

Abstract
This review summarizes data in support of the notion that the cardiac intercalated disc is the host of a protein interacting network, called “the connexome,” where molecules classically defined as belonging to one particular structure (e.g., desmosomes, gap junctions, sodium channel complex) actually interact with others, and together, control excitability, electrical coupling and intercellular adhesion in the heart. The concept of the connexome is then translated into the understanding of the mechanisms leading to two inherited arrhythmia diseases: Arrhythmogenic Cardiomyopathy, and Brugada syndrome. The cross-over points in these two diseases are addressed to then suggest that, though separate identifiable clinical entities, they represent “bookends” of a spectrum of manifestations that vary depending on the effect that a particular mutation has on the connexome as a whole.
Keywords: Brugada syndrome, Arrhythmogenic right ventricular cardiomyopathy, sodium channel, desmosomes, connexin43, plakophilin-2
The intercalated disc: structure and function
In 1877, Engelman laid the foundation for decades of work when he concluded that “cardiac cells are in direct contact during life but become independent as they die” (quoted by Weidmann, 19521). Ultrastructural studies later revealed that this “direct contact” is supported by three structures, present at the site of end-end cell apposition: gap junctions, desmosomes and adherens junctions. In the following paragraph we provide a brief historical review of the discovery of the relation between a structure (gap junctions), its function (to provide a low-resistive pathway between cells) and its primary molecule (connexins). Similar historical perspectives can be found in the literature as it pertains to the structures that provide mechanical coupling between cells (e.g.,2).
Historical overview: the puzzle of separate but connected
The way in which cardiac cells coordinate their independent activities was a fundamental point of debate in cardiac physiology for the first half of the 20th century. By the early 1950s, two seemingly contradictory results were, on the other hand, strongly supported by the data: cells were individually wrapped in a cell membrane (and therefore electrically separate from their neighbors) and yet, the space constant of the cardiac tissue was much longer than the length of the single cell1. The advent of electron microscopy opened a large window into the cardiac landscape. After the early description of the intercalated disc as an area of specialization at the site of cell-cell contact3 Dewey, Sjostrand and Andersson proposed (in 19584) that the cardiac intercalated disc was “a connecting surface,” like the one first described by Robertson in the crayfish5. Six years later, the structural observation was finally reconciled with functional studies. Vanderkloot and Dane demonstrated that the intercalated disc was a subdomain of low electrical resistance. In their article, they speculated that “One possibility which might account for the low electrical resistance is that the intercalated discs are ‘leaky’ in regions where there are desmosomes or at the regions of membrane fusion”6. In 1965, Barr, Dewey and Berger provided the first experimental evidence specifically linking that “region of membrane fusion” (at that time called the nexus) with the propagation of the action potential7. The notion that “the nexus” was formed by fused membranes7, 8 was challenged by Revel and Karnovsky who, in 19679, demonstrated that the membranes were not fused but separated by a gap with interposed junctions, leading Revel to later coin the term “gap junctions.”
The idea of one molecule-one function versus that of multi-tasking molecules
The discovery in the 1980s of the connexin gene family gave molecular identity to a well-characterized ultrastructure10–12. A molecule-structure-function association was established and set the foundation for a well-accepted principle: connexins make gap junctions, which allow for the transfer of charge between cardiac cells.
The principle stated above never intended to discard the possibility that connexins have other functions. Yet, with a few exceptions13–17, the underlying assumption has been that connexin-related events are gap junction-related (or at most, hemichannel-related) events. In contrast, evidence dating back to the early work of Ross Johnson and his colleagues18 has consistently pointed to the possibility that connexin, as a molecule, participates in cellular processes that are independent of the existence of a connexin-formed permeable channel. This notion has caught new speed partly through the finding of the perinexus19, 20 (reviewed in a separate section below), as well as the evidence that Cx43 can regulate microtubule stability independent of gap junction formation15. It is in the context of these non-canonical functions of connexin14, 21 that we build on the concept of the connexome.
Connexins are far from being the only multi-tasking molecule at the intercellular junction. Multiple examples abound, though perhaps the best studied is the case of beta-catenin, which acts as an intermediary to regulate transcription as well as cell adhesion22. In the case of desmosomes, the data suggest that they also act as signaling platforms2. Finally, evidence in cardiac, and non-cardiac cells show that cadherin-rich structures also serve as a point of capture for microtubule anchoring23, 24. Overall, the evidence indicates that individual molecules, as well as the actual structures of the intercellular junctions exert multiple functions. The idea of silos of one-molecule-one function (or one structure-one function) seems not to apply to the intercalated disc. A communal protein interacting network seems a better description of the way in which the overall function is achieved.
To review all non-canonical functions of junctional molecules would go beyond the scope of this article. On the other hand, to limit the review exclusively to intercellular contact molecules would exclude a very important component of the intercalated disc, namely, the voltage-gated sodium channel (VGSC) complex25, 26. In the next paragraphs we will concentrate on desmosomes, gap junctions and the voltage-gated sodium channel (VGSC) complex, three complexes conventionally associated with three functions: adhesion, electrical coupling and cell excitability. Our goal will be to convey the message that molecules classically defined as belonging to one of the structures are indeed relevant to the function of the others, so that intercellular adhesion is in part controlled by connexins and by components of the VGSC complex, sodium current by desmosomal molecules and by connexins and electrical coupling by the integrity of desmosomes and by the expression of ankyrin-G (AnkG), a molecule conventionally identified with sodium channel function27, 28.
The connexome
Intercellular adhesion strength depends on molecules “of the VGSC complex”
The ability of cells to adhere tightly to each other depends in part on the expression of molecules conventionally ascribed to the VGSC complex. This link was established several years ago by the Isom lab29–31. In 1981, Hartshorne and Catterall purified “the saxotoxin receptor of the sodium channel from rat brain” and identified two polipeptides, which they referred to as “α” and “β.”32 They proposed that these two subunits conformed the functional sodium channel. In 1992, Lori Isom isolated the cDNA, sequenced and functionally expressed the β-1 subunit, concluding that this protein is crucial to the overall function of the sodium channel.31 In this manner, this 22,581 dalton protein was labeled as a “β” for its “α,” a subunit merely accessory to sodium channel function. It was eight years later (in 2000) that the Isom lab demonstrated that “sodium channel beta subunits” also mediate cell adhesion,33 a fact important not only in the formation of the sodium channel complex29, 33 but also in sodium channel-independent functions such as cell migration, cell aggregation and interaction with the cytoskeleton (see30).
The work described above has been extended to show that AnkG, a well-established scaffolding protein for the sodium channel in heart as well as in the nervous system27, 34–36, is necessary for proper cell adhesion. This was shown by experiments using an assay where the contact between the cells and the extracellular matrix is disrupted by the use of dispase. If adhesion between cells is strong, the layer lifts as one sheet (cells remain attached to one another). If cell-cell adhesion is weaker, the sheet fragments. Thus, the more fragments, the weaker the mechanical coupling. Using this method, Sato et al showed that loss of Ank-G expression (causes a decrease in mechanical coupling between cells 37.
The precise contribution of sodium channel beta subunits, or of AnkG, to the overall adhesion strength of cardiac cells in situ is not known. But one can speculate that genetic alterations that displace the beta subunit or AnkG from the intercalated disc could have the potential of altering myocardial structure. This is particularly relevant in the context of data showing that a mutation in the gene coding for the alpha subunit of the sodium channels (SCN5a) associates with dilated cardiomyopathy.38 Separate studies have reported the presence of structural defects in patients with Brugada syndrome39, 40. This important link between adhesion and sodium current function will be extensively discussed in the last sections of this review.
Intercellular adhesion is impaired by loss of “gap junction proteins”
More than twenty years ago Ross Johnson and his colleagues reported that Fab fragments of antibodies to the extracellular domain of Connexin43 (Cx43), the most abundant connexin in the heart, inhibits adherens junction assembly in cells in culture 18. More recently, we implemented the dispase assay described above to compare two cell populations14: one, HEK293 cells that endogenously express Cx43 and another one, a stable HEK293 line where we used lentivirus to permanently silence Cx43 expression. Three groups were compared: untreated (UNT), treated with a virus that contains a non-silencing construct (ϕshRNA-Cx43) and a third group where Cx43 expression was prevented (shRNA-Cx43). Loss of Cx43 expression brought about a loss of intercellular adhesion strength, represented by a significant increase in the number of fragments detected 90 minutes after dispase addition14. These results show that Cx43 expression is relevant to mechanical coupling. Whether this effect is consequent to gap junctions being a physical element that provides intercellular adhesion, or whether the result involves intermolecular interactions between Cx43 and components of the mechanical junctions, remains to be determined. The results do show that intercellular adhesion strength is a function of Cx43 that extends beyond the formation of a low-resistive pathway between cells.
Electrical coupling decreases after loss or mutations in molecules “of the desmosome” or “of the VGSC complex”
Shortly after the link between desmosomal mutations and AC was established, the Saffitz group showed decreased abundance of immunoreactive Cx43 signal at the intercalated disc in heart samples derived from patients with Naxos disease,41 with Carvajal syndrome42 and in general, with AC.43 In 2007, Oxford et al confirmed that the desmosomal protein PKP2 and Cx43 co-exist in the same macromolecular complex44. Moreover, shRNA-mediated loss of PKP2 expression in cultured rat ventricular cardiomyocytes led to a reduction in Cx43 abundance at the site of cell-cell contact, as well as decreased dye coupling between cells44. The observation of an association between Cx43 and PKP2 was later confirmed by other studies45, 46. Furthermore, recent data using super-resolution fluorescence microscopy have demonstrated that Cx43 and PKP2 are in close physical proximity and in fact, PKP2 clusters are often found within the boundaries of the Cx43 plaque21. These results support the notion of direct modulation of Cx43 by PKP2. Yet, it should be noted that complete loss of PKP2 causes only a 50% decrease in gap-junction mediated intercellular coupling44, and previous work has demonstrated that this decrease in junctional coupling is not sufficient to significantly affect conduction velocity.47, 48
The extent of electrical coupling between cells is also modified by loss of expression of AnkG. Indeed, studying neonatal rat ventricular myocytes in culture, Sato et al demonstrated that loss of expression of AnkG led to reduced gap junction conductance37. These results support the notion that AnkG is much more than a scaffold for sodium channels. Its role may be key in defining the exact position of various molecules of the connexome so that they interact with each other in the appropriate subdomain. In fact, AnkG may be a key piece that controls the traffic of molecules between the perinexus and the gap junction plaque (illustrated in Fig. 1).
Figure 1.
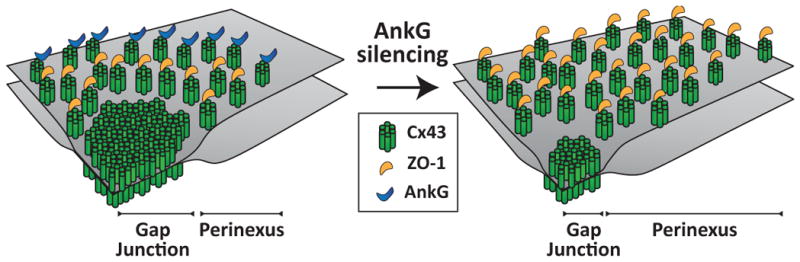
One possible model of connexome organization. The gap junction is surrounded by the perinexus where Cx43 hemichannels interact with ZO-1, which regulates the transition of connexons to the gap junction. AnkG may be localized at the border of the perinexus restricting the plaque size. When AnkG is silenced (right panel), the perinexal area expands at the expense of the actual pore-forming gap junction. This yields to larger Cx43 plaques but a reduced channel-forming domain and gap junction conductance. From21 with permission.
Sodium current amplitude is modulated by “cell adhesion molecules”
Early studies showed that the VGSC resides preferentially at the cardiac intercalated disc25, 26 and that it can be co-precipitated with Cx43 and with N-cadherin49. We therefore explored if disruption of desmosomal integrity could alter sodium current (INa). Our studies focused on the desmosomal protein PKP2, given that a) PKP2 mutations are the most common variant in AC50 and b) early-onset ventricular fibrillation and sudden death in the absence of an overt structural cardiomyopathy are often found in PKP2 mutations carriers50. In the first demonstration of a relation between PKP2 and INa, Sato et al51 showed that in cardiac myocytes lacking PKP2, INa was significantly decreased. Furthermore, loss of PKP2 expression in cardiac myocytes led to decreased abundance of immunoreactive Nav1.5 (the α-subunit of the VGSC) at sites of cell contact, and optical mapping experiments showed increased reentrant activity and significantly decreased conduction velocity in the absence of PKP2, when compared to controls51. A subsequent study showed that the cytoskeletal adaptor protein Ankyrin-G (AnkG) may play a key role in allowing for the interaction between three molecular components previously considered independent: the desmosome, the gap junction, and the VGSC complex37. These observations, limited to the cellular/molecular levels, prompted us to investigate the susceptibility to VGSC-dependent arrhythmias in hearts deficient in PKP2.
Patients affected with AC carry loss of function mutations in a heterozygous fashion. We therefore studied a mouse model with PKP2 haplo-insufficiency (PKP2-Hz), which mimics the clinical situation of patients harboring truncating mutations, with an expected ~50% of PKP2 availability52. The mouse model did not show signs of structural cardiomyopathy. Nav1.5 protein abundance was not altered and yet, the amplitude of INa in isolated ventricular cardiomyocytes was significantly decreased. Furthermore, there was a shift in gating and INa kinetics when compared to wild-type cardiomyocytes52. Thus, intrinsic, genetically mediated partial loss of PKP2 was able to affect sodium current amplitude, similarly to what was demonstrated in cells after total loss of PKP2 expression.
To further unmask the consequences of INa deficit, we challenged the PKP2-Hz mouse model with flecainide. All treated animals showed an increased sensitivity to drug-induced atrial and ventricular conduction prolongation, showing marked increased P wave, PR and QRS interval durations and increased conduction velocities in Langendorff-perfused isolated hearts. Furthermore, flecainide injection in vivo caused ventricular arrhythmias and some cases of sudden death in PKP2-Hz animals but not in the wild-types.52 These results demonstrated that PKP2 haploinsufficiency causes reduced INa in murine hearts, documenting for the first time the relationship between PKP2 and the VGSC in vivo.
Additional studies on the relation between adhesion molecules and the VGSC complex
Separate studies have confirmed the relation between desmosomal integrity, and the structure and/or function of the VGSC complex. Rizzo et al showed prolonged ventricular activation time, decreased conduction velocity, decreased upstroke velocity and decreased INa amplitude in mice over-expressing a mutation in desmoglein2.53 Gomes et al54 reported that patients with AC harboring desmoplakin mutations showed regional conduction delay and heterogeneous Nav1.5 distribution; moreover, in a collaborative immunohistochemistry study55 on heart samples from patients with AC, Noorman and colleagues showed that in most cases, Nav1.5 was reduced at the intercalated disc, even if the distribution of the N-cadherin signal remained normal. Finally, reduced sodium current amplitude has been observed in PKP2-deficient HL1 cells and in induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from a patient with PKP2 deficiency56, 57.
Though not a desmosomal molecule, the coxsackievirus and adenovirus receptor CAR is another case of an adhesion molecule that also regulates sodium current. Undoubtedly a receptor for both coxsackievirus and adenovirus, studies have demonstrated that the immunoglobulin extracellular domains of CAR are capable of homophilic binding, and participate in intercellular adhesion in epithelial cells. The role of this molecule on cell adhesion in the heart is less well defined. Interestingly, cardiac-restricted deletion of CAR causes significant slowing of A-V propagation and disruption of gap junctions at the intercalated disc.58, 59 Recent studies show that, as in the case of other molecules with immunoglobulin extracellular domains (such as the sodium channel β-subunits), CAR expression modulates sodium current function60.
Sodium current depends on “the gap junction protein” Connexin43
In the classical description of the electrophysiology of the adult ventricle, sodium channels provide the current that is necessary for the generation of an action potential, whereas Cx43 forms gap junctions to allow transfer of charge between one cell and the next. This description then separates the type of channel, with its function: sodium channels are responsible for cell excitability; gap junctions allow cell-cell passage of charge. In a recent study, however, we reported that loss of Cx43 expression brings about a loss of sodium current amplitude 61. In other words, the molecule necessary for making the gap junctions is actually necessary to maintain the complex in charge of generating the action potential. This means that loss of Cx43 expression is in fact a double-edge sword: not only will the path between cells be disrupted but also, the amount of charge that is generating by the excited cell will decrease. These results were consistent with others obtained in fetal atrial myocytes62 and led to the conclusion that loss and/or redistribution of Cx43 expression, as it happens in a number of pathological cardiac conditions (e.g.,63–65), can have complex deleterious effects on propagation.
Recent studies confirmed and expanded the importance of Cx43 in the control of sodium current amplitude. Lubkemeier et al66 developed a conditional knock-in murine model where, upon tamoxifen injection, the wild-type Cx43 was replaced, specifically in the heart, by a truncated form lacking the last five amino acids of the protein (Cx43D378stop). Interestingly, the Cx43D378stop protein forms gap junctions of normal junctional conductance, unitary conductance and permselectivity. Localization of Cx43 was not affected by the mutation, nor did it affect the localization of the ZO-1 protein (known to associate to Cx43 via its C-end). Yet, the animals died within 21 days after tamoxifen injection, and Holter recordings showed a transition from normal sinus rhythm to complex ventricular arrhythmias culminating in ventricular fibrillation and sudden death. Electrophysiological analysis revealed that, among the observed changes, there was a significant reduction in the amplitude of INa. This result clearly demonstrated that a key function of Cx43 is to maintain normal cell electrophysiology and that, even if gap junctions are present, a loss of Cx43-dependent, gap junction-independent functions can lead to severe arrhythmias and death. Studies are in progress to determine the molecular mechanism that links Cx43 and the function of the VGSC complex in the heart.
Visualizing interacting complexes at the intercalated disc
Electron microscopy
The results described above provide convincing evidence of a functional interaction between the various molecules that reside at the intercalated disc. Furthermore, it is likely that these interactions are, in fact, far more complex. Using tomographic electron microscopy (TEM) we have observed structural features that likely carry unknown functions. For example, it is common to see actual physical contact between gap junctions and the outer membrane of mitochondria67 (Fig. 2). It is also common to observe extensive vesicular activity localized specifically at the space separating desmosomes and gap junctions67 (Fig. 3) and in fact, the presence of small membranous particles in the intercellular space. The intercalated disc is, structurally and functionally, not a simple aggregate of independent, separate complexes but the host of a protein interacting network.
Figure 2.
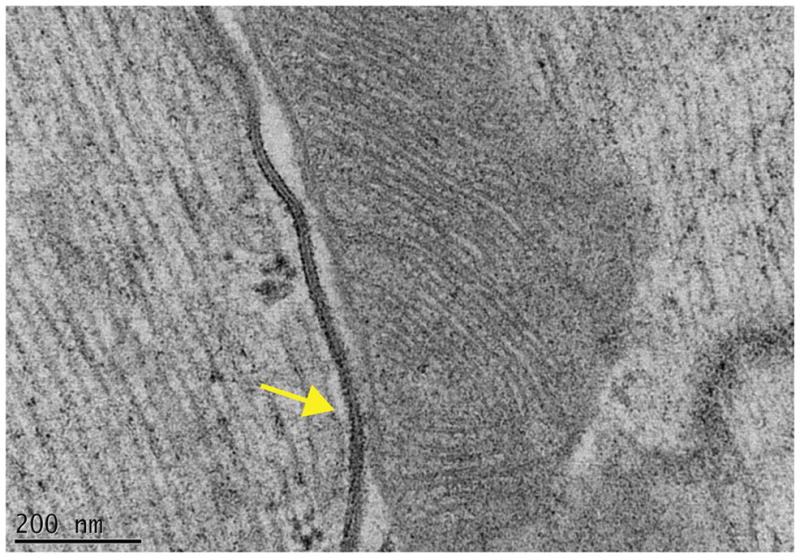
Proximity between gap junctions and mitochondria. Transmission electron micrograph image of adult murine heart tissue. The high preservation of structures was achieved by high-pressure freezing and freeze-substitution methods. Notice the close approximation and contact (yellow arrow) between the gap junction and a mitochondria. From67 with permission.
Figure 3.

Vesicular activity at the intercalated disc. Tomographic electron microscopy image of an intercalated disc region. Notice the vesicular activity in the space between the desmosomes and the gap junction, as well as in the intercellular space. From67 with permission.
Super-resolution fluorescence microscopy
A limitation of electron microscopy is that complexes that do not form electron-dense structures cannot be visualized. On the other hand, the resolution of light microscopy is limited by the diffraction of light68, 69 reaching to approximately 250 nm in the best of conditions and therefore not sufficient to assess the actual distances between molecules. To overcome this limitation, we have implemented super-resolution fluorescence microscopy methods21, 69. We built a set-up on a conventional inverted microscope using commercially available optics and applied laser illumination, reducing, and oxygen scavenging conditions to manipulate the blinking behavior of individual fluorescent reporters. Movies of blinking fluorophores were reconstructed to generate subdiffraction images at ~20 nm resolution. We mapped a molecule classically defined as a component ‘of the desmosome’ (PKP2) and another one defined as a ‘gap junction molecule’ (Cx43), and defined their spatial organization. As shown in Figure 4,21 in about half of Cx43 clusters we observed an overlay of Cx43 and PKP2 at the Cx43 plaque edge. SiRNA-mediated loss of Ankyrin-G expression yielded larger Cx43 clusters, of less regular shape, and larger Cx43-PKP2 subdomains. The Cx43-PKP2 subdomain was validated by a proximity ligation assay (PLA) and by Monte-Carlo simulations indicating an attraction between PKP2 and Cx43. Overall our studies led us to conclude that super-resolution fluorescence microscopy, complemented with Monte-Carlo simulations and PLAs, allows the study of the nanoscale organization of an interactome in cardiomyocytes. Our results further showed that PKP2 and Cx43 share a common hub that permits direct physical interaction. These results allowed us to fully support the notion that not only are molecules functionally interacting into what we dubbed a connexome, but they also occupy a common space at the intercellular junction21. The actual location of this space within the context of the overall intercalated disc architecture may be related to the studies characterizing the perinexus, as described below.
Figura 4.
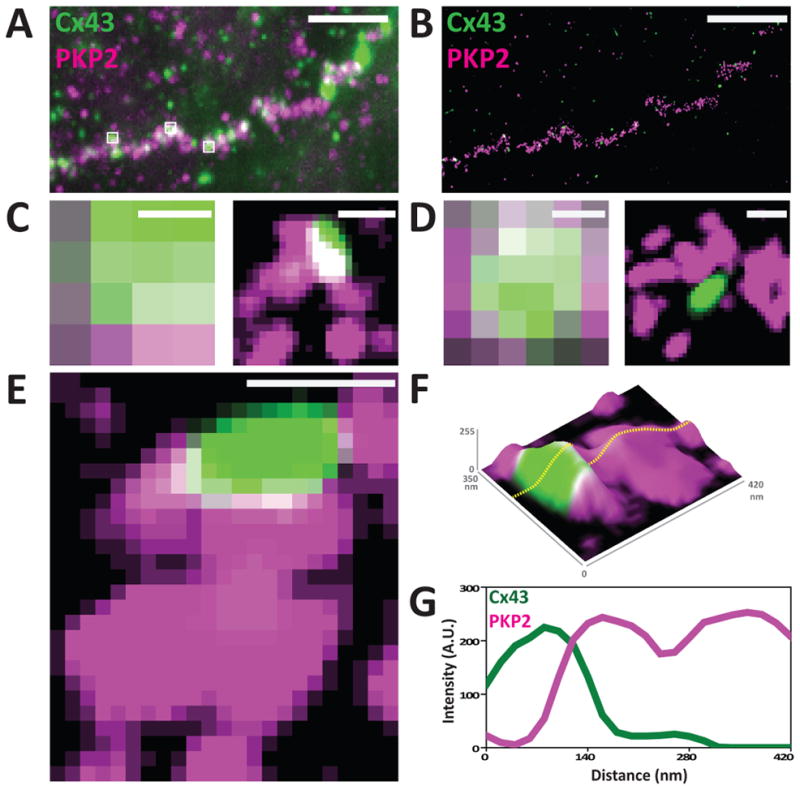
Super-resolution fluorescence microscopy: NRVMs stained for Cx43 (green) and PKP2 (magenta). Panels A and B show the same region visualized by TIRF (A) or by SRFM (B). Enlargement of the small white squares show improved resolution after reconstruction (C-E). Panel E shows a Cx43 cluster surrounded by PKP2. Same image is shown as a topological image in F (z-axis: signal intensity). Intensity plot of the dotted line across the image (G) shows the intersection of both signals. Scale bars: 5μm (A and B) and 200nm (C–E). Reproduced from21 with permission.
The perinexus and the connexome
Recent studies have shown that the association of Cx43 with its scaffolding protein ZO1 occurs in the periphery of the Cx43 plaque (the “perinexus”19, 20) and that loss of the interaction with ZO-1 leads to larger gap junction plaques, with a loss of the perinexal domain. In a recent study, the Gourdie group further proposed that the perinexus could be a site for the interaction of Cx43 with Nav1.519. From that perspective, the perinexus could be the site of the connexome. This is an interesting possibility, which in fact suggests that changes in the stability of the Cx43-ZO-1 association could actually affect the properties of INa. Defining the confines of the perinexus in adult cardiac tissue, and the molecular constrains that regulate its composition and its properties represent an important challenge for the future. It is worth noting that an additional structure in the intercalated disc that could host multiple molecules is the area composita70. Various investigators have demonstrated that these structures represent a combination of molecules classically defined as belonging to the desmosome or to the adherens junctions71, 72. We speculate that Cx43 and Nav1.5 may also be found in the area composita, and interact within that domain. These two locations (the perinexus or the area composita) are not mutually exclusive but rather, complementary sites that could ensure the proper integration of the connexome with both, molecules involved in mechanical function, and in electrical coupling between cells.
Brugada syndrome and AC: Two diseases with a common substrate
Through the writing above, we have sought to establish that the intercalated disc hosts a common protein interacting network (the connexome) that includes molecules of the desmosome and of the VGSC complex. In the clinical realm, desmosomal mutations lead to ARVC73 and sodium channel dysfunction, to Brugada syndrome74, 75. We surmised that if the molecular substrates (desmosomes; VGSC) are part of a common network, then the diseases (AC; BrS) should also share some common features. In the following section, we first give a brief description of the two diseases and then, discuss their similarities.
Arrhythmogenic Cardiomyopathy
It is generally accepted that loss of integrity of the desmosome leads to “arrhythmogenic cardiomyopathy” or “AC.” (Also called “arrhythmogenic right ventricular cardiomyopathy,” or “arrhythmogenic right ventricular dysplasia,” hence the abbreviations “ARVD” “ARVC” or ARVC/D” found in the literature to refer to this disease76, 77). Approximately 50–70% of the cases of familial AC associate with a mutation in a desmosomal gene78, 79. This condition presents with a progressive fibro-fatty infiltration, often more prominent in the right ventricular myocardium, and with a high propensity to life-threatening ventricular arrhythmias and progression toward heart failure76. It is considered one of the most relevant causes of juvenile sudden cardiac death, especially in competitive athletes, and its prevalence ranges from 1:2000 to 1:500080. It is important to emphasize, however, that a number of cases of unexpected cardiac sudden death occur during the early or “concealed” phase of the disease, in the absence of overt structural manifestations.81
Brugada syndrome
Brugada syndrome (BrS) is an inherited channelopathy characterized by ST segment elevation of coved morphology in right precordial leads, increased risk of ventricular tachycardia and ventricular fibrillation and absence of cardiac structural disease75. Mutations in the gene coding for Nav1.5, the most abundant sodium channel protein in the heart, account for ~20–25% of genotype-positive subjects82, and about 4% of patients carry mutations in the CACNA1c gene. Several other genes have been associated with sporadic cases of BrS, but each one accounts for <2% of patients; as such, current guidelines do not support routine screening on them in the general BrS population82. Overall only 25–30% of patients with clinical diagnosis of BrS have a known genotype, implying that additional, still undiscovered genes may be linked to this disease.
The intersection of BrS and AC
When BrS was initially described, some investigators proposed that this condition shared features with arrhythmogenic cardiomyopathy (AC), thus opening the possibility that they represent two poles of a common spectrum ultimately leading to increased risk of sudden death83. In fact, some BrS patients show minor RV structural abnormalities;40 furthermore, mutations in SCN5A have been associated with cases of dilated cardiomyopathy,38, 84 while desmosomal mutation carriers can experience ventricular fibrillation and sudden death without overt structural disease.41, 54, 81, 85, 86
PKP2 mutations and the BrS phenotype
This phenotypical and molecular crossover led us to investigate whether desmosomal mutations exist in cases of Brugada syndrome57. We screened by direct sequencing the PKP2 gene in a cohort of 200 patients with clinical diagnosis of BrS and no mutations on the most prevalent genes. We discovered five single amino acid substitutions in five unrelated patients57. In order to assess if this missense variant in PKP2 could affect the cardiac INa, we used an HL-1 cell line, stably silenced for the endogenous PKP2. In the absence of PKP2, these cells showed a decrease in the native INa.. Cells transiently transfected with each one of the PKP2 mutants associated with the BrS phenotype showed significantly decreased INa, when compared with cells transfected with wild type PKP257. Similar results were obtained when we used a line of human iPSC-derived cardiomyocytes from a patient lacking PKP2 at the cell membrane56, 87. In these cells, INa increased upon transfection with wild type PKP2. Transfection with one of the PKP2 mutants associated with BrS was not able to restore normal INa 57.
These data represent the first evidence that missense mutations in PKP2 can cause a decrease in cardiac INa and facilitate arrhythmias, even in the absence of a structural cardiomyopathy. We propose that PKP2 mutations provide at least part of the molecular substrate of BrS. The inclusion of PKP2 as part of routine BrS genetic testing remains premature; yet, the possibility that some patients showing signs of disease may harbor PKP2 variants should be considered when the genotype is negative for other genes associated with BrS.
ARVC, BrS and the diseases of the connexome
In summary, the experimental data support the notion that rather than controlled by “individually wrapped” separate molecular complexes, excitability, electrical coupling and intercellular adhesion are controlled by a common protein interacting network called the connexome. As the edges of the molecular complexes are blurred, so are the clinical syndromes that associate with them: Brugada syndrome is not purely arrhythmogenic (but includes a structural component), arrhythmias in AC are not only consequent to alterations in macrostructure (molecular changes in the intercalated disc subdomain, and in sodium currents, can be found), and in fact, PKP2 mutations can also be the substrate for BrS. While clinically distinguishable as individual entities, we propose that they share a common origin as diseases of the connexome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weidmann S. The electrical constants of purkinje fibres. J Physiol. 1952;118:348–360. doi: 10.1113/jphysiol.1952.sp004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harmon RM, Green KJ. Structural and functional diversity of desmosomes. Cell Commun Adhes. 2013;20:171–187. doi: 10.3109/15419061.2013.855204. [DOI] [PubMed] [Google Scholar]
- 3.Sjostrand FS, Andersson E. Electron microscopy of the intercalated discs of cardiac muscle tissue. Experientia. 1954;10:369–370. doi: 10.1007/BF02160542. [DOI] [PubMed] [Google Scholar]
- 4.Sjostrand FS, Andersson-Cedergren E, Dewey MM. The ultrastructure of the intercalated discs of frog, mouse and guinea pig cardiac muscle. J Ultrastruct Res. 1958;1:271–287. doi: 10.1016/s0022-5320(58)80008-8. [DOI] [PubMed] [Google Scholar]
- 5.Robertson JD. Ultrastructure of two invertebrate synapses. Proc Soc Exp Biol Med. 1953;82:219–223. doi: 10.3181/00379727-82-20071. [DOI] [PubMed] [Google Scholar]
- 6.Vanderkloot WG, Dane B. Conduction of the action potential in the frog ventricle. Science. 1964;146:74–75. doi: 10.1126/science.146.3640.74. [DOI] [PubMed] [Google Scholar]
- 7.Barr L, Dewey MM, Berger W. Propagation of action potentials and the structure of the nexus in cardiac muscle. The Journal of general physiology. 1965;48:797–823. doi: 10.1085/jgp.48.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewey MM, Barr L. A study of the structure and distribution of the nexus. The Journal of cell biology. 1964;23:553–585. doi: 10.1083/jcb.23.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Revel JP, Karnovsky MJ. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J Cell Biol. 1967;33:C7–C12. doi: 10.1083/jcb.33.3.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beyer EC, Paul DL, Goodenough DA. Connexin43: A protein from rat heart homologous to a gap junction protein from liver. The Journal of cell biology. 1987;105:2621–2629. doi: 10.1083/jcb.105.6.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar NM, Gilula NB. Cloning and characterization of human and rat liver cdnas coding for a gap junction protein. The Journal of cell biology. 1986;103:767–776. doi: 10.1083/jcb.103.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul DL. Molecular cloning of cdna for rat liver gap junction protein. The Journal of cell biology. 1986;103:123–134. doi: 10.1083/jcb.103.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou JZ, Jiang JX. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions - an update. FEBS letters. 2014 doi: 10.1016/j.febslet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agullo-Pascual E, Delmar M. The noncanonical functions of cx43 in the heart. The Journal of membrane biology. 2012;245:477–482. doi: 10.1007/s00232-012-9466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francis R, Xu X, Park H, Wei CJ, Chang S, Chatterjee B, Lo C. Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS One. 2011;6:e26379. doi: 10.1371/journal.pone.0026379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crespin S, Bechberger J, Mesnil M, Naus CC, Sin WC. The carboxy-terminal tail of connexin43 gap junction protein is sufficient to mediate cytoskeleton changes in human glioma cells. Journal of cellular biochemistry. 2010;110:589–597. doi: 10.1002/jcb.22554. [DOI] [PubMed] [Google Scholar]
- 17.Danik SB, Rosner G, Lader J, Gutstein DE, Fishman GI, Morley GE. Electrical remodeling contributes to complex tachyarrhythmias in connexin43-deficient mouse hearts. FASEB J. 2008;22:1204–1212. doi: 10.1096/fj.07-8974com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer RA, Laird DW, Revel JP, Johnson RG. Inhibition of gap junction and adherens junction assembly by connexin and a-cam antibodies. J Cell Biol. 1992;119:179–189. doi: 10.1083/jcb.119.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhett JM, Gourdie RG. The perinexus: A new feature of cx43 gap junction organization. Heart Rhythm. 2012;9:619–623. doi: 10.1016/j.hrthm.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–1528. doi: 10.1091/mbc.E10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agullo-Pascual E, Reid DA, Keegan S, Sidhu M, Fenyo D, Rothenberg E, Delmar M. Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc Res. 2013;100:231–240. doi: 10.1093/cvr/cvt191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gottardi CJ, Gumbiner BM. Adhesion signaling: How beta-catenin interacts with its partners. Current biology: CB. 2001;11:R792–794. doi: 10.1016/s0960-9822(01)00473-0. [DOI] [PubMed] [Google Scholar]
- 23.Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bellett G, Carter JM, Keynton J, Goldspink D, James C, Moss DK, Mogensen MM. Microtubule plus-end and minus-end capture at adherens junctions is involved in the assembly of apico-basal arrays in polarised epithelial cells. Cell motility and the cytoskeleton. 2009;66:893–908. doi: 10.1002/cm.20393. [DOI] [PubMed] [Google Scholar]
- 25.Cohen SA. Immunocytochemical localization of rh1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated disks. Circulation. 1996;94:3083–3086. doi: 10.1161/01.cir.94.12.3083. [DOI] [PubMed] [Google Scholar]
- 26.Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res. 2002;91:1176–1182. doi: 10.1161/01.res.0000046237.54156.0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowe JS, Palygin O, Bhasin N, Hund TJ, Boyden PA, Shibata E, Anderson ME, Mohler PJ. Voltage-gated nav channel targeting in the heart requires an ankyrin-g dependent cellular pathway. J Cell Biol. 2008;180:173–186. doi: 10.1083/jcb.200710107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 e1053k mutation causing brugada syndrome blocks binding to ankyrin-g and expression of nav1.5 on the surface of cardiomyocytes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303–1310. doi: 10.1161/01.cir.103.9.1303. [DOI] [PubMed] [Google Scholar]
- 30.Isom LL. Sodium channel beta subunits: Anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- 31.Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 32.Hartshorne RP, Catterall WA. Purification of the saxitoxin receptor of the sodium channel from rat brain. Proc Natl Acad Sci U S A. 1981;78:4620–4624. doi: 10.1073/pnas.78.7.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000;275:11383–11388. doi: 10.1074/jbc.275.15.11383. [DOI] [PubMed] [Google Scholar]
- 34.Grubb MS, Burrone J. Building and maintaining the axon initial segment. Current opinion in neurobiology. 2010;20:481–488. doi: 10.1016/j.conb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer JL. Nodes of ranvier and axon initial segments are ankyrin g-dependent domains that assemble by distinct mechanisms. The Journal of cell biology. 2007;177:857–870. doi: 10.1083/jcb.200612012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komada M, Soriano P. [beta]iv-spectrin regulates sodium channel clustering through ankyrin-g at axon initial segments and nodes of ranvier. The Journal of cell biology. 2002;156:337–348. doi: 10.1083/jcb.200110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-g, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201. doi: 10.1161/CIRCRESAHA.111.247023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L. Scn5a mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011;57:2160–2168. doi: 10.1016/j.jacc.2010.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frustaci A, Priori SG, Pieroni M, Chimenti C, Napolitano C, Rivolta I, Sanna T, Bellocci F, Russo MA. Cardiac histological substrate in patients with clinical phenotype of brugada syndrome. Circulation. 2005;112:3680–3687. doi: 10.1161/CIRCULATIONAHA.105.520999. [DOI] [PubMed] [Google Scholar]
- 40.Catalano O, Antonaci S, Moro G, Mussida M, Frascaroli M, Baldi M, Cobelli F, Baiardi P, Nastoli J, Bloise R, Monteforte N, Napolitano C, Priori SG. Magnetic resonance investigations in brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur Heart J. 2009;30:2241–2248. doi: 10.1093/eurheartj/ehp252. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE. Structural and molecular pathology of the heart in carvajal syndrome. Cardiovasc Pathol. 2004;13:26–32. doi: 10.1016/S1054-8807(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 43.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 44.Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007;101:703–711. doi: 10.1161/CIRCRESAHA.107.154252. [DOI] [PubMed] [Google Scholar]
- 45.Joshi-Mukherjee R, Coombs W, Musa H, Oxford E, Taffet S, Delmar M. Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (arvc)-related plakophilin-2 (pkp2) mutations. Heart Rhythm. 2008;5:1715–1723. doi: 10.1016/j.hrthm.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fidler LM, Wilson GJ, Liu F, Cui X, Scherer SW, Taylor GP, Hamilton RM. Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med. 2009;13:4219–4228. doi: 10.1111/j.1582-4934.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous cx43 knockout mice using optical mapping. Journal of cardiovascular electrophysiology. 1999;10:1361–1375. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 48.Thomas SP, Kucera JP, Bircher-Lehmann L, Rudy Y, Saffitz JE, Kleber AG. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res. 2003;92:1209–1216. doi: 10.1161/01.RES.0000074916.41221.EA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malhotra JD, Thyagarajan V, Chen C, Isom LL. Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J Biol Chem. 2004;279:40748–40754. doi: 10.1074/jbc.M407243200. [DOI] [PubMed] [Google Scholar]
- 50.van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, van der Smagt J, Boven LG, Mannens MM, van Langen IM, Hofstra RM, Otterspoor LC, Doevendans PA, Rodriguez LM, van Gelder IC, Hauer RN. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 51.Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patino GA, Taffet SM, Isom LL, Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, Birchmeier W, Mohler P, van Veen TA, van Rijen HV, Delmar M. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012;95:460–468. doi: 10.1093/cvr/cvs218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovascular research. 2012;95:409–418. doi: 10.1093/cvr/cvs219. [DOI] [PubMed] [Google Scholar]
- 54.Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, Quarta G, Nobles M, Syrris P, Chaubey S, McKenna WJ, Tinker A, Lambiase PD. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-a combined murine and human study. European heart journal. 2012;33:1942–1953. doi: 10.1093/eurheartj/ehr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10:412–419. doi: 10.1016/j.hrthm.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, Marine JE, Calkins H, Kelly DP, Judge DP, Chen HS. Studying arrhythmogenic right ventricular dysplasia with patient-specific ipscs. Nature. 2013;494:105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko-Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori S, Delmar M. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim BK, Xiong D, Dorner A, Youn TJ, Yung A, Liu TI, Gu Y, Dalton ND, Wright AT, Evans SM, Chen J, Peterson KL, McCulloch AD, Yajima T, Knowlton KU. Coxsackievirus and adenovirus receptor (car) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J Clin Invest. 2008;118:2758–2770. doi: 10.1172/JCI34777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lisewski U, Shi Y, Wrackmeyer U, Fischer R, Chen C, Schirdewan A, Juttner R, Rathjen F, Poller W, Radke MH, Gotthardt M. The tight junction protein car regulates cardiac conduction and cell-cell communication. J Exp Med. 2008;205:2369–2379. doi: 10.1084/jem.20080897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marsman RF, Bezzina CR, Freiberg F, Verkerk AO, Adriaens ME, Podliesna S, Chen C, Purfurst B, Spallek B, Koopmann TT, Baczko I, Dos Remedios CG, George AL, Jr, Bishopric NH, Lodder EM, de Bakker JM, Fischer R, Coronel R, Wilde AA, Gotthardt M, Remme CA. Coxsackie and adenovirus receptor (car) is a modifier of cardiac conduction and arrhythmia vulnerability in the setting of myocardial ischemia. Journal of the American College of Cardiology. 2013 doi: 10.1016/j.jacc.2013.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of cx43 results in decreased nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional cx43 knockout mice. Heart Rhythm. 2012;9:600–607. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desplantez T, McCain ML, Beauchamp P, Rigoli G, Rothen-Rutishauser B, Parker KK, Kleber AG. Connexin43 ablation in foetal atrial myocytes decreases electrical coupling, partner connexins, and sodium current. Cardiovasc Res. 2012;94:58–65. doi: 10.1093/cvr/cvs025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, DiSilvestre D, Tunin RS, Kass DA, Tomaselli GF. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H1223–1230. doi: 10.1152/ajpheart.00079.2007. [DOI] [PubMed] [Google Scholar]
- 64.Chkourko HS, Guerrero-Serna G, Lin X, Darwish N, Pohlmann JR, Cook KE, Martens JR, Rothenberg E, Musa H, Delmar M. Remodeling of mechanical junctions and of microtubule-associated proteins accompany cardiac connexin43 lateralization. Heart Rhythm. 2012;9:1133–1140. e1136. doi: 10.1016/j.hrthm.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qu J, Volpicelli FM, Garcia LI, Sandeep N, Zhang J, Marquez-Rosado L, Lampe PD, Fishman GI. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009;104:365–371. doi: 10.1161/CIRCRESAHA.108.184044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lubkemeier I, Requardt RP, Lin X, Sasse P, Andrie R, Schrickel JW, Chkourko H, Bukauskas FF, Kim JS, Frank M, Malan D, Zhang J, Wirth A, Dobrowolski R, Mohler PJ, Offermanns S, Fleischmann BK, Delmar M, Willecke K. Deletion of the last five c-terminal amino acid residues of connexin43 leads to lethal ventricular arrhythmias in mice without affecting coupling via gap junction channels. Basic Res Cardiol. 2013;108:348. doi: 10.1007/s00395-013-0348-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delmar M, Liang FX. Connexin43 and the regulation of intercalated disc function. Heart Rhythm. 2012;9:835–838. doi: 10.1016/j.hrthm.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drummen GP. Fluorescent probes and fluorescence (microscopy) techniques--illuminating biological and biomedical research. Molecules. 2012;17:14067–14090. doi: 10.3390/molecules171214067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van de Linde S, Loschberger A, Klein T, Heidbreder M, Wolter S, Heilemann M, Sauer M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nature protocols. 2011;6:991–1009. doi: 10.1038/nprot.2011.336. [DOI] [PubMed] [Google Scholar]
- 70.Franke WW, Borrmann CM, Grund C, Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006;85:69–82. doi: 10.1016/j.ejcb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 71.Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, Shang X, De Rycke R, van Roy F, Radice GL. Loss of alphat-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J Cell Sci. 2012;125:1058–1067. doi: 10.1242/jcs.098640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, Lorenzon A, Li Mura IE, Beffagna G, Rigato I, Vleeschouwers M, Tyberghein K, Hulpiau P, van Hamme E, Zaglia T, Corrado D, Basso C, Thiene G, Daliento L, Nava A, van Roy F, Rampazzo A. Mutations in the area composita protein alphat-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–210. doi: 10.1093/eurheartj/ehs373. [DOI] [PubMed] [Google Scholar]
- 73.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. European heart journal. 2010;31:806–814. doi: 10.1093/eurheartj/ehq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 75.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: Report of the second consensus conference: Endorsed by the heart rhythm society and the european heart rhythm association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 76.Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nature reviews. Cardiology. 2012;9:223–233. doi: 10.1038/nrcardio.2011.173. [DOI] [PubMed] [Google Scholar]
- 77.van der Zwaag PA, Jongbloed JD, van den Berg MP, van der Smagt JJ, Jongbloed R, Bikker H, Hofstra RM, van Tintelen JP. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Human mutation. 2009;30:1278–1283. doi: 10.1002/humu.21064. [DOI] [PubMed] [Google Scholar]
- 78.Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M, Bhuiyan ZA, Wiesfeld AC, Volders PG, van Langen IM, Atsma DE, Dooijes D, van den Wijngaard A, Houweling AC, Jongbloed JD, Jordaens L, Cramer MJ, Doevendans PA, de Bakker JM, Wilde AA, van Tintelen JP, Hauer RN. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 2011;123:2690–2700. doi: 10.1161/CIRCULATIONAHA.110.988287. [DOI] [PubMed] [Google Scholar]
- 79.Marcus FI, Zareba W, Calkins H, Towbin JA, Basso C, Bluemke DA, Estes NA, 3rd, Picard MH, Sanborn D, Thiene G, Wichter T, Cannom D, Wilber DJ, Scheinman M, Duff H, Daubert J, Talajic M, Krahn A, Sweeney M, Garan H, Sakaguchi S, Lerman BB, Kerr C, Kron J, Steinberg JS, Sherrill D, Gear K, Brown M, Severski P, Polonsky S, McNitt S. Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: Results from the north american multidisciplinary study. Heart rhythm: the official journal of the Heart Rhythm Society. 2009;6:984–992. doi: 10.1016/j.hrthm.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 81.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ Res. 2010;107:700–714. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- 82.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze- Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. Hrs/ehra expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the heart rhythm society (hrs) and the european heart rhythm association (ehra) Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 83.Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial st-segment elevation, and sudden death in young people. Circulation. 2001;103:710–717. doi: 10.1161/01.cir.103.5.710. [DOI] [PubMed] [Google Scholar]
- 84.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nature reviews Cardiology. 2009;6:337–348. doi: 10.1038/nrcardio.2009.44. [DOI] [PubMed] [Google Scholar]
- 85.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 86.Zhang M, Tavora F, Oliveira JB, Li L, Franco M, Fowler D, Zhao Z, Burke A. Pkp2 mutations in sudden death from arrhythmogenic right ventricular cardiomyopathy (arvc) and sudden unexpected death with negative autopsy (sudna) Circulation journal: official journal of the Japanese Circulation Society. 2012;76:189–194. doi: 10.1253/circj.cj-11-0747. [DOI] [PubMed] [Google Scholar]
- 87.Awad MM, Dalal D, Tichnell C, James C, Tucker A, Abraham T, Spevak PJ, Calkins H, Judge DP. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in pkp2. Human mutation. 2006;27:1157. doi: 10.1002/humu.9461. [DOI] [PMC free article] [PubMed] [Google Scholar]