

Abstract
Background
Atrial fibrillation (AF) risk has been associated with “leaky” ryanodine receptor (RyR2) Ca release channels. Patients with mutations in RyR2 or in the sarcoplasmic reticulum Ca binding protein calsequestrin (Casq2) display an increased risk for AF. Here we examine the underlying mechanisms of AF associated with loss of Casq2 and test mechanism-based drug therapy.
Methods and Results
Compared to wild-type mice, atrial burst pacing consistently induced atrial flutter or AF in Casq2−/− mice and in isolated Casq2−/− hearts. Atrial optical voltage maps obtained from isolated hearts revealed multiple independent activation sites arising predominantly from the pulmonary vein (PV) region. Ca and voltage mapping demonstrated sub-threshold diastolic Ca elevations (SCaE) and delayed afterdepolarizations (DADs) whenever the pacing train failed to induce AF. The dual RyR2 and Na channel inhibitor R-propafenone (3μM) significantly reduced frequency and amplitude of SCaE and DADs in atrial myocytes and intact atria and prevented induction of AF. In contrast, the S-enantiomer of propafenone, an equipotent Na channel blocker but much weaker RyR2 inhibitor, did not reduce SCaE and DADs and failed to prevent AF.
Conclusions
Loss of Casq2 increases risk of AF by promoting regional SCaE and DADs in atrial tissue, which can be prevented by RyR2 inhibition with R-propafenone. Targeting AF due to “leaky” RyR2 Ca channels with R-propafenone may be a more mechanism-based approach to treating this common arrhythmia.
Keywords: atrial fibrillation, ryanodine receptor, catecholaminergic polymorphic ventricular tachycardia, propafenone, ryanodine receptor inhibitors
Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia in the general population.1 While the etiology of AF is multifactorial, there is increasing evidence that AF is a heritable disorder especially in younger patients without underlying structural heart or systemic diseases (‘lone’ AF).2 Mutations in the cardiac ryanodine receptor (RyR2) Ca release channel or in the major sarcoplasmic reticulum (SR) Ca binding protein, cardiac calsequestrin (Casq2) have been associated with increased risk of AF.3 Patients with these mutations usually display episodes of catecholamine-induced polymorphic ventricular tachycardia (CPVT) in response to physical or emotional stress.4 Experimental work has established that ventricular arrhythmias are caused by premature Ca release from the SR that promote delayed after depolarizations (DADs) and triggered activity.5 A similar mechanism may promote AF in the atria. Consistent with this hypothesis, recent reports suggest that increased SR Ca leak (e.g., due to RyR2 mutations6 or RyR2 phosphorylation by CaMKII7) is associated with increased risk of AF in vivo. Moreover, atrial myocytes isolated from patients with chronic AF exhibit impaired intracellular Ca handling. In particular, human AF myocytes show an increased incidence of SR spontaneous Ca releases relative to SR Ca content8 comparable to what has been reported in the Casq2−/− CPVT mouse model. However, the cause-effect link between the rate of diastolic Ca release in atrial cells and incidence of AF in vivo has not been shown. This may in part be due to the difficulty of identifying and localizing sub-threshold Ca elevations in intact atria of small animal models. Here, we used voltage and Ca optical mapping in intact hearts and isolated atrial myocytes to test the hypothesis that loss of Casq2 causes spontaneous Ca release in atrial tissue and increases susceptibility to AF in structurally-normal atria by promoting DADs and triggered activity. We further hypothesized that Ca-triggered AF can be prevented by a mechanism-based approach of suppressing spontaneous Ca release with the R-enantiomer of propafenone, which is a much more potent RyR2 Ca release channel inhibitor than S-propafenone.9
Methods
Detailed methods are provided in the online supplement.
Animal use
All studies were carried out according to National Institutes of Health guidelines and were approved by the institutional animal care and use committees at Vanderbilt University.
AF induction
Casq2+/+ and Casq2−/− hearts were harvested and perfused in Langendorff mode as previously described.10 AF was induced by delivering repeated trains of atrial burst pacing (50Hz 2s). Volume conducted EKG was recorded continuously: AF was defined as rapid and fragmented atrial electrograms present for at least 150ms. Incidence and duration of AF episodes were quantified in both Casq2+/+ and Casq2−/− hearts.
Optical mapping
Isolated perfused hearts from Casq2+/+ and Casaq2−/− mice were stained with either di-4-ANEPPS or Rhod 2 dye. Both voltage and Ca maps were acquired with a RedShirt charge-coupled device camera (14-bit, 80 × 80 pixels, 1,000 fps, CardioCCD-SMQ; RedShirt Imaging), during the post pacing interval. All maps were analysed by one operator in blinded fashion with MATLAB (Mathworks, Natick, MA) using custom algorithms. Voltage maps were used to study atrial activation during both sinus rhythm and AF and to quantify the incidence of DADs in Casq2+/+ and Casq2−/− atria. In addition, activation maps of the atria during epicardial pacing at constant cycle lengths were obtained to measure atrial conduction velocity (CV) and action potential duration at 90% of repolarization levels (APD90). Incidence of spontaneous diastolic Ca elevations (SCaE) in the atria of perfused hearts was investigated by generating Ca fluorescent maps in the post pacing interval (pacing bursts at 50Hz, 2s). The amplitude of every SCaE recorded was quantified as a percentage of the Ca transient amplitude during pacing. Only elevations of at least 10% of the preceding atrial Ca transient during pacing were considered for the analysis.
To test the effect of class I antiarrhythmic drugs on SCaE and AF, Casq2−/− isolated perfused hearts stained with Rhod2 AM underwent the pacing protocol twice: first during vehicle infusion and then in the presence of R-propafenone (3μM), S-propafenone (3μM) or lidocaine (20 μM), respectively. The lidocaine concentration was chosen to achieve a similar degree of Na channel block as 3 μM of R-propafenone and S-propafenone as evidenced by a comparable increase in the QRS interval (around 25%). Drugs were continuously infused for 15 min before pacing was resumed. AF episodes as well as number and amplitude of SCaE after R-propafenone, S-propafenone or lidocaine infusion were quantified and compared to the same parameters obtained with vehicle.
Isolated myocyte studies
Atrial myocytes were isolated from Casq2+/+ and Casq2−/− atria. First, isolated hearts were perfused for 7–8 min with tyrode buffer containing collagenase type II (25mg) and protease (2mg) to obtain a primary digestion. Then, atria were removed and exposed to a secondary enzyme digestion for an additional 15–30 min. For permeabilized myocyte studies, cells were placed in laminin-coated chambers, washed with relaxing solution for 30s and chemically permeabilized by exposure to internal solution containing saponin (40 μg/ml). After one minute, the saponin solution was replaced by internal solution containing Fluo 4 pentapotassium salt (0.02 mM). The permeabilized cells were imaged in the line-scan mode after 10 minutes incubation with either vehicle or R-propafenone (10 μM) and analyzed as described.11 For intact myocyte studies, cells were loaded with Fura-2 AM and spontaneous Ca waves and trigger beats were measured as described.12
Statistical analysis
Data are presented as mean±SEM. Number and duration of AF episodes, Ca sparks and Ca wave frequency and amplitude in isolated myocytes, DADs and SCaE frequency and amplitude were compared by means of the Mann-Whitney U test. Incidence of AF was compared by Fisher exact test. A 2-tailed p value <0.05 was considered statistically significant. For statistical comparison of more than 2 groups, the Mann-Whitney U test and Fisher’s exact test were used for post-hoc analysis whenever the non-parametric analysis of variance (Kruskal–Wallis ANOVA) showed an overall statistically-significant difference.
Result
AF susceptibility of Casq2−/− mice and isolated Casq2−/− hearts
We first determined AF susceptibility in vivo by transesophageal atrial burst pacing. Consistent with other mouse models with increased Ca leak,13 atrial burst pacing resulted in a significantly higher AF burden and AF duration in Casq2−/− mice compared to wild-type Casq2+/+ littermates (Supplemental Fig. 1). To investigate the underlying mechanism for the AF susceptibility, volume-conducted EKGs and optical voltage maps were recorded simultaneously in Casq2−/− and Casq2+/+ isolated perfused hearts. Neither group displayed spontaneous episodes of AF. AF susceptibility was tested with repeated trains of atrial burst pacing (Fig. 1). Atrial burst pacing did not induce significant episodes (>150ms duration) of atrial tachyarrhythmia in wild-type hearts (0/10 hearts, Fig. 1). In Casq2−/− hearts, burst pacing induced frequent runs of atrial tachyarrhythmias resembling atrial flutter (Fig. 1B) and more disorganized forms resembling AF (Fig. 1C). For data analysis, both atrial flutter and more disorganized forms were grouped together and labelled as AF. A total of 111 AF episodes were recorded in 11 out of 12 Casq2−/− hearts (9.3±2.6 episodes/heart). Approximately 14% of all pacing trains triggered AF. Once induced, AF lasted on average for 21.9±13.1 s (Fig 1D).
Figure 1.

Casq2−/− hearts are susceptible to pacing induced AF. AF susceptibility was evaluated by atrial burst pacing (50Hz, 2s). Representative ECG records from Casq2+/+ (A) and Casq2−/− (B,C) hearts. Atrial tachyarrhythmias resembling atrial flutter (B) and AF (C) were observed in Casq2−/− hearts. For statistical analysis, episodes of AF and atrial flutter were grouped together and labelled as AF. Average rate (D) and duration (E) of AF in each group. n = 10 Casq2+/+ and 12 Casq2−/−, **P<0.01***P<0.001)
Next, we compared electrical activation in Casq2−/− atria during sinus rhythm and AF (Fig. 2). During sinus rhythm, atrial activation started from one single activation site consistent with the sinoatrial node region near the crista terminalis in the posterior right atrium (Fig. 2A). The activation wavefronts then spread simultaneously in two directions: to the right atrial appendage and across the pulmonary vein region of the left atrium towards the left atrial appendage. Atrial activation was completed within 12 ms (Fig. 2A). During AF, atrial activation occurred quasi-simultaneously from multiple independent sites (usually 2 or 3) from both atria (Fig. 2B), with independent foci observed as close as 1 mm. The ectopic activation was preceded by electrical silence. A re-entrant pattern of atrial activation was not observed.
Figure 2.

Representative optical voltage maps of Casq2−/− atria during sinus rhythm (A) and AF (B). Voltage maps were recorded from the posterior aspect of the heart. Asterisks indicate points of earliest activation. The corresponding bright-field image is in the leftmost panels. LA left atrium, RA right atrium, PV pulmonary veins, IVC inferior vena cava. The square in the ECG record indicates time of optical maps. Bottom right panels: Composite activation maps. Note that during sinus rhythm (A), only a single focal activation originates in the posterior aspect of the right atrium close to the crista terminalis where the sinoatrial node is located. Depolarization wavefronts spread towards the right appendage and the left atrium travelling across the posterior atrial walls. During AF (B), three independent depolarizations appear quasi simultaneously (two from the PV region, one near the IVC). Activation wavefronts spread only to a limited area surrounding the respective foci, followed by electrical silence (panel 15 ms), and repetitive activation from the same foci (panel 18 ms).The AF activation map (lower right panel) shows near-simultaneous activation originating from anatomically-distinct foci. C) Anatomical origin of atrial activation during AF. Left panel: Summary of atrial activation sites during AF episodes in 7 Casq2−/− hearts. Size of the circles is proportional to number of events arising from that specific area. Right panel: Distribution of atrial activation sites by region: right atrium (RA), pulmonary vein region (PV) and left atrial appendage (LA).
We quantified the ectopic activations sites observed during AF by anatomical region: right atrium, pulmonary vein region and left atrial appendage (Fig. 2C). Most of the AF activation sites were found in the pulmonary vein area (48%), especially from the lower portion of the pulmonary vein region. The remaining activation sites were equally distributed among both atrial appendages, with 25% of activation sites in the LA and 27% in the RA (Fig. 2C). Interestingly, the sinoatrial node region was spared. Taken together, the spatial and temporal activation pattern and the regional distribution suggest that focal activity (i.e., by DADs14) was responsible for triggering AF in Casq2−/− hearts.
To test this hypothesis, we examined voltage maps of pacing trains that failed to induce AF for evidence of DADs. As DADs are best observed in the pause following a pacing train, for this analysis we only examined maps with a post pacing pause of at least 100 ms before atrial or ventricular activation resumed. DADs were observed in 43 of the 95 voltage maps analyzed (45%, Fig. 3A). Consistent with the anatomical sites of atrial activation during AF (Fig. 2C), the majority (50%) of DADs were detected in the pulmonary vein region (Fig. 3B). Pacing-induced DADs were never observed in Casq2+/+ hearts (n=4).
Figure 3.
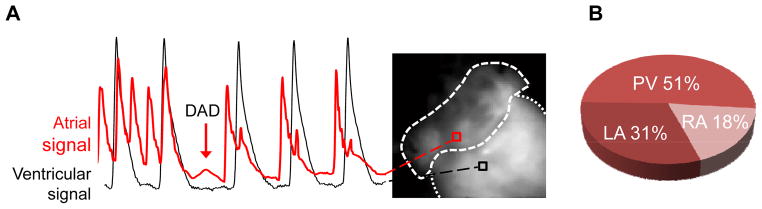
Anatomical origin of DADs in intact Casq2−/− atria. A) Representative example of a delayed afterdepolarizations (DAD). The red optical voltage record originating from the atria shows a deflection in the post pacing pause consistent with a DAD. Only deflections greater than 10% atrial of action potential amplitude were considered as DADs. No influence from ventricular fluorescence (black signal) is present during the atrial DAD. B) Anatomical distribution of DADs in Casq2−/− atria: The majority of the DADs occurred in the pulmonary vein region.
Casq2−/− atria display unchanged atrial action potential and CV
Although no clear re-entrant activation was observed in our optical maps, we next investigated whether changes in atrial action potential and/or CV might contribute to AF susceptibility in Casq2−/− mice. Action potentials were measured optically (Fig. 4A) in the right and left atrial appendage of Casq2−/− and Casq2+/+ hearts during atrial pacing at a cycle length of 100 ms. Mean action potential duration was not statistically different (Fig. 4B). Atrial CV was determined for each heart from right atrial appendage activation maps as illustrated in Fig. 4C. Mean maximum CV was not statistically different in the two groups (Fig. 4D).
Figure 4.

Action potential duration (APD) and conduction velocity (CV) of Casq2+/+ and Casq2−/− atria. A) Representative example of action potentials recorded optically from the RA appendage in Casq2+/+ and Casq2−/− hearts. B) Average APD at 90% repolarization (ADP90) measured during constant pacing at 100 ms pacing cycle length was not statistically different. C) Optical activation maps of the right atrial appendage during pacing. CV was calculated by defining isochrones where the time of maximum upstroke velocity of the fluorescent signal was the same. D) Average CV values were not statistically different between Casq2+/+ and Casq2−/− atria. N=4–5 hearts per group.
Spontaneous Ca release in isolated Casq2−/− atrial myocytes and intact Casq2−/− atria
Altered Ca homestasis with increased frequency and magnitude of Ca waves and sparks has been proposed as a cellular mechanism that can contribute to AF inducibility and maintenance. Therefore, we quantified Ca sparks and Ca waves in isolated permeabilized atrial cells. Compared to Casq2+/+, Casq2−/− atrial cells displayed a significantly higher rate of spontaneous Ca sparks (Fig. 5A). Furthermore, the frequency of propagated Ca waves was also significantly increased in Casq2−/− cells (Fig. 5B).
Figure 5.

Ca handling is impaired in isolated atrial myocytes (A, B) and intact atria (C–E) of Casq2−/− hearts. A and B left panels: Representative line scans of Ca sparks (A) and Ca waves (B) obtained in permeabilized atrial myocytes. Right panels: averaged data. N=35–45 cells per group, ***P<0.001. C) Examples of fluorescent Ca signals obtained from Ca maps of Casq2+/+ and Casq2−/− hearts. Casq2−/− atria frequently exhibit spontaneous Ca elevations (SCaE) after the rapid pacing train that are absent in Casq2+/+ atria (D). Similar to DADs, SCaE were observed only in some regions of the atria. E) Anatomical distribution of SCaE in intact atria.
To investigate Ca handling in intact atria, we obtained Ca maps from Casq2−/− and Casq2+/+ intact hearts during the post-pacing pause. A 5x5 pixel selection was used to sample different areas of both atria to obtain the corresponding Ca fluorescent trace. Diastolic spontaneous Ca elevations (SCaE) were present in 60% of pacing trains, with 100% (11/11) Casq2−/− hearts exhibiting SCaE (Fig. 5C and D). SCaE were never observed in Casq2+/+ hearts. Shape, amplitude and timing of the SCaEs varied greatly in the different areas of the atria consistent with the local nature of these events. The anatomical origin of the SCaE was defined as the atrial site where the deflection in the Ca fluorescence signal was most prominent. Only SCaEs with at least 10% of the pacing-induced Ca transient amplitude were considered for analysis. SCaE arose most frequently from the pulmonary vein area (58%) followed by the left atrial appendage (22%) and the right atrium (20%, Fig. 5 E). The anatomical distribution of SCaEs matched that of DADs (Fig. 3B) and focal activation sites during AF (Fig. 2C). These results suggest that Casq2−/− atria display increased susceptibility to spontaneous SCaE in the pulmonary vein region that can lead to DADs and ectopic triggered activity in the same area.
RyR2 block by R-Propafenone suppresses SCaE in isolated atrial myocytes and intact atria and prevents AF
R-propafenone is a class I antiarrhythmic drug with potent RyR2 blocking properties.9 Incubating Casq2−/− permeabilized atrial myocytes with R-propafenone for 10 min significantly reduced frequency, amplitude and propagation speed of Ca waves (Fig. 6A,B). In contrast, S-propafenone, which is a much weaker RyR2 channel inhibitor than R-propafenone,9 had no significant effect on Ca waves (Fig. 6A,B). We next tested R- and S-propafenone in intact atrial myocytes using burst-pacing to induced spontaneous Ca waves (SCW), DADs and triggered beats (Fig. 6C). R-propafenone significantly reduced the incidence of SCW and completely prevented triggered beats whereas S-propafenone did not (Fig. 6D).
Figure 6.

R-propafenone (R-Prop) but not S-propafenone (S-Prop) reduces the frequency of spontaneous Ca waves (SCW) in permeabilized Casq2−/− atrial myocytes and suppresses pacing-induced SCW and triggered beats in intact atrial Casq2−/− myocytes. (A) Representative line scans recorded from permeabilized Casq2−/− atrial myocytes in presence of vehicle (VEH, DMSO), R-Prop (10 μM) or S-Prop. (B) Averaged data. N=5–8 cells per group, *P<0.05, ***P<0.001. (C) Representative Ca fluorescence records from Fura2-AM loaded, intact atrial myocytes after 15 min exposure to VEH, R-Prop (3μM) or S-Prop (3μM), or Triggered Beats (TBs, arrow) and SCWs (arrowhead) were induced by a 20s pacing train (3Hz). (D) Incidence of TBs and SCWs. ***P<0.01 vs. VEH, #P<0.05 vs. S-Prop; n= 9–13 per group.
Given its efficacy in isolated myocytes, we tested R-propafenone in intact hearts. Atrial pacing induced only 4 SCaE overall in 3/8 Casq2−/− hearts pretreated with R-propafenone, resulting in a significant reduction in SCaE incidence (Fig. 7A,B). The amplitude of the few remaining SCaEs recorded in the presence of R-propafenone was also reduced compared to vehicle (Fig. 7C). The inhibition of SCW and triggered beats by R-propafenone but not S-propafenone in Casq2−/− atrial myocytes (Fig. 6) strongly suggested that propafenone’s suppression of SCaE in the intact atria is mainly related to its inhibitory effect on RyR2 rather than Na channel block. To test this hypothesis directly, we used S-propafenone and lidocaine on intact atria. S-propafenone is a much weaker RyR2 inhibitor than R-propafenone9 but blocks Na channels with equal potency.15 Lidocaine is a class IA antiarrhythmic drug with no RyR2 blocking properties. All three drugs were administered at concentrations that produced a 25% wider QRS complex, indicating a comparable degree of Na channel block (Fig. 7D). In contrast to R-propafenone, S-propafenone and lidocaine had no effect on frequency or amplitude of SCaE in intact atria (Fig. 7B–C). Furthermore, R-propafenone completely prevented pacing-induced AF (Fig. 7E), whereas both S-propafenone and lidocaine were ineffective (Fig. 7E). Collectively, these results suggest that R-propafenone’s antiarrhythmic efficacy in the Casq2−/− AF model is due to its suppression of Ca waves and SCaEs by inhibition of RyR2 channels.
Figure 7.

RyR2 channel block is necessary for prevention of pacing-induced SCaE and AF in Casq2−/− atria. (A) Representative examples of fluorescent Ca signals obtained from Ca maps of intact Casq2−/− hearts. As opposed to R-propafenone (R-Prop), administration of 3 μM of S-propafenone (S-Prop) or 20 μM of lidocaine (Lido) did not significantly reduce the rate (B) and amplitude (C) of SCaE in intact atria compared to vehicle. Despite a similar level of Na channel block evidenced by comparable QRS widening (D), S-propafenone and lidocaine failed to prevent AF in Casq2−/− hearts (E). N = 5–6 Casq2−/− hearts, **P<0.01.
Discussion
Major new findings
Our experiments demonstrate a mechanistic link between enhanced frequency of Ca waves in single atrial myocytes, SCaE and DADs in intact atria, and AF risk. An increased frequency of Ca waves has been observed in atrial cells from AF patients as well as from several AF animal models.6, 8, 16 Ca handling dysfunction may contribute both to the initiation and maintenance of chronic AF in patients.8 Although it has been speculated that increased frequency of Ca waves in atrial myocytes could be responsible for DADs and triggered AF, it has not been shown that these cellular events observed in isolated myocytes are sufficient to cause any changes in intact atria. Here, we identify and localize pacing induced SCaE in the intact atria of Casq2−/− hearts. These local events likely result from the synchronization of Ca waves in several cells, and generate DADs and triggered AF as suggested by the co-localization of SCaE, DADs and ectopic activation sites close to the pulmonary vein region. This mechanism is further support by our finding that R-propafenone, a class I anti-arrhythmic drug clinically used for the treatment of AF, not only decreases Ca wave rate in single atrial myocytes through inhibition of RyR2 Ca release channels but significantly reduces SCaE in intact atria and completely suppresses AF in Casq2−/− hearts. Importantly, S-propafenone and lidocaine, two Na channel inhibitors with either less or no RyR2 blocking properties fail to prevent SCaE and AF in intact Casq2−/− atria.
Regional SCaE in intact atria increase the risk of AF in Casq2−/− hearts
Our results show that lack of Casq2 increases susceptibility to AF in mice. Both re-entrant and triggered activation patterns have been previously described during AF in RyR2-linked CPVT models.7 Our optical mapping of AF events in Casq2−/−hearts consistently revealed 2–3 sites of near simultaneous activation that then propagated to the surrounding areas. This pattern is most consistent with focal triggered activation. Although our mouse model does not show any sign of macroscopic structural abnormality or fibrosis in atria or ventricles,5 we cannot completely exclude the possibility that micro re-entrant circuits are responsible for sustaining AF. However, when we measured APD90, and CV, two main parameters implicated in the risk of functional re-entry,17 we found them unchanged compared to Casq2+/+ controls. Consistent with SCaE triggering focal activation in Casq2−/− atria, spontaneous SR Ca releases in the form of both Ca sparks (local Ca releases) and Ca waves (propagated subcellular Ca releases) were significantly more frequent in Casq2−/− isolated atrial myocytes than in controls. Our results support the hypothesis that when Ca waves occur in a sufficient number of adjacent cells at the same time, the phenomenon can be observed at the tissue level and can cause local DADs and triggered activity, as suggested previously.18 Moreover, their co-localisation on voltage and Ca maps in intact atria suggests a causal link between the regional Ca waves, DADs and triggered ectopic activity. The finding that R-propafenone not only reduces Ca waves in myocytes and SCaE in intact atria but also completely prevents AF in Casq2−/− hearts further supports the hypothesis that AF in Casq2−/− hearts is generated by Ca-induced focal activity.
Casq2−/− mice represent a model of AF triggered by Ca-mediated enhanced automaticity
Increased intracellular spontaneous Ca releases have been observed in atrial myocytes of AF patients without any known mutations in intracellular Ca handling proteins.16 However, several other electrophysiological and ultrastructural abnormalities are present in these cells,19 thus making it challenging to determine whether Ca handling dysfunction is the cause of AF or is secondary to disease progression.20 The Casq2−/− mouse AF model reported here can be used to address the role of frequent SCaE in the pathogenesis of AF in the absence of other confounding intracellular perturbations. In addition, reduction of functional Casq2 has been observed in some chronic acquired heart diseases that are associated with increased risk of AF, such as congestive heart failure.21, 22 It is possible that acquired Casq2 reduction might promote AF in these patients as a complication of a pre-existing chronic cardiac disease. In this context, acquired Ca handling dysfunction could provide local triggers to initiate paroxysmal re-entrant AF sustained by anatomical substrates. Similarly leakiness of the SR could contribute to post-operative AF where the beta adrenergic stimulation after surgery may increase SR Ca content and spontaneous Ca releases. One limitation of the current study is that AF was induced acutely in mouse hearts by rapid atrial pacing, which may not be relevant to persistent or chronic forms of AF in humans. However, in the clinical electrophysiology laboratory atrial pacing is often performed to induce paroxysmal AF in patients. As over 70% of paroxysmal AF is thought to be driven by enhanced spontaneous activity originating in the pulmonary veins, rapid atrial pacing may be most relevant to unmasking increased susceptibility to paroxysmal AF in humans.
RyR2 block by R-propafenone prevents AF in Casq2−/− isolated hearts
Currently, the drug management of AF in clinical practice varies considerably, in part due to the lack of reliably effective therapies for maintaining sinus rhythm, and the toxicities associated with antiarrhythmic drugs. Drug efficacy could be improved by targeting the underlying pathophysiology of AF.2 For example, increased PKA-phosphorylation of RyR223, 24 as well as altered phosphatase and phosphodiesterase-4 activity25, 26 have all been proposed as mechanisms contributing to Ca triggered AF. The experimental compound S107, a RyR2 channel modulator, has been successfully used to prevent AF in a variety of RyR2 mutant mouse models6. Calcium/calmodulin-dependent protein kinase-II (CaMKII) has also been identified as a potential therapeutic target as it appears to contribute to the risk of chronic AF by altering RyR2 permeability to Ca; experimentally, CaMKII inhibition reduces SCaE in isolated atrial myocytes from chronic AF patients.8, 27 However, these compounds are not yet ready for patient treatment and will require further testing and clinical trials. Hence, we tested the R-enantiomer of propafenone (the racemic mixture of propafenone is routinely used in clinical practice) and investigated its mode of action in our model of Ca-triggered AF. We previously reported the efficacy of R-propafenone in suppressing ventricular arrhythmias in CPVT due to its dual action as both Na and RyR2 channel blockers.9 Our new results reported here show that R-propafenone can also completely prevent AF in Casq2−/− mice, likely through a significant reduction of diastolic SCaE in intact atria. Importantly, Na channel block in the absence of RyR2 inhibition, as obtained with S-propafenone or lidocaine, was not sufficient to prevent SCaE and AF in our model of Ca-triggered AF (Fig. 7). However, we cannot exclude that block of transient outward K currents28 or L-type Ca currents29 by propofenone contributed to suppression of SCaE and AF in Casq2 KO mice.
Propafenone is clinically used as a racemic mixture of R- and S-enantiomers to treat recurrent symptomatic AF as well as the pill in the pocket treatment for patients with sporadic paroxysmal AF.30 However, up to 20% of patients are not responsive to propafenone.30 Incomplete efficacy of the drug could be due to heterogeneity of the underlying AF pathophysiological mechanisms. We speculate that in patients with atrial remodelling and fixed reentry circuits, propafenone may not be as effective because it mainly targets AF caused by Ca-triggered activity. Another possible explanation for the lack of propafenone efficacy in a subset of AF patients is the use of racemic propafenone. R-propafenone inhibits the metabolism and elimination of S-propafenone, leading to accumulation of S-propafenone during chronic administration.15 Since S-propafenone was not effective for preventing Ca-triggered AF (our results), and since “leaky” RyR2s have been observed in many forms of AF,16 we speculate that R-propafenone might be a more effective mechanism-based treatment than the propafenone racemate currently used in clinical practice. R-propafenone could be considered for clinical drug development.
Supplementary Material
Acknowledgments
Funding Sources: This work was supported in part by NIH grants HL88635 and HL71670 (BCK), HL108173 (BCK, PJK), HL092217 (DD) and by an AHA Innovative Research Grant (BCK).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, D’Agostino RB, Massaro JM, Beiser A, Wolf PA, Benjamin EJ. Lifetime risk for development of atrial fibrillation: The framingham heart study. Circulation. 2004;110:1042–1046. doi: 10.1161/01.CIR.0000140263.20897.42. [DOI] [PubMed] [Google Scholar]
- 2.Darbar D, Roden DM. Genetic mechanisms of atrial fibrillation: Impact on response to treatment. Nat Rev Cardiol. 2013;10:317–329. doi: 10.1038/nrcardio.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sumitomo N, Sakurada H, Taniguchi K, Matsumura M, Abe O, Miyashita M, Kanamaru H, Karasawa K, Ayusawa M, Fukamizu S, Nagaoka I, Horie M, Harada K, Hiraoka M. Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ J. 2007;71:1606–1609. doi: 10.1253/circj.71.1606. [DOI] [PubMed] [Google Scholar]
- 4.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 5.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, Marks AR. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2012;111:708–717. doi: 10.1161/CIRCRESAHA.112.273342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase ii-mediated sarcoplasmic reticulum ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D. Enhanced sarcoplasmic reticulum ca2+ leak and increased na+-ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, Yin H, Knollmann BC. Inhibition of cardiac ca2+ release channels (ryr2) determines efficacy of class i antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011;4:128–135. doi: 10.1161/CIRCEP.110.959916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galimberti ES, Knollmann BC. Efficacy and potency of class i antiarrhythmic drugs for suppression of ca2+ waves in permeabilized myocytes lacking calsequestrin. J Mol Cell Cardiol. 2011;51:760–768. doi: 10.1016/j.yjmcc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, Yin H, Knollmann BC. Inhibition of cardiac ca2+ release channels (ryr2) determines efficacy of class i antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 4:128–135. doi: 10.1161/CIRCEP.110.959916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH. Intracellular calcium leak due to fkbp12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. 2008;5:1047–1054. doi: 10.1016/j.hrthm.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: Mechanistic insights and therapeutic opportunities. Eur Heart J. 2012;33:1870–1877. doi: 10.1093/eurheartj/ehs079. [DOI] [PubMed] [Google Scholar]
- 15.Kroemer HK, Funck-Brentano C, Silberstein DJ, Wood AJ, Eichelbaum M, Woosley RL, Roden DM. Stereoselective disposition and pharmacologic activity of propafenone enantiomers. Circulation. 1989;79:1068–1076. doi: 10.1161/01.cir.79.5.1068. [DOI] [PubMed] [Google Scholar]
- 16.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- 17.Krummen DE, Bayer JD, Ho J, Ho G, Smetak MR, Clopton P, Trayanova NA, Narayan SM. Mechanisms for human atrial fibrillation initiation: Clinical and computational studies of repolarization restitution and activation latency. Circ Arrhythm Electrophysiol. 2012;5:1149–1159. doi: 10.1161/CIRCEP.111.969022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: Requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ Arrhythm Electrophysiol. 2008;1:62–73. doi: 10.1161/CIRCEP.107.754564. [DOI] [PubMed] [Google Scholar]
- 20.Greiser M, Lederer WJ, Schotten U. Alterations of atrial ca(2+) handling as cause and consequence of atrial fibrillation. Cardiovasc Res. 2011;89:722–733. doi: 10.1093/cvr/cvq389. [DOI] [PubMed] [Google Scholar]
- 21.Kiarash A, Kelly CE, Phinney BS, Valdivia HH, Abrams J, Cala SE. Defective glycosylation of calsequestrin in heart failure. Cardiovasc Res. 2004;63:264–272. doi: 10.1016/j.cardiores.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 22.McFarland TP, Milstein ML, Cala SE. Rough endoplasmic reticulum to junctional sarcoplasmic reticulum trafficking of calsequestrin in adult cardiomyocytes. J Mol Cell Cardiol. 2010;49:556–564. doi: 10.1016/j.yjmcc.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- 24.Llach A, Molina CE, Prat-Vidal C, Fernandes J, Casado V, Ciruela F, Lluis C, Franco R, Cinca J, Hove-Madsen L. Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine a2a receptors. Eur Heart J. 2011;32:721–729. doi: 10.1093/eurheartj/ehq464. [DOI] [PubMed] [Google Scholar]
- 25.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–680. doi: 10.1161/CIRCULATIONAHA.106.636845. [DOI] [PubMed] [Google Scholar]
- 26.Van Wagoner DR, Lindsay BD. Phosphodiesterase-4 activity: A critical modulator of atrial contractility and arrhythmogenesis. J Am Coll Cardiol. 2012;59:2191–2192. doi: 10.1016/j.jacc.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–1144. doi: 10.1161/CIRCRESAHA.109.203836. [DOI] [PubMed] [Google Scholar]
- 28.Seki A, Hagiwara N, Kasanuki H. Effects of propafenone on k currents in human atrial myocytes. Br J Pharmacol. 1999;126:1153–1162. doi: 10.1038/sj.bjp.0702428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fei L, Gill JS, McKenna WJ, Camm AJ. Effects of propafenone on calcium currents in single ventricular myocytes of guinea-pig. Br J Pharmacol. 1993;109:178–182. doi: 10.1111/j.1476-5381.1993.tb13550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alboni P, Botto GL, Baldi N, Luzi M, Russo V, Gianfranchi L, Marchi P, Calzolari M, Solano A, Baroffio R, Gaggioli G. Outpatient treatment of recent-onset atrial fibrillation with the “pill-in-the-pocket” approach. N Engl J Med. 2004;351:2384–2391. doi: 10.1056/NEJMoa041233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.