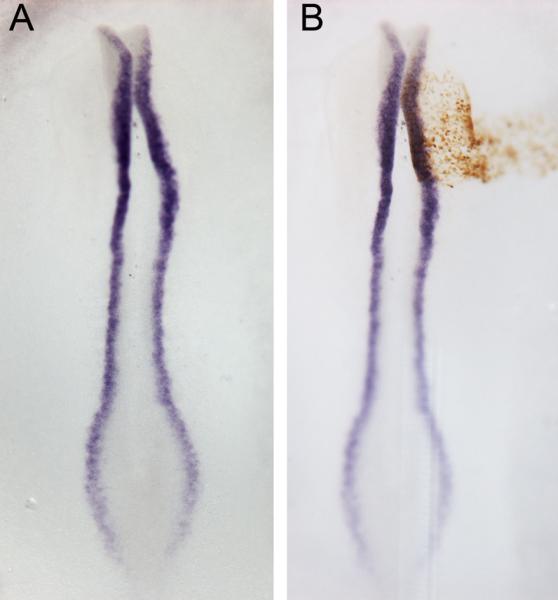
(A) Wholemount in situ hybridisation using Pax7 antisense probe in a chick embryo (HH8) (purple). (B) Visualisation of control MO-electroporated cells by DAB reaction (brown). See section 7.2 for details.
Acknowledgements
This work was supported by the BBSRC, NIH and a Fight for Sight Studentship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest The authors declare no conflict of interest.
13. References
- 1.Malpighi . De formatione pulli in ovo. Royal Society; London: 1672. [Google Scholar]
- 2.McGrew MJ, et al. Efficient production of germline transgenic chickens using lentiviral vectors. EMBO rep. 2004;5(7):728–33. doi: 10.1038/sj.embor.7400171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman SC, et al. Ubiquitous GFP expression in transgenic chickens using a lentiviral vector. Development. 2005;132(5):935–40. doi: 10.1242/dev.01652. [DOI] [PubMed] [Google Scholar]
- 4.Voiculescu O, Papanayotou C, Stern CD. Spatially and temporally controlled electroporation of early chick embryos. Nat Protoc. 2008;3(3):419–26. doi: 10.1038/nprot.2008.10. [DOI] [PubMed] [Google Scholar]
- 5.Muramatsu T, et al. Comparison of three nonviral transfection methods for foreign gene expression in early chicken embryos in ovo. Biochem Biophys Res Commun. 1997;230(2):376–80. doi: 10.1006/bbrc.1996.5882. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura H, et al. Gain- and loss-of-function in chick embryos by electroporation. Mech Dev. 2004;121(9):1137–43. doi: 10.1016/j.mod.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura H, Funahashi J. Dev Growth Differ. 2012. Electroporation: Past, present and future. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura H, Funahashi J. Introduction of DNA into chick embryos by in ovo electroporation. Methods. 2001;24(1):43–8. doi: 10.1006/meth.2001.1155. [DOI] [PubMed] [Google Scholar]
- 9.Bron R, et al. Functional knockdown of neuropilin-1 in the developing chick nervous system by siRNA hairpins phenocopies genetic ablation in the mouse. Dev Dyn. 2004;230(2):299–308. doi: 10.1002/dvdy.20043. [DOI] [PubMed] [Google Scholar]
- 10.Das RM, et al. A robust system for RNA interference in the chicken using a modified microRNA operon. Dev Biol. 2006;294(2):554–63. doi: 10.1016/j.ydbio.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 11.Mende M, Christophorou NA, Streit A. Specific and effective gene knock-down in early chick embryos using morpholinos but not pRFPRNAi vectors. Mech Dev. 2008;125(11–12):947–62. doi: 10.1016/j.mod.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Corey DR, Abrams JM. Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol. 2001;2(5):REVIEWS1015. doi: 10.1186/gb-2001-2-5-reviews1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kos R, et al. Methods for introducing morpholinos into the chicken embryo. Dev Dyn. 2003;226(3):470–7. doi: 10.1002/dvdy.10254. [DOI] [PubMed] [Google Scholar]
- 14.Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999;1489(1):141–58. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- 15.Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre- mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. 2001;30(3):154–6. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- 16.Ekker SC, Larson JD. Morphant technology in model developmental systems. Genesis. 2001;30(3):89–93. doi: 10.1002/gene.1038. [DOI] [PubMed] [Google Scholar]
- 17.Schmajuk G, Sierakowska H, Kole R. Antisense oligonucleotides with different backbones. Modification of splicing pathways and efficacy of uptake. J Biol Chem. 1999;274(31):21783–9. doi: 10.1074/jbc.274.31.21783. [DOI] [PubMed] [Google Scholar]
- 18.Eisen JS, Smith JC. Controlling morpholino experiments: don't stop making antisense. Development. 2008;135(10):1735–43. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- 19.Scaal M, et al. In ovo electroporation of avian somites. Dev Dyn. 2004;229(3):643–50. doi: 10.1002/dvdy.10433. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, et al. Stable, conditional, and muscle-fiber-specific expression of electroporated transgenes in chick limb muscle cells. Dev Dyn. 2011;240(5):1223–32. doi: 10.1002/dvdy.22498. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi JK, et al. Tbx5 and Tbx4 genes determine the wing/leg identity of limb buds. Nature. 1999;398(6730):810–4. doi: 10.1038/19762. [DOI] [PubMed] [Google Scholar]
- 22.Ohta S, et al. Microbubble-enhanced sonoporation: efficient gene transduction technique for chick embryos. Genesis. 2003;37(2):91–101. doi: 10.1002/gene.10232. [DOI] [PubMed] [Google Scholar]
- 23.Ohta S, et al. Sonoporation for gene transfer into embryos. Cold Spring Harb Protoc. 2011;2011(3):prot5581. doi: 10.1101/pdb.prot5581. [DOI] [PubMed] [Google Scholar]
- 24.G.L P, Panda T. Electroporation: basic principles, practical considerations and applications in molecular biology. Bioprocess Engineering. 1997;16:261–264. [Google Scholar]
- 25.Stern CD, Ireland GW. An integrated experimental study of endoderm formation in avian embryos. Anat Embryol (Berl) 1981;163(3):245–63. doi: 10.1007/BF00315703. [DOI] [PubMed] [Google Scholar]
- 26.New DAT. A new technique for the cultivation of the chick embryo in vitro. J. Embryol. Exp. Morph. 1955;(3):326–31. [Google Scholar]
- 27.Eyal-Giladi H, Kochav S. From cleavage to primitive streak formation: a complementary normal table and a new look at the first stages of the development of the chick. I. General morphology. Dev Biol. 1976;49(2):321–37. doi: 10.1016/0012-1606(76)90178-0. [DOI] [PubMed] [Google Scholar]
- 28.Hamburger V.a.H., L H. A series of normal stages in the development of the chick embryo. J. Morph. 1951;88:49–92. [PubMed] [Google Scholar]
- 29.Luo J, Redies C. Ex ovo electroporation for gene transfer into older chicken embryos. Dev Dyn. 2005;233(4):1470–7. doi: 10.1002/dvdy.20454. [DOI] [PubMed] [Google Scholar]
- 30.Chapman SC, et al. Improved method for chick whole-embryo culture using a filter paper carrier. Dev Dyn. 2001;220(3):284–9. doi: 10.1002/1097-0177(20010301)220:3<284::AID-DVDY1102>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 31.Sheng G, dos Reis M, Stern CD. Churchill, a zinc finger transcriptional activator, regulates the transition between gastrulation and neurulation. Cell. 2003;115(5):603–13. doi: 10.1016/s0092-8674(03)00927-9. [DOI] [PubMed] [Google Scholar]
- 32.Kos R, et al. The winged-helix transcription factor FoxD3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development. 2001;128(8):1467–79. doi: 10.1242/dev.128.8.1467. [DOI] [PubMed] [Google Scholar]
- 33.Geetha-Loganathan P, et al. Development of high-concentration lipoplexes for in vivo gene function studies in vertebrate embryos. Dev Dyn. 2011;240(9):2108–19. doi: 10.1002/dvdy.22708. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi Y, et al. Transposon-mediated stable integration and tetracycline-inducible expression of electroporated transgenes in chicken embryos. Methods Cell Biol. 2008;87:271–80. doi: 10.1016/S0091-679X(08)00214-8. [DOI] [PubMed] [Google Scholar]
- 35.Sugiyama S, Nakamura H. The role of Grg4 in tectal laminar formation. Development. 2003;130(3):451–62. doi: 10.1242/dev.00232. [DOI] [PubMed] [Google Scholar]
- 36.Tucker RP. Abnormal neural crest cell migration after the in vivo knockdown of tenascin-C expression with morpholino antisense oligonucleotides. Dev Dyn. 2001;222(1):115–9. doi: 10.1002/dvdy.1171. [DOI] [PubMed] [Google Scholar]
- 37.Moftah MZ, et al. Ectodermal FGFs induce perinodular inhibition of limb chondrogenesis in vitro and in vivo via FGF receptor 2. Dev Biol. 2002;249(2):270–82. doi: 10.1006/dbio.2002.0766. [DOI] [PubMed] [Google Scholar]
- 38.Granata A, Quaderi NA. The Opitz syndrome gene MID1 is essential for establishing asymmetric gene expression in Hensen's node. Dev Biol. 2003;258(2):397–405. doi: 10.1016/s0012-1606(03)00131-3. [DOI] [PubMed] [Google Scholar]
- 39.Hu N, et al. DNA methyltransferase3A as a molecular switch mediating the neural tube-to-neural crest fate transition. Genes Dev. 2012;26(21):2380–5. doi: 10.1101/gad.198747.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakai N, et al. Axon Sorting within the Spinal Cord Marginal Zone via Robo-Mediated Inhibition of N-Cadherin Controls Spinocerebellar Tract Formation. J Neurosci. 2012;32(44):15377–87. doi: 10.1523/JNEUROSCI.2225-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeltser LM, Larsen CW, Lumsden A. A new developmental compartment in the forebrain regulated by Lunatic fringe. Nat Neurosci. 2001;4(7):683–4. doi: 10.1038/89455. [DOI] [PubMed] [Google Scholar]
- 42.New DAT. A new technique for the cultivation of the chick embryo in vitro. J. Embryol. Exp. Morph. 1955;(3):326–31. [Google Scholar]
- 43.Kulesa PM, et al. In ovo live imaging of avian embryos. Cold Spring Harb Protoc. 2010;2010(6):pdb prot5446. doi: 10.1101/pdb.prot5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakamoto K, et al. Ectopic expression of lunatic Fringe leads to downregulation of Serrate-1 in the developing chick neural tube; analysis using in ovo electroporation transfection technique. FEBS Lett. 1998;426(3):337–41. doi: 10.1016/s0014-5793(98)00369-x. [DOI] [PubMed] [Google Scholar]
- 45.Stern CD, P.W.H.H. Essential Developmental Biology: A Practical Application. Oxford University Press Inc.; New York: 1993. [Google Scholar]
- 46.Streit A, Stern CD. Combined whole-mount in situ hybridization and immunohistochemistry in avian embryos. Methods. 2001;23(4):339–44. doi: 10.1006/meth.2000.1146. [DOI] [PubMed] [Google Scholar]
- 47.Gene Tools LLC.
- 48.Heasman J. Morpholino oligos: making sense of antisense? Dev Biol. 2002;243(2):209–14. doi: 10.1006/dbio.2001.0565. [DOI] [PubMed] [Google Scholar]
- 49.Craven SE, et al. Gata2 specifies serotonergic neurons downstream of sonic hedgehog. Development. 2004;131(5):1165–73. doi: 10.1242/dev.01024. [DOI] [PubMed] [Google Scholar]
- 50.Goljanek-Whysall K, et al. MicroRNA regulation of the paired-box transcription factor Pax3 confers robustness to developmental timing of myogenesis. Proc Natl Acad Sci U S A. 2011;108(29):11936–41. doi: 10.1073/pnas.1105362108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Levin M. Left-right asymmetry in the chick embryo requires core planar cell polarity protein Vangl2. Genesis. 2009;47(11):719–28. doi: 10.1002/dvg.20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robu ME, et al. p53 activation by knockdown technologies. PLoS Genet. 2007;3(5):e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cole LS, Ross LS. Apoptosis in the developing zebrafish embryo. Dev Biol. 2001;240(1):123–42. doi: 10.1006/dbio.2001.0432. [DOI] [PubMed] [Google Scholar]
- 54.Liu TX, et al. Knockdown of zebrafish Fancd2 causes developmental abnormalities via p53-dependent apoptosis. Dev Cell. 2003;5(6):903–14. doi: 10.1016/s1534-5807(03)00339-3. [DOI] [PubMed] [Google Scholar]
- 55.Gerety SS, Wilkinson DG. Morpholino artifacts provide pitfalls and reveal a novel role for pro-apoptotic genes in hindbrain boundary development. Dev Biol. 2011;350(2):279–89. doi: 10.1016/j.ydbio.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]