

Abstract
Cardiac macrophages are abundant in the healthy heart and after myocardial infarction (MI). Different macrophage phenotypes likely promote myocardial health vs. disease. Infarct macrophages are inflammatory and derive from circulating monocytes produced by the haematopoietic system. These cells are centrally involved in inflammatory tissue remodelling, resolution of inflammation during post-MI healing, and left ventricular remodelling. Presumably, macrophages interact with myocytes, endothelial cells, and fibroblasts. Although macrophages are primarily recruited to the ischaemic myocardium, the remote non-ischaemic myocardium macrophage population changes dynamically after MI. Macrophages' known roles in defending the steady state and their pathological actions in other disease contexts provide a road map for exploring cardiac macrophages and their phenotypes, functions, and therapeutic potential. In our review, we summarize recent insights into the role of cardiac macrophages, focus on their actions after ischaemia, and highlight emerging research topics.
Keywords: Macrophage, Myocardial infarction, Heart failure, Bone marrow, Spleen
1. Introduction
The role of inflammation in cardiovascular disease has been under-appreciated in the past, possibly because cardiologists deal with infection (as in endocarditis) and autoimmunity (as in myocarditis) less frequently than with ischaemic or hypertensive heart disease. Today, however, this situation is rapidly changing as cardiovascular science and immunology merge their efforts. Increasing numbers of immunologists work on ischaemic heart disease while cardiologists are more interested in the intricacies of the immune system. The observation that atherosclerosis is not merely a lipid storage disorder but is also propelled by immune system dysfunction initiated a cardiology–immunology convergence. Research on immune cells in cardiovascular disorders now moves beyond the arterial wall to yield data on leucocytes in the heart. For instance, like the arterial wall, the healthy myocardium hosts a considerable number of macrophages. In fact, macrophages are among the largest cardiac resident cell populations, trailing only fibroblasts, myocytes, and endothelial cells. This review summarizes recent work on heart macrophage fate and function in steady state and disease. We will highlight open questions that merit exploration. Whenever data on the heart are lacking, we look to other organs and pathologies to learn from potential parallels and formulate hypotheses. We believe that heart macrophages deserve attention because they are system-wide actors that perform numerous tasks in wound healing, regeneration, and tissue remodelling. They are recognized as central operators in cancer, infection, rheumatoid arthritis, diabetes, obesity, atherosclerosis, and stroke. Exciting recent studies of macrophages and related cells reveal properties that go far beyond their classical phagocytotic function. For instance, skin macrophages are involved in blood pressure regulation,1 and macrophages may promote insulin resistance.2 If future research confirms that macrophages pursue analogous specialized tasks in the myocardium, then these cells may be attractive targets for cardiovascular therapy.
2. Macrophage phenotypes and functions
Macrophages are large extravascular immune cells with diverse phenotypes. Their amoeboid cell bodies may extend dendrite-like protrusions to survey their surroundings. The cells are numerous and distribute throughout the body to take up residence in many tissues. The life span of macrophages may be measured in months during steady state but can be limited to mere hours during disease. Typically, we study tissue macrophages by staining for cell surface markers in histology or multicolour flow cytometry. These markers include CD45, F4/80, MAC-3, CD11b, CD11c, CD68, and CD115. Some macrophage markers are also shared by other leucocyte classes, e.g. neutrophils or dendritic cells. The Immunological Genome Project (www.immgen.org) provides an excellent resource for further information.3
Macrophages have a plethora of divergent functions. As part of the innate immune system, macrophages provide a first line of defence during infection and injury. These macrophages may use phagocytosis to take up bacteria or foreign bodies, which they destroy by enzymatic digestion in acidic phagolysosomes. Cytotoxic macrophages deliver oxidative bursts or digest matrix by secreting proteases. Inflammatory macrophages are often referred to as classical or M1 macrophages. In addition, there are a number of macrophage phenotypes that pursue highly specialized functions, including osteoclasts (bone remodelling), Kupffer cells (iron homeostasis), microglia (synaptic pruning), and CD169+ macrophages in the haematopoietic niche (regulating blood cell production).4 Less inflammatory macrophages are commonly called alternatively activated or M2 macrophages. The scientific community agrees that this classification, while helpful, is an oversimplification and should be used with caution, especially when discussing in vivo phenotypes.
Two macrophage functions make the cells particularly interesting for cardiovascular health: (i) their propensity to ingest lipoproteins and give rise to foam cells in atherosclerotic plaque and (ii) their vigorous response to ischaemia. The role of macrophages in atherosclerosis was recently summarized elsewhere5–7 and is not the subject of this review.
3. Sources of macrophages
The last decade brought about renewed interest in studying the origin of tissue macrophages. The current view describes a dichotomy, with different sources of macrophage renewal depending on cellular function and phenotype. In the steady state, the majority of macrophages, with the exception of those in the intestine,8 do not derive from monocytes but rather from local progenitors that arise from the embryonic yolk sac. Macrophages produced by these local progenitors take up residence in their destination tissues prior to birth, as proved experimentally for microglia in the brain,9 lung macrophages, Langerhans cells in the skin, and Kupffer cells in the liver.10 Macrophages in the healthy gut, however, derive from circulating monocytes.8 It is not currently known whether steady-state heart macrophages are renewed independently of circulating monocytes.
Although most macrophages derive from local progenitors in steady state, tissue inflammation changes macrophages' source. During such inflammation, most macrophages derive from inflammatory monocytes (Ly6Chigh in the mouse, CD14+CD16− in humans) that are recruited to the site of inflammation from the blood pool. These monocytes are the progeny of haematopoietic progenitors and, ultimately, hamatopoietic stem cells (HSCs) residing in the bone marrow. HSCs are rare, with a frequency of only 1 in 10 000 bone marrow cells. The HSC majority is quiescent, as only ∼5% cycle in the steady state.11 After injury, haematopoiesis may accelerate to ramp up production of an increasingly differentiated tree of progenitors (Common Myeloid Progenitors, Granulocyte Macrophage Progenitors, Macrophage Monocyte Progenitor, Common Monocyte Progenitor), which in turn give rise to monocytes in the bone marrow.12 The signals that control HSC proliferation and differentiation to myeloid cells and, specifically, monocytes have been studied in the steady state and during many pathologies, but little is known about how the bone marrow adapts to cardiovascular disease. A number of housekeeping cells reside in the haematopoietic niche and regulate the blood cell production in the bone marrow by supplying soluble signals and adhesion to HSCs. These regulatory and supply cells include mesenchymal stem cells, endothelial cells, macrophages, nerve cells, and osteoblasts, and they provide fate-regulating signals and growth factors such as Stem Cell Factor, CXCL12, angiopoietin-1, and G-CSF.13,14 The above list is incomplete, and the precise cellular sources of signals that drive HSC activity are still being identified. Additionally, HSCs express receptors, e.g. toll like and interferon receptors, to directly sense circulating danger signals.15 An emerging research area aims to identify factors that modulate haematopoietic activity after ischaemic injury and in individuals with atherosclerosis, as increased production and supply of inflammatory monocytes likely enlarge the pool of inflammatory macrophages in the heart and in atherosclerotic plaque. Mature cells follow specific cues for liberation from the bone marrow into the blood stream. Experiments in mice challenged with lipopolysacharide, a component of the bacterial cell membrane, show that monocyte release from the bone marrow into the blood pool depends on CCR2 chemokine receptor signalling.16
In addition to the bone marrow, the spleen may also contribute blood monocytes (Figure 1). There is a substantial splenic reservoir of ready-made monocytes.17 Once this population is exhausted, the spleen produces new monocytes by hosting extramedullary haematopoiesis. Splenic haematopoiesis occurs in many different disease settings in the mouse, including infection, cancer,19 atherosclerosis,20 and after MI.21 During embryogenesis, the spleen and liver are sites of haematopoiesis, but these organs cease haematopoietic activity, at least in the steady state, after birth. The monocyte release from the spleen is independent of CCR2 but caused by angiotensin-2 signalling.17 Splenic monocyte mobilization can be attenuated with ACE inhibitor therapy in the mouse.22
Figure 1.
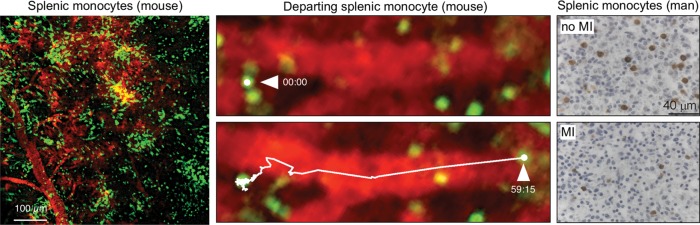
The spleen as a source of macrophages. The left panel shows clusters of GFP+ monocytes residing in the sub-capsular red pulp of CX3XR1GFP/+ mice. The middle panel shows a monocyte departing after MI in two movie frames acquired in the spleen after coronary ligation. Adapted from reference.17 The right panel shows immunohistochemical monocyte staining in the spleens of a control patient and a patient after myocardial infarction. The comparison verifies the existence of a splenic monocyte reservoir in humans.18
After coronary ligation in mice, sympathetic nervous signalling alarms the haematopoietic system.23 Beta-3 adrenoreceptors expressed by niche cells in the bone marrow translate noradrenalin release into a modified micro-milieu;24 for instance, CXCL12 levels decrease after MI.23 Lower levels of this retention factor liberate haematopoietic progenitor cells, which then migrate to the spleen where, supported by increased interleukin-1β levels and stem cell factor (SCF), the cells are retained in a VLA-4-dependent manner and produce monocytes that consequently migrate to the infarct to supply the macrophage population in the ischaemic myocardium.21,23 The phenotype of monocytes made in the spleen is inflammatory;23 however, less inflammatory Ly6Clow monocytes can also be found in the organ.17 Other research shows that Wnt, which is thought to regulate HSC self-renewal and expansion,25 increases signalling in the bone marrow after MI.26
4. Macrophages in the healthy heart
FACS analysis27 and histology using CX3CR1GFP/+ mice reveal abundant extravascular macrophages in the healthy heart (Figure 2) located in direct contact with myocytes and endothelial cells.28 In the steady state, heart macrophages are not particularly inflammatory. The cells sparsely express the surface marker Ly6C and have a set of 22 genes associated with alternatively activated M2 macrophages translated at high levels.28 That steady-state cardiac macrophages also express some inflammatory genes, including interleukin-1β, highlights the limitations of M1/M2 classification. The function of these cardiac macrophages in the steady state is largely unknown, and data from macrophage ablation studies are currently not available. We speculate that these cells may have typical tissue resident macrophage tasks: they guard against infection, regulate angiogenesis, and direct matrix turnover. A potential role in regulating cardiac capillary density and collagen production should be explored, as both processes are involved not only in steady state but also in the genesis of heart failure. In other tissues, such as adipose tissue, there is active crosstalk between resident macrophages and stromal cells, e.g. adipocytes.30 It remains to be seen whether cardiac macrophages, which are abundant in the vicinity of myocytes, interfere with metabolism, contraction, or survival of cardiomyocytes in the steady state and/or disease.
Figure 2.
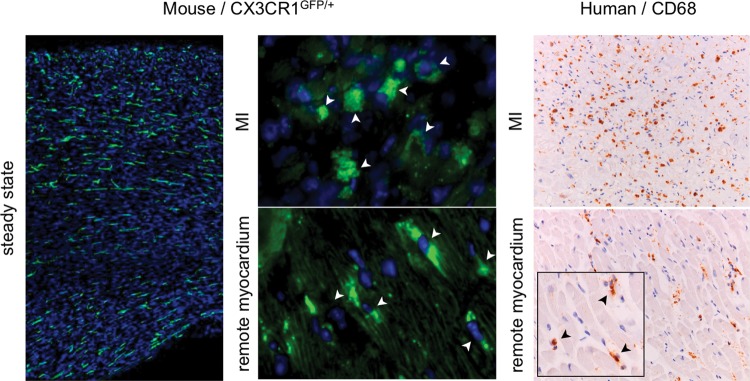
Macrophages in the heart. The left panel shows the steady-state myocardium in a CX3CR1GFP/+ mouse (adapted from reference28). The middle panel shows macrophage presence in the infarct and the remote myocardium after coronary ligation (adapted from reference29). The right panel shows the presence of monocytes in the infarct and in the remote myocardium of a patient with acute MI (adapted from reference29).
The source of macrophages in the healthy heart is currently unknown. Given that other tissues, such as the skin and the liver, do not replenish macrophages from circulating monocytes during steady state, it seems reasonable that cardiac macrophages similarly derive from local progenitors. In other words, macrophages most likely proliferate in the heart.
Though we do not yet know the origins of cardiac macrophages, research is already mapping the roles monocytes play in steady-state cardiac health. Intravital microscopy shows patrolling monocytes in small arterioles in the heart.31 These Ly6Clow monocytes crawl along the endothelial surface of vessels, sometimes even against the blood stream. In other tissues, patrolling Ly6Clow monocytes contribute to endothelium maintenance by removing apoptotic endothelial cells.32 The function of Ly6Clow monocytes in the heart remains to be explored.
5. Monocytes and macrophages in acute myocardial infarction
Soon after myocardial infarction (MI), ischaemic tissue attracts inflammatory Ly6Chigh monocytes (Figure 3). As early as 30 min after coronary ligation, these cells are abundantly recruited, initially outpacing even neutrophils.31 Neutrophil tissue numbers peak 24 h after onset of ischaemia, whereas inflammatory monocytes peak around day 3.27 Ly6Chigh monocyte recruitment relies on MCP-1/CCR2 chemokine/chemokine receptor interaction.27,33 Further, the endothelium increases expression of adhesion molecules such as selectins and vascular cell adhesion molecule-1.29 Once recruited, many monocytes may differentiate to inflammatory macrophages, as the marker F4/80 is expressed at high levels for several days after MI. Both monocytes and macrophages generate inflammatory cytokines, cathepsins and matrix metalloproteinases. Presumably, the main function of these monocytes and macrophages is to remove debris, dead myocytes, and apoptotic neutrophils in order to prepare for tissue rebuilding and regeneration.
Figure 3.

The biphasic monocyte/macrophage response after myocardial infarction.
Monocytes and macrophages initially accumulate in the infarct border zone,18 probably because vascular access is still intact in the myocardium adjacent to the ischaemic tissue. Over time, macrophages migrate into the infarct core. This migration has yet to be observed directly; however, monocyte numbers increase in the infarct core later than in the border zone, according to human histology studies.18 If reperfusion occurs and if microvascular obstruction is absent, monocytes may directly distribute to the infarct core shortly after ischaemic injury.34
Initial inflammation is followed by a proliferation phase during which new matrix is generated to provide mechanical stability to the left ventricle. If this phase is compromised, the infarct scar may rupture or expand, leading to sudden death and left ventricular dilation. At this point, the surface marker Ly6C is less abundant.27 Macrophages active during days 4–7 post-MI are less inflammatory and express genes associated with prototypical M2 macrophages.35 These cells help rebuild tissue while likely regulating angiogenesis via VEGF and myofibroblast activity via TGF beta.27 Clodronate liposome depletion targeting either the early Ly6Chigh or the late Ly6Clow phase shows that both are essential for infarct healing.27,36,37 When myeloid cells were depleted, histologic healing biomarkers indicated impaired dead myocyte removal, slower matrix rebuilding, and lower microvessel counts.27 Macrophage depletion also led to high incidence of left ventricular mural thrombi attached to the infarct. This may be clinically relevant, as patients with a left ventricular thrombus exhibit a relative decrease of the CD14+CD16+ monocyte subset in blood,37 a decrease that corresponds to patrolling Ly6Clow monocytes in mice. It is conceivable that the lack of patrolling Ly6Clow monocytes and infarct macrophages compromise ventricular endothelial layer maintenance, thereby exposing thrombogenic infarct tissue to the blood pool.
The exact lineage relationship of Ly6Chigh and Ly6Clow monocytes to M1 and M2 macrophages has yet to be determined with in vivo lineage tracing tools. These experiments are complex because isolation and adoptive transfer of monocytes expose cells to stress and plastic surfaces, both of which may confound the cell's phenotype. It is safe to assume, however, that in the early days after MI inflammatory Ly6Chigh monocytes give rise to inflammatory M1 macrophages.
The described biphasic monocyte/macrophage response, which likely occurs in many wounds, was first described in rodent models of permanent coronary ligation.27,35 A similar time course was subsequently found for temporal ischaemia followed by reperfusion injury38 and in human patients after MI.18 The first clinical study described a biphasic monocyte pattern in the blood of patients after MI, with inflammatory CD14+CD16− monocytes dominating first.39 A recent autopsy study confirmed the kinetics in infarct tissue, with early abundance of CD14+CD16− monocytes while CD14+CD16+ monocytes were found only in the infarcts of patients who died at later time points.18 The same study also reported a precipitous drop in monocyte levels within the bone marrow and the spleen of infarct patients, suggesting that, as previously described in mice and rats with coronary ligation,17 these organs supply monocytes to the infarct in humans.
6. Macrophages in remote, non-ischaemic myocardium after MI
The remote, non-ischaemic myocardium also changes after MI, as capillaries in the remote zone increase recruiting chemokine expression and attract monocytes.29 When compared with the ischaemic myocardium, the remote myocardium accumulates monocytes and macrophages more slowly, as remote zone macrophage numbers peak around day 10 after coronary ligation.29 Although the total numbers of recruited cells per mg myocardium remain lower than in the ischaemic zone, it is safe to assume that the non-ischaemic myocardium is also ‘inflamed’ after MI (Figure 2). The increased presence of myeloid cells in the non-infarcted remote zone likely contributes to elevated protease activity, including matrix metalloproteinases40 and cathepsins.29 These proteases may attack the extracellular matrix and promote left ventricular dilation. Even months after coronary ligation in mice, macrophage numbers in the heart remain high.41 Macrophages' precise function in remote myocardium within the chronically remodelling heart are under investigation in several laboratories. By extrapolating the role of macrophages in chronic inflammation, e.g. pro-fibrotic macrophage action in liver disease,42 or their matrix-destabilizing function in atherosclerotic plaque,43 we postulate substantial cross talk between macrophage and resident cell impacts post-MI myocardium remodelling. Further, increased myocardial macrophage presence also occurs after aortic constriction in mice.44 We suspect that macrophages may influence myocardial capillary density or fibrosis during the chronic remodelling process. Given that inflammatory macrophages kill microbes, we should also investigate potential cross talk with myocytes, which undergo apoptosis at higher rates during post-MI remodelling45 and in heart failure.46 The hypothesis that inflammatory macrophages may directly harm myocytes is further supported by macrophages' ability to produce TNFα, a cytokine that induces myocyte apoptosis.47
7. Macrophages and resolution of infarct inflammation
Emerging awareness of macrophages' role in inflammation after MI indicates that while these cells are absolutely necessary to adequate wound healing and generating a stable scar that preserves left ventricular geometry, overabundant pro-inflammatory macrophages are also harmful. Timely resolution of inflammation, i.e. transition from a Ly6Chigh to a Ly6Clow monocyte/macrophage phenotype in the infarct, is a prerequisite for regenerative processes. If these two phases are corrupted and infarct inflammation lingers, then healing derails and heart failure occurs. This concept is supported by pre-clinical data obtained in ApoE−/− mice after coronary ligation.48 The rationale for these experiments lies in the limitations of coronary ligation in otherwise healthy wild-type mice. This widely used protocol fails to account for atherosclerosis triggering MI in almost all patients. ApoE−/− mice on a high-fat diet develop atherosclerosis and blood monocytosis49 due to haematopoietic monocyte overproduction.20,50 These monocytes give rise to inflammatory macrophages in atherosclerotic plaque. In ApoE−/− mice, the circulating monocyte pool and monocyte production expand, particularly the inflammatory monocyte subset.49 This is the exact same cell type that gives rise to inflammatory infarct macrophages. Inducing MI in the setting of pre-existing chronic inflammation may change macrophage supply dynamics and consequently alter cardiac wound healing. Indeed, in ApoE−/− mice infarct inflammation resolution is disturbed by oversupplied inflammatory Ly6Chigh cells. Compared with wild-type mice, ApoE−/− infarcts recruit more monocytes, and the transition between the Ly6Chigh and Ly6Clow phases is delayed.48 Disrupted inflammation resolution inhibits regenerative processes and accelerates post-MI left ventricular dilation, possibly due to higher protease activity in the heart.51 Clinical data show a correlation between blood monocytosis and outcome in cardiovascular disease. A patient cohort with high CD14+CD16− blood monocyte levels at the time of acute MI had larger left ventricles at follow-up,39 and monocyte and white blood counts correlate with heart failure progression.52,53
The mechanisms that regulate monocyte production, migration, and macrophage supply after MI have not yet been understood completely. Of particular interest is the biphasic infarct macrophage phenotype regulation. Dendritic cells, part of the adaptive immune arm but closely related to macrophages, actively collaborate with T cells in the setting of infection. Likewise, there may be interaction of lymphocytes with monocytes and macrophages after ischaemic injury. In mice that genetically lack CD4 and therefore T-lymphocytes, the infarct recruits a higher number of leucocytes, including Ly6Chigh monocytes/macrophages. Resolution of inflammation in the infarct is disrupted, leading to impaired healing and accelerated left ventricular remodelling.54 Although T-lymphocytes increase in the infarct, the innate immune cell population remains larger, and their relative numbers suggest that T-lymphocytes have a regulatory rather than executive role. B-lymphocytes likewise interfere with monocyte supply post-MI.55 These cells increase systemic levels of the chemokine CCL7, which binds to the receptor CCR2 on monocytes and enables their mobilization into circulation.55 A previous study in mice with sepsis also implied a role for CCR2 in monocyte bone marrow exit, although in this case mesenchymal stem cells provided a different CCR2 ligand, namely CCL2.56 These studies demonstrate the need for further examination of myeloid cell interaction with adaptive immune cells, despite the lack of exogenous antigens after ischaemic sterile injury.
8. Acute and chronic macrophage function in the heart and arterial wall
As discussed above, pre-existing atherosclerosis may influence macrophage phenotype in acute MI. Conversely, acute MI may also influence macrophage biology in atherosclerotic plaque. Epidemiology data show that re-infarction is common,57 and patients are >50% likely to experience ischaemic complications in the first year after first infarct.58 Does acute MI change the supply of monocytes to atherosclerotic lesions? Indeed, MI (and ischaemic stroke) accelerates macrophage progenitor production by increasing splenic monocyte output.23 Heightened sympathetic bone marrow tone immediately after MI changes the haematopoietic microenvironment and enables haematopoietic progenitor cells to transfer to the spleen, where outsourced monocyte production feeds monocyte recruitment to atherosclerotic plaque. This process leads to higher plaque macrophage numbers and increased plaque protease activity in ApoE−/− mice with MI. These pathophysiologic phenomena23 may also occur in patients with more than one vulnerable plaque, especially after tissue ischaemia accelerates system-wide monocyte supply to atherosclerotic lesions.59
9. Macrophages and stem cells
Regeneration of myocardium by circulating and resident progenitor cells continues to be a subject of intense investigation, with the ultimate goal of regrowing myocardium lost to ischaemia. Clinical studies investigating whole bone marrow injection showed only minor and rather temporary beneficial effects. Currently, the most promising experimental avenues concern cardiac stem cells, which are local progenitors that support low-level myocyte regeneration even in the healthy state.60 The SCIPIO trial explored this cell population via in vitro expansion and autologous reinfusion of purified cardiac progenitors.61 Other data suggest that injecting mesenchymal stromal cells (MSCs) might increase the percentage of reparative M2 macrophages in the heart. Depleting macrophages ablated beneficial effects of MSC injection.62 Reprogramming cardiac resident fibroblasts into myocytes is another interesting in vivo approach,63 although its efficiency remains low. Encouraging experiments in zebrafish64 and newborn mice65 indicate that regrowing myocardium from endogenous sources is possible in principle, but the mechanisms involved are currently not well understood.
Macrophages may have a role in regenerative processes in the heart, just as they support tissue regeneration in other organs. Macrophages are an essential element of the haematopoietic stem cell niche.13 For instance, a specialized CD169+ macrophage is involved in retaining haematopoietic stem cells in the bone marrow. Depletion of CD169+ macrophages impairs red blood cell regeneration.66 Further evidence for macrophages' role in forming regenerative niches is that salamanders injected with macrophage-depleting clodronate liposomes lose their ability to regrow amputated limbs.67 The amputation wounds close, but limb regeneration fails after systemic macrophage depletion. If the wounds are refreshed at a later time point, when the systemic monocyte population has recovered, the limb regains the capacity to regenerate. Thus, monocytes and macrophages may inform the microenvironment and instruct tissue resident progenitors. We speculate that manipulating macrophage phenotype or number may also aid efforts to regenerate myocardial tissue.
10. Macrophages as a therapeutic target
Cardiac macrophages have not been explored as therapeutic targets. This does not come as a surprise, given that they were only recently acknowledged as a major cell population in the heart. Once we better understand the cells' roles in the heart, specifically targeting macrophages, their progenitors or macrophage subtypes may be feasible. This endeavour must avoid pitfalls arising from targeting inflammation. Because macrophages are crucial to maintaining the steady state and defending against infection, any therapy must be specific to ‘harmful’ macrophage functions or subsets, precisely dosed and closely monitored to avoid collateral damage.
Despite the challenges, targeting macrophages presents interesting opportunities. Nanoparticles may be particularly well suited to delivering drugs to macrophages. Using lipidoid nanoparticle encapsulation, systemically injected siRNA is faithfully delivered to monocytes, resulting in silencing of the chemokine CCR2.68 Consequently, inflammatory monocyte recruitment, which depends on interaction between CCL2 and CCR2, decreases in ischaemia reperfusion injury68 and in non-reperfused MI.69 When ApoE−/− mice with MI are injected with nanoparticle-loaded siRNA targeting CCR2, the resolution of inflammation and infarct healing improves, and post-MI heart failure attenuates.69 This approach likely does not interfere with sessile immune cells, nor should it inhibit proliferation of cardiac resident cells.
As angiotensin 2 receptor signalling regulates monocyte motility and release from the splenic reservoir,17 ACE inhibitors and angiotensin receptor blockers may influence monocyte migration to the site of inflammatory activity.22 In addition to many other beneficial actions, this drug class may have anti-inflammatory properties that can support infarct healing.
Manipulating the macrophage phenotype may be another therapeutic option. Once we understand the mechanisms chaperoning the transition from inflammatory to non-inflammatory macrophage phenotypes, we can begin to harness them as therapeutic targets. One master phenotype switch may be the transcription factor IRF5.70 In isolated macrophages, silencing IRF5 shifts inflammatory M1 macrophages towards the alternatively activated M2 phenotype.71 While intracellular molecules are often difficult to target with small molecules and biologicals, siRNA therapy may overcome this hurdle. Targeting the transcription factor IRF5 in mice with MI and in mice with skin wounds led to down-regulation of inflammatory M1 associated genes in infarct and wound macrophages.72 The expression of M2 genes did not change. RNAi macrophage reprogramming supported the resolution of infarct inflammation and accelerated wound healing. Skin wounds closed faster and post-MI heart failure was less likely to occur.72 Interestingly, potentially beneficial paracrine effects of mesenchymal stem cell treatment after acute MI may also come from changing macrophage polarization from M1 towards M2.62
11. Conclusion
Our review summarizes the opportunities and challenges presented by the study of cardiac macrophages. Though immunology colleagues have investigated inflammatory and tissue macrophages in various organ systems in the past decades, much less is known about macrophages in the heart, possibly because rapid cardiac motion has made it difficult to follow these cells in vivo. Newer imaging tools73,74 now allow us to investigate macrophage numbers, fate, and function at different scales, from the cellular resolution provided by intravital microscopy in mouse hearts75 to the infarct macrophage population probed with iron oxide nanoparticle MRI in patients.76,77 Multichannel intravital microscopy of the heart in live mice that express reporter proteins for macrophages31 and spectrally different colored reporter proteins for other cardiac resident cells will help identify in vivo cell-cell cross talk. Emerging molecular imaging probes that either target macrophages directly or follow their specific functions74 enable basic research but are also poised to facilitate clinical trials by adopting molecular imaging read-outs as end points. Companion imaging strategies help test new drugs by imaging a drug's target, thus enabling faster and more economical clinical trials. Imaging cardiac macrophages in patients with acute MI is already possible, as recently demonstrated in two clinical trials.76,77
To understand complex biology requires in-depth knowledge spanning multiple research fields. When cardiovascular scientists team up with immunologists, it enables the application of cutting-edge knowledge and technique, for instance identifying and phenotyping macrophages by employing advanced macrophage surface markers. This type of synergy has already led to increased awareness of cardiac resident macrophages through adopting reporter strategies such as the CX3CR1GFP/+ mouse (Figures 1 and 2). In addition, macrophage-specific gene knock outs and macrophage ablation strategies are now being used to investigate the role of cardiac macrophages in heart disease.
The cardiovascular system is not isolated; rather, it intimately interconnects with the entire organism. Many cardiac interactions result in risk factors for heart disease: e.g. with metabolism (obesity), the endocrine system (diabetes), the central nervous system (stress, depression), autoimmunity (rheumatism), and the haematopoietic system (anaemia). We hypothesize that the innate immune system, and specifically the macrophage, may act as an interface between the cardiovascular and other organ systems (Figure 4). Macrophages reside in haematopoietic tissue, the heart, the vascular wall, atherosclerotic plaque, fat, skeletal muscle, bone marrow, lymphatic system, and in the brain. We know macrophages and their progenitors receive instructions from many other organs, including the nervous and endocrine systems.7 Investigation into how these forces functionally connect requires a systems approach and will, we believe, bring about preventive and therapeutic innovation.
Figure 4.

The role of macrophages in cardiovascular disease and intersections with other organ systems. (A) After myocardial infarction, sympathetic nervous signalling changes the micro-milieu in the bone marrow, leading to haematopoietic stem cell activation (HSC). (B) Macrophages are part of the haematopoietic stem cell niche and provide signals that regulate the blood cell production. (C) The haematopoietic niche provides monocytes, which are progenitors of inflammatory macrophages. (D) After MI, haematopoietic stem and progenitor cells (HSPC) are released into the blood stream and seed the spleen, where they initiate accelerated monocyte production. (E) The spleen is also a site of blood cell recycling. (F) Macrophages are involved in regulating blood pressure. (G) Macrophages reside in fat tissue and obtain inflammatory phenotypes during obesity. They also promote diabetes. (H) Monocytes recruited to atherosclerotic lesions differentiate into macrophages and foam cells, which promote plaque growth and rupture. Macrophages respond to ischaemic injury in the heart (i) and the brain (j), regulating tissue repair and outcomes.
Funding
This project has been funded in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN268201000044C, and grants R01-HL095629, R01-HL114477, R01-HL117829 (M.N.), as well as grants from the Bundesministerium für Bildung und Forschung (BMBF01 EO1004) and Deutsche Forschungsgemeinschaft (SFB688 TP A10) (S.F.).
Conflict of interest: none declared.
References
- 1.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 2.Li P, Spann NJ, Kaikkonen MU, Lu M, Oh da Y, Fox JN, et al. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell. 2013;155:200–214. doi: 10.1016/j.cell.2013.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zigmond E, Jung S. Intestinal macrophages: well educated exceptions from the rule. Trends Immunol. 2013;34:162–168. doi: 10.1016/j.it.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 11.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med. 2011;208:421–428. doi: 10.1084/jem.20110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 15.Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 17.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Laan AM, Ter Horst EN, Delewi R, Begieneman MP, Krijnen PA, Hirsch A, et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J. 2013;35:376–385. doi: 10.1093/eurheartj/eht331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci USA. 2012;109:2491–2496. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–137. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leuschner F, Panizzi P, Chico-Calero I, Lee WW, Ueno T, Cortez-Retamozo V, et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ Res. 2010;107:1364–1373. doi: 10.1161/CIRCRESAHA.110.227454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 25.Lo Celso C, Scadden DT. The haematopoietic stem cell niche at a glance. J Cell Sci. 2011;124:3529–3535. doi: 10.1242/jcs.074112. (Pt 21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Assmus B, Iwasaki M, Schachinger V, Roexe T, Koyanagi M, Iekushi K, et al. Acute myocardial infarction activates progenitor cells and increases Wnt signalling in the bone marrow. Eur Heart J. 2012;33:1911–1919. doi: 10.1093/eurheartj/ehr388. [DOI] [PubMed] [Google Scholar]
- 27.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One. 2012;7:e36814. doi: 10.1371/journal.pone.0036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee WW, Marinelli B, van der Laan AM, Sena BF, Gorbatov R, Leuschner F, et al. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol. 2012;59:153–163. doi: 10.1016/j.jacc.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang CP, Han S, Senokuchi T, Tall AR. The macrophage at the crossroads of insulin resistance and atherosclerosis. Circ Res. 2007;100:1546–1555. doi: 10.1161/CIRCRESAHA.107.152165. [DOI] [PubMed] [Google Scholar]
- 31.Jung K, Kim P, Leuschner F, Gorbatov R, Kim JK, Ueno T, et al. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ Res. 2013;112:891–899. doi: 10.1161/CIRCRESAHA.111.300484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153:362–375. doi: 10.1016/j.cell.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 34.Ye YX, Basse-Lusebrink TC, Arias-Loza PA, Kocoski V, Kampf T, Gan Q, et al. Monitoring of monocyte recruitment in reperfused myocardial infarction with intramyocardial hemorrhage and microvascular obstruction by combined fluorine 19 and proton cardiac magnetic resonance imaging. Circulation. 2013;128:1878–1888. doi: 10.1161/CIRCULATIONAHA.113.000731. [DOI] [PubMed] [Google Scholar]
- 35.Troidl C, Mollmann H, Nef H, Masseli F, Voss S, Szardien S, et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J Cell Mol Med. 2009;13:3485–3496. doi: 10.1111/j.1582-4934.2009.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frantz S, Hofmann U, Fraccarollo D, Schafer A, Kranepuhl S, Hagedorn I, et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 2013;27:871–881. doi: 10.1096/fj.12-214049. [DOI] [PubMed] [Google Scholar]
- 38.Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, et al. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J Immunol. 2013;191:4838–4848. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54:130–138. doi: 10.1016/j.jacc.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 40.Sahul ZH, Mukherjee R, Song J, McAteer J, Stroud RE, Dione DP, et al. Targeted imaging of the spatial and temporal variation of matrix metalloproteinase activity in a porcine model of postinfarct remodeling: relationship to myocardial dysfunction. Circ Cardiovasc Imaging. 2011;4:381–391. doi: 10.1161/CIRCIMAGING.110.961854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the Mononuclear Phagocyte Network Underlies Chronic Inflammation and Disease Progression in Heart Failure: Critical Importance of the Cardiosplenic Axis. Circ Res. 2014;114:266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 44.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anversa P, Olivetti G, Leri A, Liu Y, Kajstura J. Myocyte cell death and ventricular remodeling. Curr Opin Nephrol Hypertens. 1997;6:169–176. doi: 10.1097/00041552-199703000-00011. [DOI] [PubMed] [Google Scholar]
- 46.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 47.Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007;117:2692–2701. doi: 10.1172/JCI29134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–1638. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tall AR, Yvan-Charvet L, Westerterp M, Murphy AJ. Cholesterol efflux: a novel regulator of myelopoiesis and atherogenesis. Arterioscler Thromb Vasc Biol. 2012;32:2547–2552. doi: 10.1161/ATVBAHA.112.300134. [DOI] [PubMed] [Google Scholar]
- 51.Lindsey ML, Weintraub ST, Lange RA. Using extracellular matrix proteomics to understand left ventricular remodeling. Circ Cardiovasc Genet. 2012;5:o1–o7. doi: 10.1161/CIRCGENETICS.110.957803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engstrom G, Melander O, Hedblad B. Leukocyte count and incidence of hospitalizations due to heart failure. Circ Heart Fail. 2009;2:217–222. doi: 10.1161/CIRCHEARTFAILURE.108.827071. [DOI] [PubMed] [Google Scholar]
- 53.Maekawa Y, Anzai T, Yoshikawa T, Asakura Y, Takahashi T, Ishikawa S, et al. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: a possible role for left ventricular remodeling. J Am Coll Cardiol. 2002;39:241–246. doi: 10.1016/s0735-1097(01)01721-1. [DOI] [PubMed] [Google Scholar]
- 54.Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652–1663. doi: 10.1161/CIRCULATIONAHA.111.044164. [DOI] [PubMed] [Google Scholar]
- 55.Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273–1280. doi: 10.1038/nm.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Milonas C, Jernberg T, Lindback J, Agewall S, Wallentin L, Stenestrand U. Effect of angiotensin-converting enzyme inhibition on one-year mortality and frequency of repeat acute myocardial infarction in patients with acute myocardial infarction. Am J Cardiol. 2010;105:1229–1234. doi: 10.1016/j.amjcard.2009.12.032. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein JA, Demetriou D, Grines CL, Pica M, Shoukfeh M, O'Neill WW. Multiple complex coronary plaques in patients with acute myocardial infarction. N Engl J Med. 2000;343:915–922. doi: 10.1056/NEJM200009283431303. [DOI] [PubMed] [Google Scholar]
- 59.Naghavi M, Falk E, Hecht HS, Jamieson MJ, Kaul S, Berman D, et al. From vulnerable plaque to vulnerable patient—Part III: executive summary of the Screening for Heart Attack Prevention and Education (SHAPE) Task Force report. Am J Cardiol. 2006;98:2H–15H. doi: 10.1016/j.amjcard.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 60.Anversa P, Kajstura J, Rota M, Leri A. Regenerating new heart with stem cells. J Clin Invest. 2013;123:62–70. doi: 10.1172/JCI63068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Chugh AR, Beache GM, Loughran JH, Mewton N, Elmore JB, Kajstura J, et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126(11 Suppl 1):S54–S64. doi: 10.1161/CIRCULATIONAHA.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ben-Mordechai T, Holbova R, Landa-Rouben N, Harel-Adar T, Feinberg MS, Elrahman IA, et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 2013;62:1890–1901. doi: 10.1016/j.jacc.2013.07.057. [DOI] [PubMed] [Google Scholar]
- 63.Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 65.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med. 2013;19:695–703. doi: 10.1038/nm.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Godwin JW, Pinto AR, Rosenthal NA. Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci USA. 2013;110:9415–9420. doi: 10.1073/pnas.1300290110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation. 2013;127:2038–2046. doi: 10.1161/CIRCULATIONAHA.112.000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 71.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 72.Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2013 doi: 10.1016/j.jacc.2013.11.023. doi:10.1016/j.jacc.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanz J, Fayad ZA. Imaging of atherosclerotic cardiovascular disease. Nature. 2008;451:953–957. doi: 10.1038/nature06803. [DOI] [PubMed] [Google Scholar]
- 74.Leuschner F, Nahrendorf M. Molecular imaging of coronary atherosclerosis and myocardial infarction: considerations for the bench and perspectives for the clinic. Circ Res. 2011;108:593–606. doi: 10.1161/CIRCRESAHA.110.232678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee S, Vinegoni C, Feruglio PF, Fexon L, Gorbatov R, Pivoravov M, et al. Real-time in vivo imaging of the beating mouse heart at microscopic resolution. Nat Commun. 2012;3:1054. doi: 10.1038/ncomms2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alam SR, Shah AS, Richards J, Lang NN, Barnes G, Joshi N, et al. Ultrasmall superparamagnetic particles of iron oxide in patients with acute myocardial infarction: early clinical experience. Circ Cardiovasc Imaging. 2012;5:559–565. doi: 10.1161/CIRCIMAGING.112.974907. [DOI] [PubMed] [Google Scholar]
- 77.Yilmaz A, Dengler MA, van der Kuip H, Yildiz H, Rosch S, Klumpp S, et al. Imaging of myocardial infarction using ultrasmall superparamagnetic iron oxide nanoparticles: a human study using a multi-parametric cardiovascular magnetic resonance imaging approach. Eur Heart J. 2013;34:462–475. doi: 10.1093/eurheartj/ehs366. [DOI] [PubMed] [Google Scholar]