

Abstract
Purpose
Recent studies suggested that AKT activation might confer poor prognosis in acute myeloid leukemia (AML), providing the rationale for therapeutic targeting of this signaling pathway. We therefore explored the preclinical and clinical anti-AML activity of an oral AKT inhibitor, MK-2206.
Experimental Methods
We first studied the effects of MK-2206 in human AML cell lines and primary AML specimens in vitro. Subsequently, we conducted a phase 2 trial of MK-2206 (200 mg weekly) in adults requiring second salvage therapy for relapsed/refractory AML, and assessed target inhibition via reverse phase protein array (RPPA).
Results
In preclinical studies, MK-2206 dose-dependently inhibited growth and induced apoptosis in AML cell lines and primary AML blasts. We then treated 19 patients with MK-2206 but, among 18 evaluable participants, observed only 1 (95% CI: 0–17%) response (complete remission with incomplete platelet count recovery), leading to early study termination. The most common grade 3/4 drug-related toxicity was a pruritic rash in 6/18 patients. Nevertheless, despite the use of MK-2206 at maximum tolerated doses, RPPA analyses indicated only modest decreases in Ser473 AKT (median 28%; range, 12–45%) and limited inhibition of downstream targets.
Conclusions
While preclinical activity of MK-2206 can be demonstrated, this inhibitor has insufficient clinical anti-leukemia activity when given alone at tolerated doses, and alternative approaches to block AKT signaling should be explored.
Keywords: Acute myeloid leukemia, MK-2206, AKT pathway
Introduction
In adults, acute myeloid leukemia (AML) remains difficult to treat (1, 2). The need for novel therapies is thus unquestioned. With increasing availability of specific small molecule inhibitors, considerable effort now focuses on targeting signaling pathways that are pivotal for leukemic cells as a means to improve therapeutic outcomes. Emerging evidence suggests that the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway (3, 4) could provide a rational drug target in AML. Constitutive activation of AKT, as indicated by phosphorylation at Thr308 and/or Ser473, is observed in 50–85% of patients with AML (5–12) and has been correlated with adverse cytogenetics as well as inferior relapse-free and overall survival (11, 13, 14). Activated AKT has been implicated in proliferation, myeloid transformation, cell survival, and chemoresistance of AML cells (6, 9, 10); in fact, induction of PI3K/AKT signaling may be one of the mechanisms by which bone marrow stroma cells confer protection from chemotherapy (15). Conversely, inhibition of the PI3K/AKT signaling pathway restores apoptosis in AML cells in vitro (8).
MK-2206 is an oral, highly specific allosteric inhibitor for all three isoforms of human AKT (16). Preclinical studies confirmed efficient AKT dephosphorylation and demonstrated growth inhibition of various human solid tumor cell lines (17–22); consequently, MK-2206 has entered clinical testing for patients with solid tumors (23). On the other hand, the effects of MK-2206 on malignant hematopoietic cells are poorly explored so far except for recent studies, which indicated significant cytotoxic activity against diffuse large B-cell lymphoma and T-cell acute lymphoblastic leukemia (ALL) cells in vitro (24, 25). In the current study, we have investigated the anti-tumor activity of MK-2206 against human AML cell lines and primary AML blasts. To begin testing this compound clinically, we then conducted a phase 1/2 trial in adults with poor-prognosis AML to determine the drug’s tolerability and obtain preliminary data on its efficacy of AKT inhibition. Weekly (34) rather than every-other-day (23) dosing of MK-2206 was explored following recommendations of the Cancer Therapy Evaluation Program at the National Cancer Institute (CTEP/NCI; see Treatment, Methods).
Methods
In Vitro Investigations
Materials
All reagents were purchased from commercial sources unless otherwise stated. MK-2206 was partially provided by Merck & Co, Inc. (Whitehouse Station, NJ) and partially obtained from LC Laboratories (Woburn, MA).
AML cell lines and primary AML cells
OCI-AML3 cells were kindly provided by M. D. Minden (Ontario Cancer Institute, Toronto, ON, Canada). HL60, U937, and MOLM13 cells were obtained from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). THP-1 and MO7e were purchased from the American Type Culture Collection (Manassas, VA). MOLM14 cells were kindly provided by Dr. Mark Levis (Johns Hopkins University, Baltimore, MD). Cells were maintained in RPMI 1640 supplemented with 5% fetal bovine serum and 5% bovine calf serum at 37°C in 5% CO2. Pperipheral blood specimens containing >40% blasts were obtained from patients with newly diagnosed or recurrent AML. Informed consent was obtained following institutional guidelines. Mononuclear cells were isolated via Ficoll density gradients (Sigma-Aldrich, St. Louis, MO). Samples from healthy bone marrow donors were selected for CD34+ cells using a MiniMacs Separator (Miltenyi Biotec, Auburn, CA) as directed by the manufacturer.
Analysis of cell viability and apoptosis
Cells were treated with various doses of MK-2206 for up to 72 hours. Cell viability and cell numbers were quantified by trypan blue dye exclusion assay using a Vicell. To determine the mechanism of cell death, cells were washed in phosphate-buffered saline, and resuspended in binding buffer containing Annexin V (Roche Diagnostics, Indianapolis, IN). Apoptotic cells were identified by positive Annexin V staining using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA).
Western blot analysis
OCI-AML3, MOLM13, or primary AML blasts were sonicated in lysis buffer (62.5 mM Tris (pH 8.0), 2% SDS, 10% glycerol, 100 µM AEBSF, 80nM Aprotinin, 5µM Bestatin, 1.5 µM E-64, 2 µM leupeptin, 1 µM Pepstatin, 500 µM sodium orthovanadate, 500 µM glycerol phosphate, 500 µM sodium pyrophosphate and 50 µM DTT), and protein (5 × 105 cell equivalents) was subjected to electrophoresis using 10–14% acrylamide/0.1% SDS gels. Proteins were transferred onto nitrocellulose, and membranes were probed with monoclonal antibodies against pAKT Thr308 and Ser473, phospho-S6, S6 (all from Cell Signaling Technology, Danvers, MA), and Tubulin (Sigma-Aldrich).
Clinical Trial
Study population
A phase 2 study with MK-2206 was conducted at MD Anderson Cancer Center (MDACC) and Fred Hutchinson Cancer Research Center (FHCRC) between October 2010 and October 2012. Patients ≥18 years of age were eligible if they had persistent or relapsing AML (other than acute promyelocytic leukemia [APL]) (31) requiring 2nd salvage therapy (i.e. treatment for second or higher relapse or for primary refractory disease after failure of two prior treatment regimens) provided they had a prior complete remission (CR) duration <12 months. At MDACC, patients ≥60 years were also eligible with <2 prior regimens if they did not have favorable-risk cytogenetics and were not candidates, or refused, standard chemotherapy. Other inclusion criteria included: an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2; total bilirubin ≤2.0 × Upper Limit of Normal (ULN) unless elevation was due to hepatic infiltration by AML, Gilbert’s syndrome, or hemolysis; SGOT/SPGT ≤2.5 × ULN unless elevation was due to hepatic infiltration by AML; serum creatinine ≤1.5 × ULN; fasting glucose ≤150 mg/dL, and HbA1c ≤9%. Exclusion criteria were: use of other investigational agents; major surgery within 4 weeks prior to treatment without complete recovery; uncontrolled systemic infection; systemic chemotherapy within14 days; central nervous system (CNS) disease; history of clinically significant heart disease; QTc prolongation >480 msec; uncontrolled hypertension; pregnancy and breast-feeding; HIV infection with CD4 cells prior to leukemia onset ≤400 cells/mm3 or AIDS-defining illness; and active hepatitis B or C. Cytogenetic risk-group assignment was done according to the refined NCRI/MRC criteria (32). Treatment responses were defined according to standard criteria (2, 33). The institutional review boards at MDACC and FHCRC approved this study (ClinicalTrials.gov: NCT01253447), and patients gave consent in accordance with the Declaration of Helsinki.
Treatment plan
Treatment consisted of MK-2206 200 mg orally once weekly, the maximum tolerated dose in patients with solid tumors (34). The weekly dosing was recommended by CTEP based on Phase I data demonstrating pAKT suppression in platelet-rich plasma up to 96 hours post-dose (34). With the exception of hydroxyurea during the first month of therapy, concomitant cytotoxic medications were prohibited. Patients were assessed for response after every 28-day cycle and were allowed to continue study treatment if there was no evidence of progressive disease or clinically significant toxicities. Patients achieving either CR, CR with incomplete platelet count recovery (CRp), or partial remission (PR) after 3 cycles of therapy could continue study treatment for up to 12 cycles. Patients not achieving an objective response but obtaining clinical benefit from the trial drug could also remain on study beyond 3 months if no significant toxicities occurred.
Statistical considerations
The study’s primary objective was to determine the proportion of patients achieving CR, CRp, or PR as best response within 3 cycles of therapy; secondary objectives included the estimation of disease-free survival of patients who achieved CR/CRp, and the establishment of the toxicity profile of MK-2206. A Simon two stage optimum design (35) was used to test the null hypothesis that the response rate (i.e. CR, CRp, or PR) at week 12 was ≤10% vs. the alternative that the response rate is ≥25% at an α level of .05 with 80% power. Patients who were removed from study therapy for reasons other than disease progression (e.g. non-compliance, fatal infections) before completion of 3 cycles of therapy were not included in the efficacy assessment, and additional patients were enrolled. With 3 or more responses in the first 18 patients, the trial would continue to accrue up to 43 patients; otherwise, the trial would stop early and the drug would be rejected for further study. With ≥8 responses among 43 patients enrolled, the drug would be considered effective. With this design, the probability of early termination was 73% if the true response rate was only 10%. With the concern of treatment-related toxicity, the non-hematologic toxicity (≥grade 3, with the exception of hyperglycemia) would also be continuously monitored during the study using established Bayesian-based methods (36). The stopping rule was set such that the trial would be halted if, at any time during the study, there was a >90% chance that the toxicity rate exceeded 30%.
Biomarker analyses
Biomarkers studies were optional and only performed in samples from patients consenting to these studies. Peripheral blood samples were collected before and 24 hours after the first administration of MK-2206 during the initial treatment cycle, and bone marrow samples were collected at baseline and before the second treatment cycle. Mononuclear cells were separated by Ficoll gradients, and cell lysates were subjected to reverse phase protein array (RPPA) analysis following previously described and validated methods (26–28). Slides were probed with 157 primary antibodies, including two recognizing pAKT (Thr308 and Ser473; both Cell Signaling Technology), a secondary antibody for signal amplification, and a stable dye for precipitation (29). The slides were scanned, and images quantified using Microvigene Version 2.9 software (Vigene Tech, Carlisle, MA). Raw signal intensity data was processed with SuperCurve to estimate relative protein concentrations, with further data normalization to adjust loading bias by median-centering each marker and median-centering each sample (30, 37). We used paired t-tests employing the Benjamini-Hochberg method for adjustment to control the false discovery rate (FDR) (38) to detect differential protein expression or phosphorylation of each protein in the following three subsets: (a) the target itself (pAKT Ser473 and Thr308); (b) selected proteins associated with AKT inhibition (4E-BP1-pS65, 4E-BP1-pT37-T46, Bim, Cyclin D1, FOXO3a pS318-S321, GSK3α-β pS21-S9, IRS1, Mcl1, mTOR pS2448, NF-kB-p65 pS536, p70S6K-pT389, PRAS40-pT246, PTEN, S6 pS235-S236, S6 pS240-S244, Survivin, and XIAP); and (c) all 157 proteins. In subsets (a) and (b), we used one-sided tests because we could specify the expected directions of change a priori. Heat maps of selected markers and samples were plotted in Supplemental Figure 4, where the data were median-centered for each marker.
Results
In Vitro Investigations of MK-2206 in Human AML Cells
In line with previous investigations in solid tumor cancer as well as ALL cells (18, 20, 39–41), treatment of human AML cell lines (OCI-AML3 and U937) with MK-2206 dose-dependently reduced Ser473 AKT phosphorylation but left total AKT levels unchanged (Figure 1A). Reflective of loss of AKT activity, both cell lines exhibited reduced phosphorylation of PRAS40 and the mTOR targets, S6 kinase (at Ser240) and 4EBP1 (Thr37/45) in response to MK-2206, albeit inhibition of these downstream targets required higher concentrations of MK-2206. Similar findings were seen in FLT3-mutated MOLM13 and MOLM14 cells (Supplemental Fig. 1). Consistent with these cell line data, treatment with MK-2206 suppressed Ser473 AKT phosphorylation, pPRAS40, and p4EPBP1 (Thr37/46) in 3 primary AML samples tested (Figure 1B).
Figure 1. Effects of MK-2206 on AKT signaling in Human AML Cells.
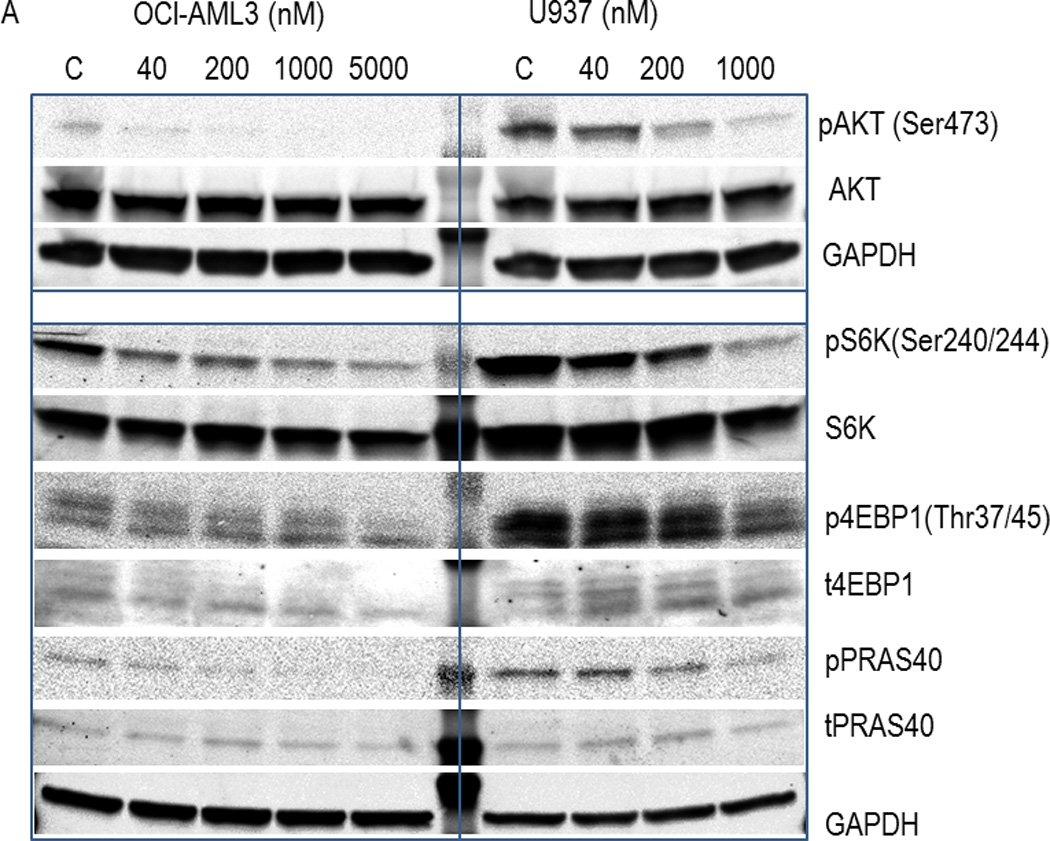
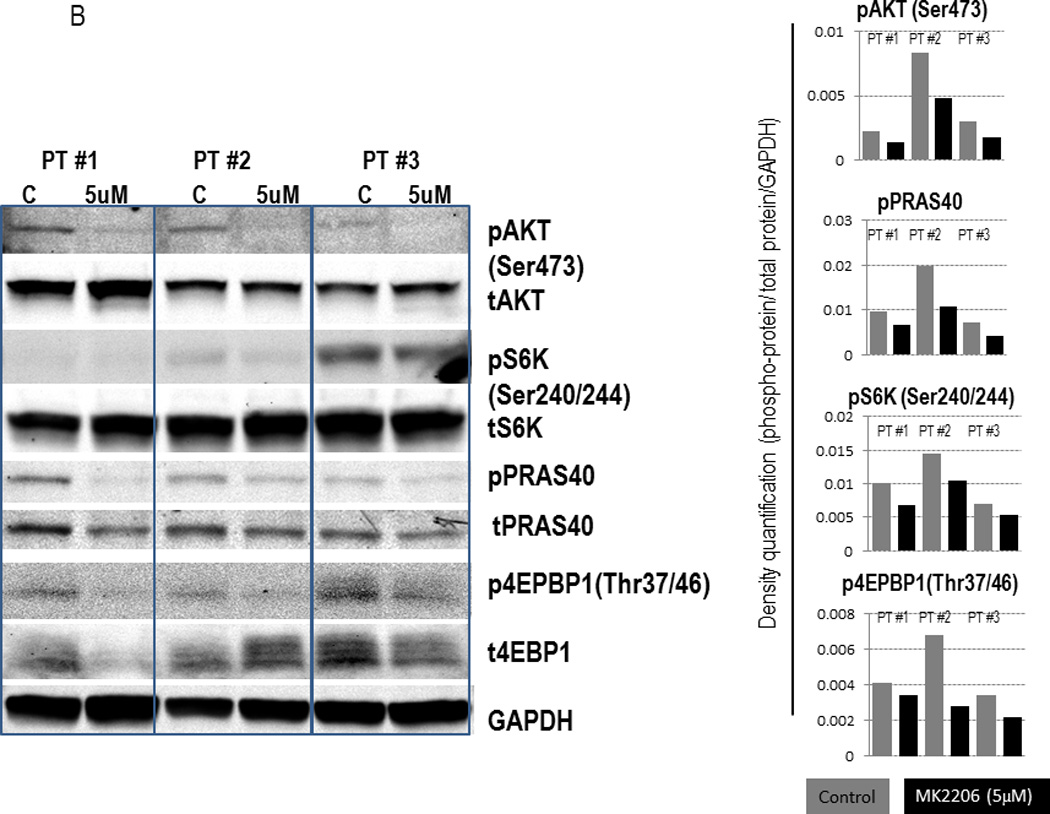
(A) OCI-AML3 and U937 cells were exposed to the indicated concentrations of MK-2206 for 6 hours. Whole cell lysates were subjected to immunoblot analyses with the indicated antibodies. (B) Mononuclear cells from AML patients (pt) with high (>50%) blast count were treated with MK-2206 (5 µM) for 24 hours. Expression of AKT signaling proteins was analyzed by immunoblotting (left) and quantified by densitometry (right). Pt#1, refractory AML, N-Ras mutated, del(12p); pt#2, refractory AML, diploid; pt#3, newly diagnosed AML arising from antecedent MDS, N-Ras mutated, del(5q) and -7.
Having confirmed target inhibition with MK-2206, we then investigated the drug’s effects on viability of various human AML cell lines. As shown in Table 1 and Supplemental Figure 2A, MK-2206 at 1 µM effectively suppressed cell growth in all cell lines except Mo7e cells after 72 hours, with OCI-AML3, MV4;11, MOLM14, THP1 but not U937 or MOLM13 cells exhibiting growth inhibition with as little as 0.1–0.2 µM of MK-2206. In turn, induction of apoptosis was only seen at higher doses (5–10 µM) of MK-2206 (Supplemental Figure 2A). Cell cycle analyses in OCI-AML3 cells indicated that MK-2206 induced G1 arrest, particularly at higher drug concentrations (Supplemental Figure 2B). Finally, we assessed the effects of MK-2206 in 2 primary AML specimens harboring FLT3 mutations and found blockade of AKT signaling, as indicated by inhibition of 4EBP1 phosphorylation and S6 (Figure 2). Furthermore, as shown in Figure 2, MK-2206 induced apoptosis in both samples after 48 hours. Together, these in vitro studies indicate that MK-2206 causes cell cycle arrest, growth inhibition, and – at higher drug concentrations – apoptosis in human AML cell lines and primary AML blasts.
TABLE 1.
Effects of MK-2206 on cell growth and survival in human AML cell lines.
| Cell line | Characteristics | IC50 (µM) |
|---|---|---|
| MOLM13 | FLT3-mutant | 1.16 |
| OCI-AML3 | NPM1-mutant N-Ras-mutant |
1 |
| U937 | PTEN-mutant | 1.48 |
| THP1 | N-Ras-mutant | 4.46 |
| Mo7e | N-Ras-mutant | >5 |
| MV4;11 | t(4;11), MLL-rearranged FLT3-mutant |
3.92 |
| MOLM14 | FLT3-mutant | 0.42 |
AML cells were cultured with increasing concentrations of MK-2206 (from 40nM up to 10µM, in triplicates)for 48 hours, after which effects on cell growth were determined by viable cell counts. IC50 values were calculated using Calcusyn software.
Figure 2. Effects of MK-2206 on cell growth and survival of primary AML cells.
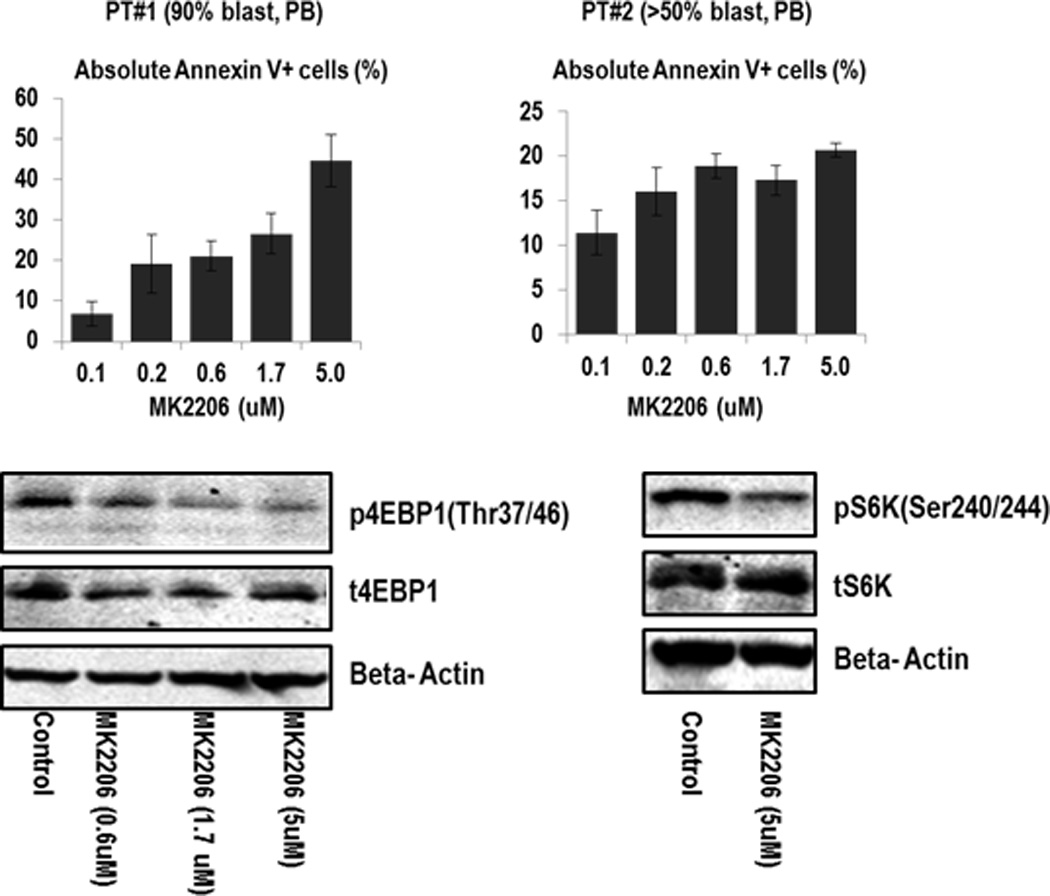
Primary AML cells from two AML patients were treated with MK-2206, and effects on apoptosis induction as examined by annexin V staining. Both patients had diploid karyotype; pt#1 had FLT3-ITD mutation, and pt#2 FLT3 D835 gene mutation. The extent of drug-specific apoptosis was assessed by the formula: % specific apoptosis = (test – control) × 100 / (100 – control).(49)
Clinical Trial
Given the above findings, we conducted a phase 2 trial of MK-2206 in adults with poor-prognosis AML to begin testing its clinical efficacy and tolerability in this disease. While initial clinical studies explored alternate dosing schedules (23), preclinical data indicated that weekly dosing would be at least as efficacious. Thus, later clinical studies investigated MK-2206 given once weekly (34). Building upon this experience, we administered MK-2006 at 200 mg weekly, the maximum tolerated dose established in patients with solid tumors (34).
Over a period of 2 years, 19 adults with a median age of 70 years were enrolled in this study and received at least 1 dose of MK-2206 (Table 2). The median number of cycles administered was 2, with 4 patients completing ≥3 cycles. Eighteen patients were eligible for efficacy and toxicity assessments. One patient was not included in these analyses because of study removal after 1 cycle due to non-compliance; at that time, however, this patient had stable disease and no evidence of drug-associated toxicities. Among the 18 eligible patients, only 1 CRp was observed (5.6%; exact 95% confidence interval: 0–17%). This patient was a 77-year old male with normal-karyotype secondary AML without FLT3, KIT or RAS mutations that arose from antecedent chronic myelomonocytic leukemia. He initially achieved CR with clofarabine/low-dose cytarabine but then relapsed and failed a first salvage therapy with twice-daily fludarabine and cytarabine. He achieved a CRp after the first cycle of MK-2206 with a decrease in bone marrow blasts from 13% to <5%. Subsequently, his leukemia burden fluctuated over the next several months of therapy between 4% and 15%, and he was removed from study after completion of 11 treatment cycles. In contrast, the 17 non-responders were removed from study for disease progression after 1 cycle (n=7), 2 cycles (n=8), 3 cycles (n=1), or 4 cycles (n=1), respectively.
TABLE 2.
Characteristics of Study Cohort
| Parameter | n=19 |
|---|---|
| Median Age (range), years | 70 (31–86) |
| Sex (male/female), n | 12/7 |
| Disease Stage, n (%) | |
| 1st Relapse | 10 (52.6%) |
| 2nd Relapse | 9 (47.4%) |
| Disease Manifestation, n (%) | |
| Bone marrow relapse | 18 (94.7%) |
| Isolated extramedullary relapse | 1 (5.3%) |
| Performance Status, n (%) | |
| 1 | 17 (89.5%) |
| 2 | 2 (10.5%) |
| Cytogenetic Risk Group, n (%)* | |
| Favorable | 0 |
| Intermediate | 13 |
| Unfavorable | 6 |
| Secondary AML, n (%) | 9 (47.4%) |
| Laboratory Findings at Baseline, median (range) | |
| WBC (× 103/µL)* | 1.7 (0.5–11.5) |
| Hemoglobin (g/dL) | 9.6 (8.4–13.2) |
| Platelets (× 103/µL) | 39 (11–1887) |
| LDH (U/L) | 455 (153–1222) |
| Creatinine (mg/dL) | 0.91 (0.6–1.69) |
| Total Bilirubin (mg/dL) | 0.6 (0.3–1.3) |
| SGOT (U/L) | 22 (11–42) |
| SGPT (U/L) | 27 (12–64) |
Based on revised MRC prognostic classification(32)
In general, MK-2206 monotherapy was reasonably well tolerated. However, as the most common grade 3/4 adverse event related to study drug, a pruritic rash occurred in 6/18 evaluable patients (33.3%); this side effect led to dose-reduction in 3 patients (135 mg/weekly) and discontinuation of drug in 1. Besides milder (grade 1/2) rashes in another 4 patients (22.2%), other adverse events were hyperglycemia (n=12 [66.7%], all grade 1/2 except for grade 4 in one patient), and grade 1 QTc prolongation (n=4 [22.2%]), respectively (Table 3). Ultimately, the study was terminated early after inclusion of 18 eligible patients because of insufficient drug efficacy, as per predefined stopping rules.
TABLE3.
Tolerability and Safety of Study Therapy
| Parameter | Grade 1–2 | Grade 3–4 |
|---|---|---|
| Fever, Infection | ||
| Neutropenic Fever | 2 | |
| Documented Infection | 6 | |
| Pneumonia | 2 | |
| Fever of Unknown Origin | 2 | |
| Cardiac | ||
| QTc prolongation | 4 | |
| Rash maculo-papular | 4 | 6 |
| Metabolic | ||
| Hyperglycemia | 11 | 1 |
| Other | ||
| PRES* syndrome | 1 |
Posterior reversible encephalopathy syndrome
Finally, we conducted proteomic analyses to investigate the degree of target inhibition with MK-2206 utilizing RPPA in paired peripheral blood specimens – obtained prior to and 24 hours after the first dose of MK-2206 – in 8 patients, as well as in paired bone marrow specimens - obtained prior to and 28 days after the first dose of MK-2206 – in 5 patients, respectively. Two patients (#1 and #17, the latter with extramedullary disease only) who had low pAKT levels in the baseline samples (less than −4.0 on log2 scale) were excluded from this analysis and, unfortunately, the sole responding patient did not consent to these optional biomarker studies. Considering AKT alone, the decrease in Ser473 AKT was significant (P=0.018). However, the larger sets (i.e. testing direct AKT targets or the entire 157 proteins) showed no significant changes after adjusting for multiple testing. Of note, a decrease in Ser473 AKT (median 28%; range, 12–45%) was seen in 5/7 peripheral blood and 3/4 bone marrow specimens, while a reduction in Thr308 AKT phosphorylation was found in 5/7 peripheral blood as well as 3/4 bone marrow samples, respectively (Figure 3A and 3B). In turn, changes in AKT targets were less evident (downregulation of pFOX3A in 3/7, pPRAS40 in 2/7, pS6K in 4/7 and p4EBP1 in 2/7 PB samples), possibly due to incomplete AKT inhibition. In samples with sufficient amounts of protein available, confirmatory immunoblotting was performed, which showed findings consistent with RPPA. Specifically, ≥50% downregulation of pAKT-473 was seen in peripheral blood samples from subjects #2, 3, 7 and in bone marrow sample #6, with lesser decreases in the mTOR targets, pS6 and p4EBP1 (Figure 4A and 4B), with changes in pAKT being highly concordant between the two techniques (P=0.01, Supplemental Figure 3). With regard to downstream targets or other proteins including phosphorylated MAPK, no significant changes were found after adjustment for multiple testing by RPPA (Figure 3A, last column). We further analyzed a subset of proteins recently reported to be upregulated in a compensatory fashion upon PI3K/AKT blockade. RPPA analysis indeed demonstrated upregulation of a subset of these proteins, such as Bcl-2 (P=0.036), Smad3 (P=0.028), HER2 (P=0.064), pY705 Stat-3 (P=0.033), p38 MAPK (P=0.039) and MEK1 (P=0.01), although these changes did not remain significant after accounting for multiple comparisons (Supplemental Figure 4).
Figure 3. Modulation of protein expression in AML cells by MK-2206 in peripheral blood (PB, 3A) or bone marrow (BM, 3B).
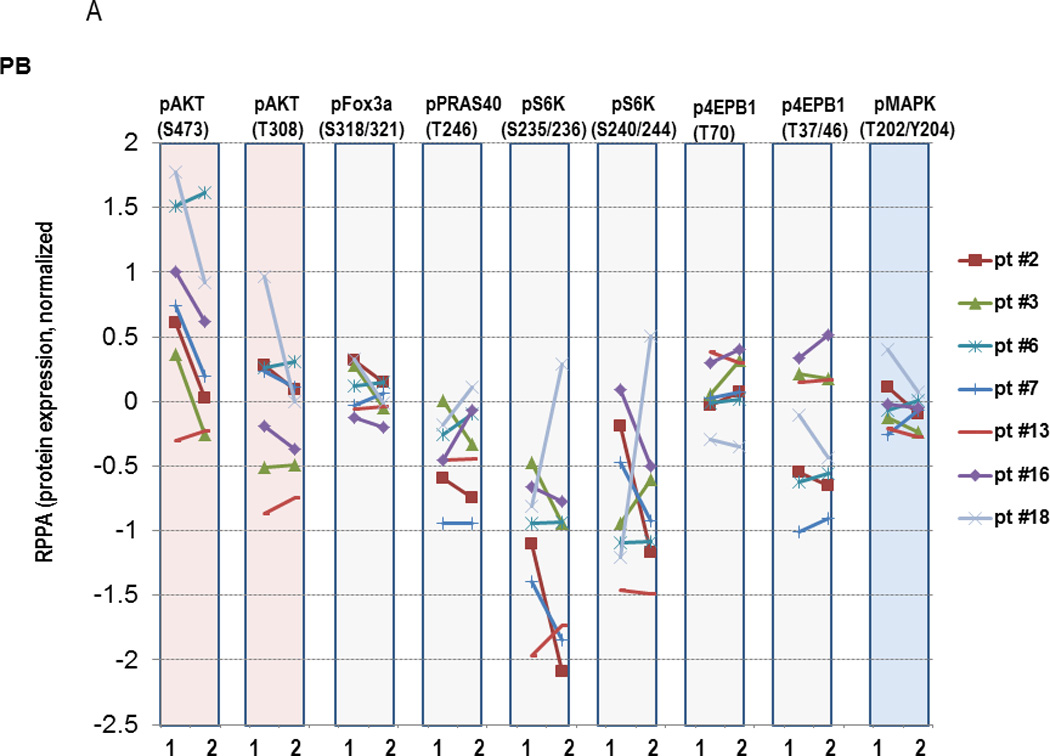
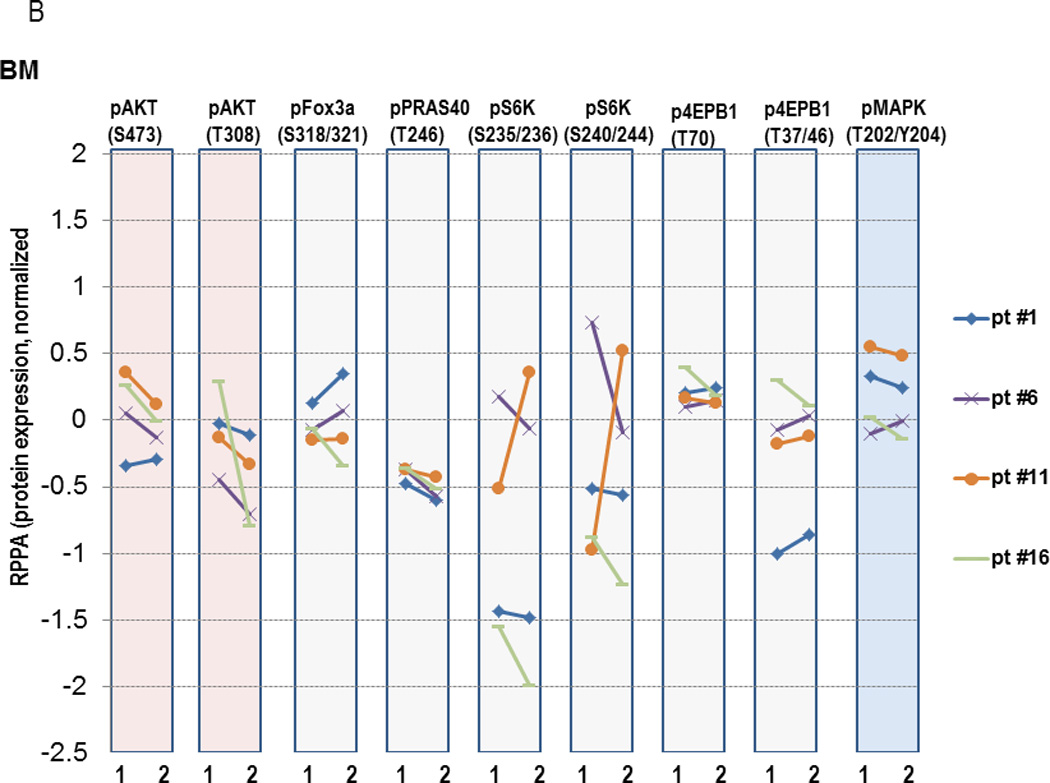
PB or BM mononuclear cells were collected prior to treatment (“1”) or at selected time-points after initiation of MK-2206 (“2”), which was 24 hours for PB samples and at the completion of the 1st cycle of therapy for BM samples. Protein lysates were subjected to RPPA analysis. Y-axis, normalized log2-transformed relative values of protein expression for the indicated proteins.
Figure 4. Immunoblot analysis of PB samples prior to and after MK-2206 exposure in selected patients treated on the trial.
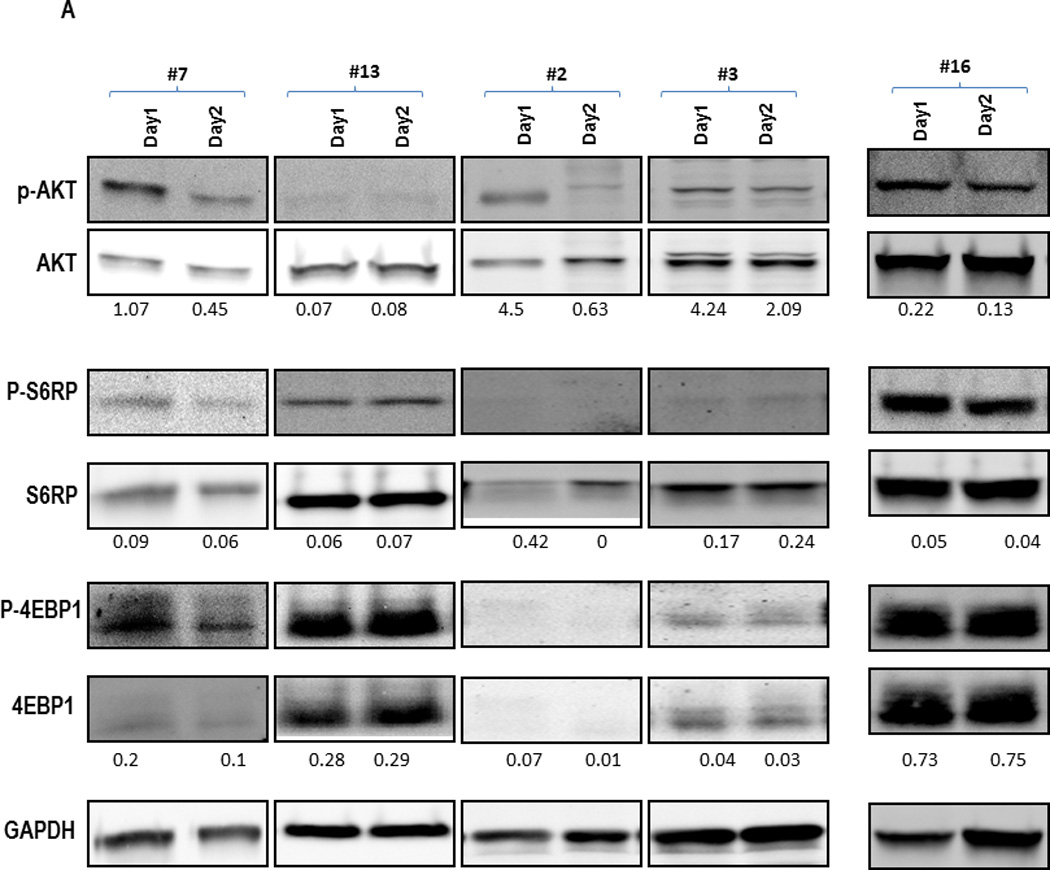
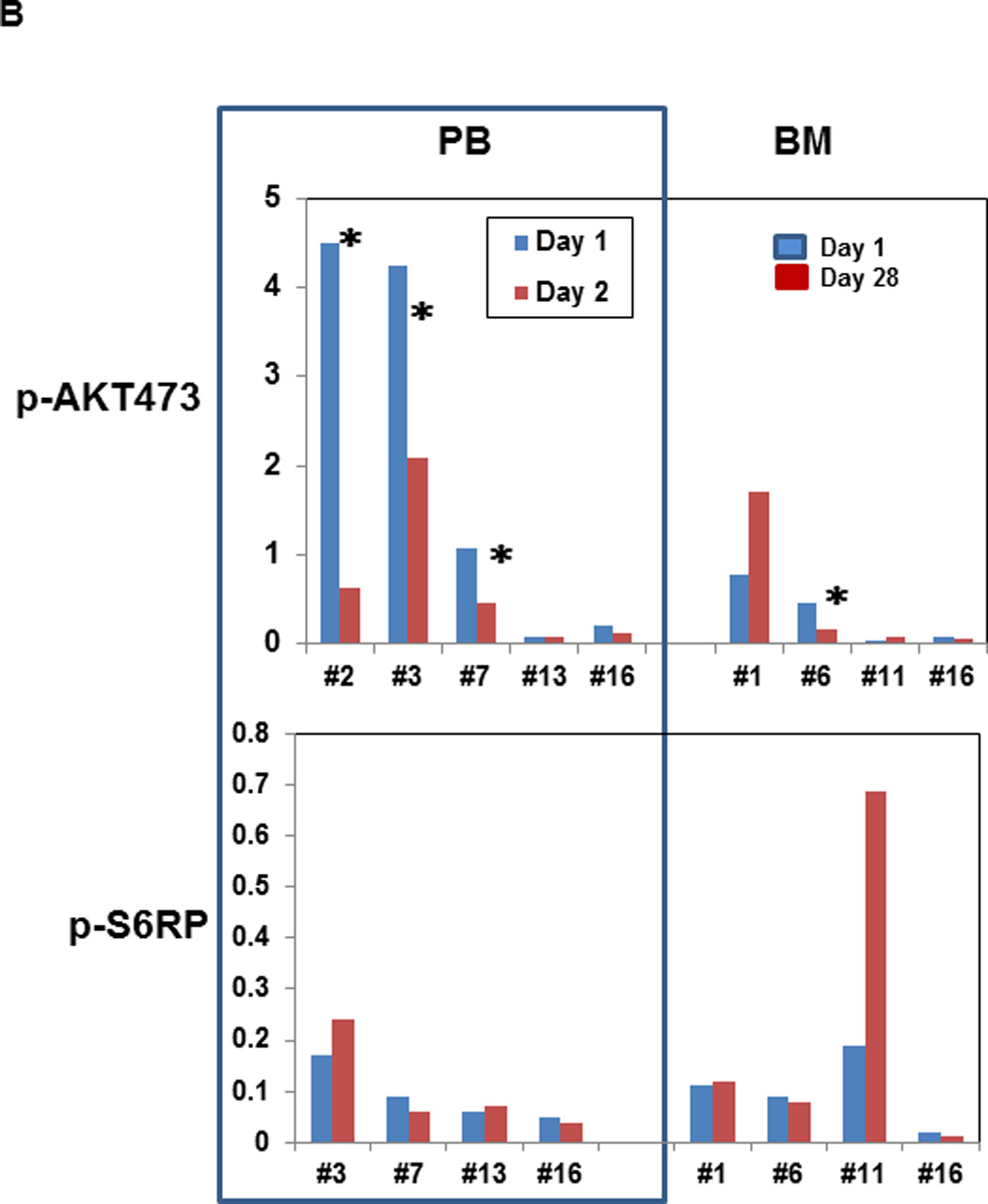
Peripheral blood samples were collected before and 24 hours after the first administration of MK-2206 during the initial treatment cycle. Mononuclear cells were separated by Ficoll-Hypaque (Sigma-Aldrich, St Louis, MO) density gradient centrifugation, cells were lysed in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA), transferred to Hybond-P membranes (GE Healthcare, Little Chalfont, United Kingdom), probed with the appropriate antibodies (AKT, Ser 473–phosphorylated AKT, S6 Ribosomal Protein, Ser240/244-phosphorylated S6Ribosomal Protein, 4E-BP1, Thr37/46-phosphorylated 4E-BP1 from Cell Signaling Technologies [Beverly, MA]) and visualized using LI-COR Odyssey system(LI-COR Biosciences, Lincoln, Nebraska). (A) Examples of Western blot for selected AKT targets. (B) Intensity of the immunoblot signals was quantified and the relative intensity compared to total protein (AKT and S6K, respectively) calculated. *Indicates patients with ≥50% decrease in the density of the protein.
Discussion
The in vitro data presented in this study showing that AKT inhibition impairs the survival of AML cells provide further rationale for selecting this signaling pathway as pharmacologic target in AML. However, our findings also indicate that the oral AKT inhibitor, MK-2206, has insufficient activity as a single agent in patients with relapsed/refractory AML at the chosen dose. While initial studies with MK-2206 were conducted using every-other-day drug dosing, a weekly dosing schedule was explored because of the drug’s long half-life (60–80h) and an attempt to ameliorate feedback activation of alternative signaling pathways in response to continuous AKT inhibition. Despite the fact that MK-2206 was reasonably well tolerated in our study cohort, the frequent occurrence of grade 3/4 rash suggests that dose-escalation at the weekly schedule may not be feasible. Skin toxicity was also a prominent toxicity in solid tumor patients who received MK-2206 at doses higher than 60 mg every other day (23), a finding that disabled escalation of drug dosing in those patients.
In our study population, an objective response was documented in only one patient, indicating suboptimal activity of MK-2206. Failure of targeted anti-cancer agents has been attributed to multiple causes, including (a) use in the “wrong” patient population, e.g. because tumor cells do not depend on the specific pathway; (b) rapid activation of the compensatory survival pathways; and (c) incomplete target inhibition. In AML, activation of the AKT pathway is generally attributed to mutation(s) and hyperactivation of the upstream receptor tyrosine kinases (RTKs) such as KIT, FLT3, or RAS, without activating mutations in PI3K or AKT itself. If activating mutations in AKT are present, cells may become hypersensitive to MK-2206 (42). Since the present study did not include patients with such mutations, potential activity of MK-2206 in selected patient populations may have been missed.
To assess the response to AKT inhibition and the potential for compensatory activation of other pathways, we interrogated intracellular signaling in AML blasts using RPPA. Recent evidence suggests that the acquired resistance to PI3K/AKT inhibition stems from the upregulation and activation of RTKs through mechanisms that involve FOXO-regulated transcription (43) and of other cellular survival proteins, including anti-apoptotic proteins (Bcl-2, XIAP1) and transcription factors (p-STAT3, p-STAT6, p-c-Jun, p-SMAD3), in part through cap-independent translation (44). RPPA analysis indeed demonstrated upregulation of a subset of pro-survival proteins, including Bcl-2, Smad3, pY705 Stat-3, p38 MAPK and MEK1. However, MK-2206 failed to substantially downregulate p-FOXO3A and did not significantly modulate other FOXO targets, indicating FOXO-independent upregulation of these proteins. Further studies with MK-2206 or other PI3K/AKT inhibitors may help characterize AML-specific compensatory pathways induced in response to AKT blockade, paving the way for further combination strategies. Our correlative studies indicate that the main clinical limitation of MK-2206 may be incomplete target inhibition, with <50% decrease in pAKT expression at 24 hours. Although the degree of AKT inhibition was likely higher at earlier time-points, we purposefully chose a 24-hour time-point to estimate sustained AKT inhibition, consistent with prior PD assessments (23, 34). Not surprisingly, non-sustained AKT blockade translated into insubstantial inhibition of known AKT downstream targets. Our in vitro studies indicated that MK-2206 could inhibit AKT phosphorylation at low concentrations (of 40–200 nM). However, higher doses (1–5 µM) were required for complete inhibition of downstream targets, and only such higher doses translated into AML growth suppression. While our study did not include pharmacokinetic assessments, prior Phase I data (34) showed Cmax concentrations ranging from approximately 150 nM to 400 nM at 200 mg weekly dosing. Combined, these data indicate below-target exposures in vivo at tolerable drug doses, indicating limited potential of further development of MK-2206 as a single agent. It is interesting to speculate whether a different dosing schedule (e.g. one that is optimized based on pharmacokinetic/dynamic measurements) could provide better results. While such a strategy might improve target inhibition, it will need to be approached carefully as the clinical experience obtained with MK-2206 thus far indicates significant toxicities, which may worsen as target inhibition is improved. Alternatively, our findings might suggest that other (e.g. combinatory) approaches to block AKT signaling should be explored in AML. As such, additive or synergistic anti-tumor effects were observed upon combination of MK-2206 with standard chemotherapeutic agents and targeted agents (17, 18, 43, 46–48).
In summary, while our preclinical data support further development of AKT targeting agents in AML, this first AML trial with an allosteric AKT inhibitor failed to show clinical benefit at tolerated doses. Evaluation of MK-2206 together with other agents affecting the AKT signaling pathway and/or other AKT inhibitors alone or in combination, coupled with detailed biomarker analyses, may be necessary to define the potential utility of such small molecule inhibitors in the treatment armamentarium for AML.
Supplementary Material
Translational Relevance.
Outcomes for patients with acute myeloid leukemia (AML) remain largely unsatisfactory. Recent studies have suggested that AKT activation is common in AML and may be associated with worse survival, providing the rationale for therapeutically targeting this signaling pathway. In this report, we establish the preclinical activity of the AKT inhibitor, MK-2206, against human AML cells and describe its efficacy in AML patients requiring second salvage treatment for relapsed/refractory disease. Our study fails to demonstrate the desired clinical benefit but reveals insufficient target modulation at maximally tolerated drug doses. These results emphasize the importance of pharmacodynamic monitoring in clinical trials of targeted agents and support investigation of more potent AKT pathway modulators in AML.
Acknowledgements
We acknowledge the provision of MK-2206 by NCI/CTEP.
Grant Support
This work was supported by grants R21-CA159285 (M.Y.K. and R.B.W.), P30-CA015704-35S6 (R.B.W.) and MDACC Core grant CA16672 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests.
Authors’ Contributions
Conception and design: MY Konopleva, RB Walter
Development of methodology: MY Konopleva, RB Walter, Z Zeng, PP Ruvolo, SM Kornblau, M Andreeff, EH Estey
Acquisition of data: MY Konopleva, RB Walter, SH Faderl, EJ Jabbour, G Borthakur, TM Kadia, JA Burger, EH Estey
Analysis and interpretation of data: X Huang, W Liu, KA Baggerly
Writing, review, and/or revision of the manuscript: RB Walter, MY Konopleva, LA Doyle, HM Kantarjian, EH Estey
Administrative, technical, or material support: JB Feliu
Study supervision: HM Kantarjian, EH Estey, LA Doyle
References
- 1.Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 3.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 4.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martelli AM, Evangelisti C, Chiarini F, McCubrey JA. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1:89–103. doi: 10.18632/oncotarget.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102:972–980. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- 7.Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK, et al. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2003;17:995–997. doi: 10.1038/sj.leu.2402874. [DOI] [PubMed] [Google Scholar]
- 8.Zhao S, Konopleva M, Cabreira-Hansen M, Xie Z, Hu W, Milella M, et al. Inhibition of phosphatidylinositol 3-kinase dephosphorylates BAD and promotes apoptosis in myeloid leukemias. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2004;18:267–275. doi: 10.1038/sj.leu.2403220. [DOI] [PubMed] [Google Scholar]
- 9.Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwable J, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer research. 2005;65:9643–9650. doi: 10.1158/0008-5472.CAN-05-0422. [DOI] [PubMed] [Google Scholar]
- 10.Grandage VL, Gale RE, Linch DC, Khwaja A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2005;19:586–594. doi: 10.1038/sj.leu.2403653. [DOI] [PubMed] [Google Scholar]
- 11.Gallay N, Dos Santos C, Cuzin L, Bousquet M, Simmonet Gouy V, Chaussade C, et al. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2009;23:1029–1038. doi: 10.1038/leu.2008.395. [DOI] [PubMed] [Google Scholar]
- 12.Muranyi AL, Dedhar S, Hogge DE. Combined inhibition of integrin linked kinase and FMS-like tyrosine kinase 3 is cytotoxic to acute myeloid leukemia progenitor cells. Experimental hematology. 2009;37:450–460. doi: 10.1016/j.exphem.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS, et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer research. 2004;64:5225–5231. doi: 10.1158/0008-5472.CAN-04-0174. [DOI] [PubMed] [Google Scholar]
- 14.Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M, et al. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108:2358–2365. doi: 10.1182/blood-2006-02-003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, McQueen T, Priebe W, et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer research. 2007;67:684–694. doi: 10.1158/0008-5472.CAN-06-3166. [DOI] [PubMed] [Google Scholar]
- 16.Yan L. MK-2206: a potent oral allosteric AKT inhibitor [abstract #DDT01-1]. 2009 AACR Annual Meeting. Denver, CO: 2009. [Google Scholar]
- 17.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Molecular cancer therapeutics. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 18.Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PloS one. 2010;5:e14124. doi: 10.1371/journal.pone.0014124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen KF, Chen HL, Tai WT, Feng WC, Hsu CH, Chen PJ, et al. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. The Journal of pharmacology and experimental therapeutics. 2011;337:155–161. doi: 10.1124/jpet.110.175786. [DOI] [PubMed] [Google Scholar]
- 20.Liu R, Liu D, Trink E, Bojdani E, Ning G, Xing M. The Akt-specific inhibitor MK2206 selectively inhibits thyroid cancer cells harboring mutations that can activate the PI3K/Akt pathway. The Journal of clinical endocrinology and metabolism. 2011;96:E577–E585. doi: 10.1210/jc.2010-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pant A, Lee II, Lu Z, Rueda BR, Schink J, Kim JJ. Inhibition of AKT with the orally active allosteric AKT inhibitor, MK-2206, sensitizes endometrial cancer cells to progestin. PloS one. 2012;7:e41593. doi: 10.1371/journal.pone.0041593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, et al. Biomarkers of Response to Akt Inhibitor MK-2206 in Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:5816–5828. doi: 10.1158/1078-0432.CCR-12-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 24.Petrich AM, Leshchenko V, Kuo PY, Xia B, Thirukonda VK, Ulahannan N, et al. Akt inhibitors MK-2206 and nelfinavir overcome mTOR inhibitor resistance in diffuse large B-cell lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:2534–2544. doi: 10.1158/1078-0432.CCR-11-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2012 doi: 10.1038/leu.2012.136. [DOI] [PubMed] [Google Scholar]
- 26.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Molecular cancer therapeutics. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 27.Kornblau SM, Tibes R, Qiu YH, Chen W, Kantarjian HM, Andreeff M, et al. Functional proteomic profiling of AML predicts response and survival. Blood. 2009;113:154–164. doi: 10.1182/blood-2007-10-119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornblau SM, Qiu YH, Zhang N, Singh N, Faderl S, Ferrajoli A, et al. Abnormal expression of FLI1 protein is an adverse prognostic factor in acute myeloid leukemia. Blood. 2011;118:5604–5612. doi: 10.1182/blood-2011-04-348052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunyady B, Krempels K, Harta G, Mezey E. Immunohistochemical signal amplification by catalyzed reporter deposition and its application in double immunostaining. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1996;44:1353–1362. doi: 10.1177/44.12.8985127. [DOI] [PubMed] [Google Scholar]
- 30.Hu J, He X, Baggerly KA, Coombes KR, Hennessy BT, Mills GB. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- 31.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 32.Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–365. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- 33.Cheson BD, Bennett JM, Kopecky KJ, Büchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 34.Biondo A, Yap TA, Yan L, Patnaik A, Fearen I, Baird RD, et al. Phase I clinical trial of an allosteric AKT inhibitor, MK-2206, using a once weekly (QW) dose regimen in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29 doi: 10.1200/JCO.2011.35.5263. abstract #3037. [DOI] [PubMed] [Google Scholar]
- 35.Simon R. Optimal two-stage designs for phase II clinical trials. Controlled clinical trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 36.Thall PF, Simon RM, Estey EH. Bayesian sequential monitoring designs for single-arm clinical trials with multiple outcomes. Statistics in medicine. 1995;14:357–379. doi: 10.1002/sim.4780140404. [DOI] [PubMed] [Google Scholar]
- 37.Coombes KR, Liu W, Ju Z, Roebuck P. SuperCurve. 2012. Version 1.4.3:[Available from: http://bioinformatics.mdanderson.org/main/SuperCurve:Overview. [Google Scholar]
- 38.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 39.Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X, et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Molecular cancer therapeutics. 2012;11:154–164. doi: 10.1158/1535-7163.MCT-11-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bressanin D, Evangelisti C, Ricci F, Tabellini G, Chiarini F, Tazzari PL, et al. Harnessing the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia: Eliminating activity by targeting at different levels. Oncotarget. 2012;3:811–823. doi: 10.18632/oncotarget.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stegeman H, Kaanders JH, Wheeler DL, van der Kogel AJ, Verheijen MM, Waaijer SJ, et al. Activation of AKT by hypoxia: a potential target for hypoxic tumors of the head and neck. BMC cancer. 2012;12:463. doi: 10.1186/1471-2407-12-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorlick R, Maris JM, Houghton PJ, Lock R, Carol H, Kurmasheva RT, et al. Testing of the Akt/PKB inhibitor MK-2206 by the Pediatric Preclinical Testing Program. Pediatric blood & cancer. 2012;59:518–524. doi: 10.1002/pbc.23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Speranza G, Kinders RJ, Khin S, Weil MK, Do KT, Horneffer Y, et al. Pharmacodynamic biomarker-driven trial of MK-2206, an AKT inhibitor, with AZD6244 (selumetinib), a MEK inhibitor, in patients with advanced colorectal carcinoma (CRC) J Clin Oncol. 2012;30 abstract#3529. [Google Scholar]
- 46.Floc'h N, Kinkade CW, Kobayashi T, Aytes A, Lefebvre C, Mitrofanova A, et al. Dual targeting of the Akt/mTOR signaling pathway inhibits castration-resistant prostate cancer in a genetically engineered mouse model. Cancer research. 2012;72:4483–4493. doi: 10.1158/0008-5472.CAN-12-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grabinski N, Ewald F, Hofmann BT, Staufer K, Schumacher U, Nashan B, et al. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Molecular cancer. 2012;11:85. doi: 10.1186/1476-4598-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ewald F, Grabinski N, Grottke A, Windhorst S, Norz D, Carstensen L, et al. Combined targeting of AKT and mTOR using MK-2206 and RAD001 is synergistic in the treatment of cholangiocarcinoma. International journal of cancer Journal international du cancer. 2013 doi: 10.1002/ijc.28214. [DOI] [PubMed] [Google Scholar]
- 49.Kojima K, Konopleva M, McQueen T, O'Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108:993–1000. doi: 10.1182/blood-2005-12-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.