

Abstract
Mollusca is the second most diverse group of animals in the world. Despite their perceived importance, omics-level studies have seldom been applied to this group of animals largely due to a paucity of genomic resources. Here, we report the first large-scale gene-associated marker development and evaluation for a bivalve mollusc, Chlamys farreri. More than 21,000 putative single-nucleotide polymorphisms (SNPs) were identified from the C. farreri transcriptome. Primers and probes were designed and synthesized for 4500 SNPs, and 1492 polymorphic markers were successfully developed using a high-resolution melting genotyping platform. These markers are particularly suitable for population genomic analysis due to high polymorphism within and across populations, a low frequency of null alleles, and conformation to neutral expectations. Unexpectedly, high cross-species transferability was observed, suggesting that the transferable SNPs may largely represent ancestral genetic variations that have been preserved differentially among subfamilies of Pectinidae. Gene annotations were available for 73% of the markers, and 65% could be anchored to the recently released Pacific oyster genome. Large-scale association analysis revealed key candidate genes responsible for scallop growth regulation, and provided markers for further genetic improvement of C. farreri in breeding programmes.
Keywords: mollusca, single-nucleotide polymorphism (SNP), transcriptome, high resolution melting (HRM), genome-wide association (GWAS)
1. Introduction
Mollusca is the second most speciose animal phylum, containing more than 1 00 000 extant species distributed across eight major lineages.1 Molluscs play vital roles in the structure and functioning of aquatic and terrestrial ecosystems. Many molluscs are important fishery and aquaculture species, and some also serve as models for studying neurobiology, biomineralization, and adaptive evolution in response to climate change.2,3 Despite their perceived importance, systematic studies of molluscan biology and evolution remain very limited largely due to a paucity of genomic resources.
Recent advances in next-generation sequencing (NGS) technologies (e.g. Roche's 454 and Illumina's Solexa) now allow for rapid generation of extensive genomic resources at affordable cost for any organism, thus opening up opportunities for conducting omics-level analyses in molluscs. NGS-based genome sequencing has recently been performed for two bivalve molluscs, i.e. Pacific oyster (Crassostrea gigas)4 and pearl oyster (Pinctada fucata),5 providing the first insights into molluscan genome architecture and the genetic basis of stress adaptation, shell formation, and pearl biosynthesis. However, whole-genome de novo sequencing is currently still costly even using NGS platforms and remains out of reach for most laboratories focusing on non-model organisms. As an attractive alternative, transcriptome sequencing represents a cost-effective approach to rapidly expand gene resources, and it has been widely applied in many non-model organisms.6 The extensive gene resources generated by transcriptome sequencing are very useful not only for transcriptome-wide gene characterization and expression profiling, but also for large-scale single-nucleotide polymorphism (SNP) discovery. Such ‘functional’ SNPs are particularly valuable for quantitative genetic and evolutionary studies, because they have a great potential for quickly identifying causal genes responsible for either complex traits or adaptive evolution.
Genome-wide scans for detecting quantitative trait loci (QTL) for traits of interest or genetic determinants for local adaptation usually require a large number of genetic markers. SNPs have attracted significant attention, as they represent the most abundant class of genetic variations in eukaryotic genomes and have already become the marker of choice in large-scale genotyping applications, such as high-resolution linkage and association mapping, genomic selection, and comparative genome analysis. However, SNP markers have been insufficiently developed for molluscs in comparison with well-studied model organisms, such as mouse, nematode worm, and zebrafish. Even for the molluscs that are well characterized at the molecular level (e.g. Pacific oyster), only hundreds of markers are available. Transcriptome sequencing has recently been conducted in many molluscs,7–11 providing extensive resources for large-scale gene-associated SNP mining. High-throughput SNP screening and marker development rely on an efficient genotyping platform. High resolution melting (HRM) has proved to be an extremely powerful technique for rapid profiling of genetic variation within PCR amplicons.12,13 HRM has several advantages over other genotyping methods, such as its simplicity, low cost, high sensitivity, and specificity, and has been widely applied to many non-model species for marker development at medium-to-large scale.14,15
Our group has recently released a large amount of transcriptome data via 454 sequencing for a bivalve mollusc, Chlamys farreri (Jones et Preston 1904).16 Chlamys farreri is naturally distributed along the seacoasts of China, Japan, and Korea and is also an important aquaculture species in China. To date, <100 SNP markers have been developed for this species,17,18 which is not sufficient for large-scale genomic analyses, such as high-resolution linkage and QTL mapping, association mapping, and comparative genome analysis. Here, we conducted the first large-scale gene-associated SNP marker development for C. farreri. The 1492 SNP markers developed in this study were further evaluated for their usefulness in population genomic, comparative genomic, and association analyses.
2. Materials and Methods
2.1. Transcriptome sequences, assembly, and SNP mining
The C. farreri transcriptome sequences (SRA accession no. SRA030509) were produced by 454 sequencing of cDNA libraries prepared from diverse developmental stages and adult tissues. The details of sample collection, library preparation, and 454 sequencing have been described in a previous study.16 Briefly, for larval samples, approximately 1000 parents were used for artificial fertilization, while adult tissues were collected from 30 individuals. To reduce the risk of identifying artificial SNPs arising from sequencing errors or misassembly of paralogous sequences, 454 reads were reassembled using the CAP3 programme19 under very stringent assembly criteria (overlap setting: 100 bp and 95% similarity). Using the QualitySNP programme,20 putative SNPs were identified in the assembled contigs that were covered by at least four reads and had at least two reads for each allele.
2.2. SNP marker development based on the HRM genotyping platform
SNP markers were developed using a cost-effective HRM method14 that used two PCR primers and one unlabelled probe. SNP markers were named as follows: C followed by a contig ID, and then S followed by the SNP position (bp) within the contig. HRM primers and probes were designed following the rules described by Wang et al.14 Primers and probes were synthesized (Sangon Biotech) and evaluated using six C. farreri individuals. PCR amplifications were performed in a 10-µl volume composed of 20 ng genomic DNA, 0.1 µM forward primer, 0.5 µM reverse primer, 1.5 mM MgCl2, 0.2 mM dNTPs (Invitrogen), 1× LCGreen Plus (Idaho Technology), 1× PCR buffer, and 0.5 U Taq DNA polymerase (TaKaRa). Thermal cycling began with an initial denaturing step at 95°C for 5 min, followed by 60 cycles of 95°C for 40 s, 63°C for 40 s, and 72°C for 40 s with a final extension at 72°C for 5 min. The corresponding probe was added to each PCR to a final concentration of 3 µM, and the reaction mixture was denatured at 95°C for 10 min and then slowly cooled to 4°C. HRM genotyping was performed on a Light-Scanner instrument (Idaho Technology) with continuous melting curve acquisition (10 acquisitions per °C) during a 0.1°C/s ramp from 40 to 95°C. All primer and probe sequences of the developed SNP markers are provided in Supplementary Table S1.
2.3. Population genetic analysis
The developed SNP markers were evaluated using four wild, geographical populations. In total, 54 C. farreri individuals were used for this evaluation, of which 24 were collected from the Jiaonan (JN) population, 12 from the Changdao (CD) population, 12 from the Rongcheng (RC) population, and 6 from the Dalian (DL) population. The collection details of these samples were described in a previous study.21 Marker polymorphisms were evaluated within and among populations. For each marker, allele frequency, observed heterozygosity (Ho), and expected heterozygosity (He), along with tests for neutrality, Hardy–Weinberg equilibrium, and linkage disequilibrium, were calculated using POPGENE22 or GENEPOP 4.0 program.23 For Hardy–Weinberg equilibrium and linkage disequilibrium tests, Bonferroni correction was also applied to account for multiple testing.
2.4. Test for cross-species transferability
In total, 34 markers targeting 24 synonymous and 10 non-synonymous SNPs were selected for evaluating cross-species transferability in five scallop species from three Pectinidae subfamilies, i.e. Chlamydinae (Chlamys nobilis and Patinopecten yessoensis), Pectininae (Amusium pleuronectes), and Aequipectini (Argopecten irradians and Argopecten purpuratus). All species except A. purpuratus were purchased from local markets in China. Argopecten purpuratus samples were kindly provided by Dr Chunde Wang (Qingdao Agriculture University, China). For each species, eight individuals were used to evaluate polymorphisms.
2.5. Marker annotation and gene comparison with Pacific oyster
To provide functional annotations for the SNP markers, relevant contig sequences were compared against the Swiss-Prot and Nr protein databases using BlastX, with an e-value threshold of 1e−5. Gene names were assigned to each marker based on the best hit. The BlastX results were imported into the Blast2GO software24 for gene ontology (GO) analysis. GO terms were assigned to query sequences based on three ontology classifications, i.e. biological process, molecular function, and cellular component. To gain an overview of gene pathways, KEGG analysis was also performed using the KEGG Automatic Annotation Server.25 The bi-directional best hit method was used to obtain KEGG orthology assignments. The contig sequences containing the SNP markers were also compared against the oyster protein database using BlastX, with an e-value threshold of 1e−4.
2.6. Large-scale marker associations with growth traits
Large-scale marker associations with growth traits were conducted on an elite variety of C. farreri named the ‘Penglai-Red’ scallop (Aquacultural Variety Registration Number of the Ministry of Agriculture of China: GS02-001-2005), which was developed by our group and has been under continuous artificial selection for red shell colour and fast growth for multiple generations. A large breeding population was established in May 2010 by artificial fertilization of more than 3000 sexually mature ‘Penglai-Red’ scallops at the hatchery of the Xunshan Aquatic Group Corporation (Shandong Province, China). After rearing for 2 months, half the juvenile scallops were moved to another hatchery that is ∼250 km from the original hatchery. In May 2012, approximately 1000 2-year-old scallops were randomly sampled from each hatchery, constituting two target populations for the association analyses in this study. To reduce the genotyping cost, a selective genotyping strategy was adopted. For each population, 40 individuals with large body sizes and 40 ones with small body sizes were chosen for genotyping with the 1492 SNP markers. For an initial scan of the whole marker set, a DNA pool was established for each group by mixing equal amount of genomic DNA prepared from individuals within each group. The resultant DNA pools were subject to HRM analysis to search for allele frequency differences between groups. Markers showing large between-group differences in allele frequencies were subject to further validation by genotyping all individuals in both groups; statistical significance was determined using a χ2 test.
3. Results
3.1. Transcriptome assembly and SNP mining
In total, 1 099 254 clean reads were used for transcriptome assembly. The final assembly consisted of 50 741 contigs and 406 825 singletons. Contig sizes ranged from 60 to 6063 bp, with an average of 541 bp. The average read and sequencing depth coverage across all contigs were 13.6 and 7.5, respectively. The relatively low sequencing depth limits our ability to detect less common or rare SNPs; nevertheless, this transcriptomic resource enabled us to identify a large number of candidate SNPs for marker development. A total of 21 813 putative SNPs were identified from 18 780 contigs. The distribution of SNP coverage is shown in Fig. 1. The overall SNP frequency was one SNP per 1258 bp. Of these SNPs, 14 641(67%) were transitions, whereas 7172 (33%) were transversions. A total of 7338 SNPs were identified from contigs for which gene annotation information was available.
Figure 1.

Distribution of sequencing coverage for the SNPs detected in the C. farreri transcriptome.
3.2. Marker development based on the HRM genotyping platform
The entire marker development and evaluation procedure are depicted in Fig. 2. All putative SNPs were evaluated for HRM primer and probe design, which led to a successful set of primers and probes for 4500 SNPs with a wide range of minor allele frequencies (MAFs; Table 1). All primers and probes were synthesized and evaluated using six C. farreri individuals. A total of 3042 primer pairs produced strong single bands with expected sizes. Although the expected amplicon length was restricted to ∼100 bp during primer design, 616 primer pairs still produced bands larger than the expected sizes, indicating potential introns in the vicinity of those SNPs. Longer amplicons can greatly diminish the sensitivity of HRM analysis, so these SNPs were excluded from further consideration. Two hundred and forty-seven primer pairs produced more than one band possibly due to non-specific amplifications. The remaining primer pairs resulted in poor amplifications, i.e. very weak or no PCR product, and were excluded from further consideration. Probe testing was subsequently performed for primer pairs with successful amplifications. In total, 2231 probes generated well-recognizable melting curve profiles with a distinct peak for each allele. Of these, 1492 loci were polymorphic across all assayed individuals, whereas 739 loci were monomorphic possibly due to the small number of assayed individuals. The SNP validation rate was correlated with the MAF in the initial discovery panel. A higher MAF tended to lead to a higher validation rate (Table 1). For example, at the MAF interval of 0.4–0.5, 75% of markers were polymorphic. In contrast, only 42% were polymorphic at the MAF interval of 0–0.1, and the low validation rate may be largely attribute to the relatively low quality of predicted SNPs (i.e. SNPs that were difficult to distinguish from sequencing errors). Information pertaining to all 1492 SNP loci has been submitted to the dbSNP database (http://www.ncbi.nlm.nih.gov/snp/) under accession numbers rs831881546–rs831883035 and rs831883061–rs831883062.
Figure 2.

A schematic workflow describing SNP marker development and evaluation in C. farreri.
Table 1.
Overview of the efficiency of HRM-based SNP marker development in C. farreri
| Minor allele frequency ranges | No. of primers designed | No. of primer validated (% of designed) | No. of probe validated (% of designed) | No. of polymorphic loci (% of designed) |
|---|---|---|---|---|
| 0.4–0.5 | 1538 | 1056 (69%) | 761 (49%) | 571 (37%) |
| 0.3–0.4 | 1422 | 954 (67%) | 701 (49%) | 506 (36%) |
| 0.2–0.3 | 832 | 559 (67%) | 422 (51%) | 266 (32%) |
| 0.1–0.2 | 636 | 418 (66%) | 309 (49%) | 133 (21%) |
| 0–0.1 | 72 | 55 (76%) | 38 (53%) | 16 (22%) |
3.3. Evaluation of SNP markers for population genomic analysis
The suitability of the 1492 polymorphic markers for population genomic analysis was evaluated using four geographical populations. PCR success rates across all individuals ranged from 48 to 100% with an average of 93%, suggesting that null alleles are indeed present at very low frequencies in these populations as expected for gene-derived markers. All the markers showed polymorphisms across the four populations and within each population. Marker polymorphism was 100% for the JN population, 96% for the CD population, 97% for the RC population, and 88% for the DL population. Population genetic parameters were estimated for the JN population (Supplementary Table S1), because this population had a relatively large sample size compared with the other populations, and therefore, genetic parameters could be estimated more reliably. For all markers, MAF ranged from 0.02 to 0.50. The Ho and He were 0.34 and 0.37, respectively; these values are relatively low compared with the previous estimates for the same population using microsatellite markers.21 In total, 1422 markers passed the neutrality test, suggesting that these markers are not under strong selection and are suitable for population genomic analyses that usually require neutral markers. The majority of markers were in Hardy–Weinberg equilibrium, and only 81 markers showed significant departures after Bonferroni correction. Significant linkage disequilibrium was detected in 102 marker pairs. These markers are valuable for detecting recent admixture and migration events in natural populations.26
3.4. Cross-species transferability test
To assess the cross-species transferability of the developed SNP markers, 34 markers targeting 24 synonymous and 10 non-synonymous SNPs were selected for HRM genotyping in five scallop species deriving from three Pectinidae subfamilies. A total of 23 were successfully amplified in at least one species, of which 16 were polymorphic in at least one species. The highest transferability (up to four species) was observed for marker C10745S115_CG (Table 2). Unexpectedly, the total number of transferable markers was quite similar (6–7 markers) for the three Pectinidae subfamilies, which does not reflect the difference in their phylogenetic relationships to C. farreri (i.e. C. farreri belongs to Chlamydinae, which is more closely related to Pectininae than to Aequipectini27). The markers that could be transferred to at least three species were all synonymous SNPs; while all but one of the non-synonymous SNPs were transferable only to a single species.
Table 2.
The cross-species transferability of 16 C. farreri SNP markers
| Marker name | Species name |
||||
|---|---|---|---|---|---|
| Chlamys nobilis | Patinopecten yessoensis | Amusium pleuronectes | Argopecten irradians | Argopecten purpuratus | |
| C10745S115_CG | √ | √ | √ | √ | |
| C12208S460_TC | √ | √ | √ | ||
| C49010S592_AG | √ | √ | √ | ||
| C49829S586_AC | √ | √ | √ | ||
| C30426S483_TC | √ | √ | |||
| C47021S446_CA | √ | √ | |||
| C34305S164_CG | √ | √ | |||
| C15644S268_AG | √ | ||||
| C13134S327_TC | √ | ||||
| C37392S686_GA | √ | ||||
| C20740S340_TC | √ | ||||
| C11312S559_GA | √ | ||||
| C20815S313_AT | √ | ||||
| C19522S376_TG | √ | ||||
| C30287S1286_TG | √ | ||||
| C15676S348_AG | √ | ||||
Non-synonymous SNP makers are indicated in grey.
3.5. Functional annotation and gene comparison with Pacific oyster
Gene annotation was performed for the 1492 markers using BlastX comparison against the Swiss-Prot and Nr protein databases. A total of 1095 (73.4%) markers had significant matches to known proteins in these databases, corresponding to 953 unique accessions (Supplementary Table S1). GO analysis revealed that one or more GO terms could be assigned to 466 markers for a total of 923 GO assignments. The annotated markers were involved in diverse biological processes and functions, and their GO composition largely resembled that summarized for the total set of contigs (Supplementary Fig. S1). KEGG pathway analysis revealed that 420 markers were involved in 123 different pathways (Supplementary Table S2). In particular, 21 markers were involved in the immune system, and these markers are worthy of further evaluation to determine whether any are associated with disease/pathogen resistance.
To evaluate the utility of the 1492 SNP markers for future comparative analysis with the oyster genome, the contig sequences of these markers were compared against the oyster protein database. A total of 963 C. farreri contigs containing 1034 SNP markers showed significant sequence homology to oyster proteins (e-value of < 1e−4; Supplementary Table S1). One example showing the correspondence between eight SNP markers and oyster scaffold419 is presented in Fig. 3. These oyster proteins are distributed on 514 genomic scaffolds (genome assembly v9; Table 3). The lengths of these scaffolds ranged from 2 to 1965 kb. A total of 235 scaffolds were matched by at least two C. farreri markers, and 39 scaffolds were matched by at least five markers (Table 3). These markers and matching related oyster scaffolds provide an important basis for further genome comparison between the two species.
Figure 3.
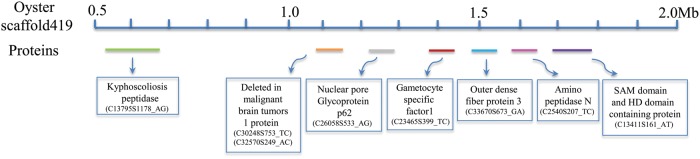
Schematic representation of the correspondence between eight C. farreri SNP markers and Pacific oyster scaffold419 (genome assembly v9). Oyster proteins and the related C. farreri SNPs are indicated below the scaffold.
Table 3.
Summary of the oyster scaffolds matched by C. farreri SNP markers
| Total marker no. matched to each scaffold | No. of matched oyster scaffolds | Length of matched scaffolds (kb) |
|---|---|---|
| 12 | 1 | 1620 |
| 10 | 1 | 1193 |
| 9 | 1 | 1726 |
| 8 | 7 | 649–1965 |
| 7 | 4 | 969–1120 |
| 6 | 10 | 650–1697 |
| 5 | 15 | 538–1855 |
| 4 | 25 | 63–1372 |
| 3 | 63 | 58–1861 |
| 2 | 108 | 31–1727 |
| 1 | 279 | 2–1100 |
3.6. Large-scale marker associations with growth traits
Based on the developed marker set, we further conducted a large-scale association analysis of growth traits using two C. farreri populations that were derived from a large breeding population of an elite C. farreri variety (‘Penglai-Red’ scallop), but were reared in two different geographic locations. Two populations were used in the association analysis to ensure identification of SNP loci linked with the genetic variance of traits but not the environmental effects. To reduce the genotyping cost, a selective genotyping approach was adopted, in which only two groups of individuals sampled from the two tails of the trait distribution were used for HRM analysis (Fig. 4). An initial scan of the whole marker set on DNA pools revealed that five markers showed prominent allele frequency difference between the two groups in both populations (Fig. 5). Statistical significance of the observed allele frequency difference was further confirmed for the five markers by genotyping each individual within each group for both populations (Table 4). After Bonferroni correction, four markers remained significant in at least one population. Gene annotation information was available for only one marker, C7493S233_CT (SDF4, 45 kDa calcium-binding protein, 7e−19).
Figure 4.

The shell-height distribution of approximately 1000 C. farreri individuals collected from each of two C. farreri populations. For the large body size (LBS) groups, the average trait value was 75.59 ± 2.92 mm for population a and 72.02 ± 2.57 mm for population b, whereas for small body size (SBS) groups, it was 49.93 ± 3.84 and 53.04 ± 3.41 mm for populations a and b, respectively. For each population, 40 individuals were sampled from each group for association analysis.
Figure 5.

Example HRM profiles for two growth-related markers, C10973S307_GA and C7493S233_CT, identified by genotyping DNA pools prepared from both the LBS and SBS groups. Clear differences in allele frequencies were observed between groups.
Table 4.
Comparison of the allele and genotype frequency of five growth-related markers between LBS and SBS groups sampled from two C. farreri populations.
| Maker name | LBS group (population 1/population 2) |
SBS group (population 1/population 2) |
Allele frequency P-value | Genotype frequency P-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele frequency |
Genotype frequency |
Allele frequency |
Genotype frequency |
|||||||||
| C10973S307_GA | 0.51/0.29 (A) | 0.49/0.71 (G) | 0.36/0.07 (AA) | 0.31/0.44 (GA) | 0.33/0.49 (GG) | 0.86/0.74 (A) | 0.14/0.26 (G) | 0.76/0.68 (AA) | 0.18/0.12 (GA) | 0.05/0.20 (GG) | 8.86e−06a/1.10 e−07a | 5.00 e−04/5.68 e−07a |
| C6068S140_CA | 0.49/0.41 (A) | 0.51/0.59 (C) | 0.33/0.24 (AA) | 0.31/0.33 (CA) | 0.36/0.43 (CC) | 0.81/0.80 (A) | 0.19/0.20 (C) | 0.70/0.76 (AA) | 0.22/0.08 (CA) | 0.08/0.16 (CC) | 3.98 e−03/1.12 e−06a | 2.24 e−03/5.27 e−05 |
| C13555S183_TG | 0.45/0.32 (G) | 0.55/0.68 (T) | 0.28/0.13 (GG) | 0.34/0.38 (TG) | 0.38/0.49 (TT) | 0.77/0.74 (G) | 0.23/0.26 (T) | 0.69/0.64 (GG) | 0.17/0.18 (TG) | 0.14/0.18 (TT) | 6.10 e−05/2.55 e−07a | 1.70 e−03/1.20 e−05a |
| C17803S726_GA | 0.55/0.76 (A) | 0.45/0.24 (G) | 0.40/0.72 (AA) | 0.30/0.08 (GA) | 0.30/0.20 (GG) | 0.25/0.47 (A) | 0.75/0.53 (G) | 0.10/0.36 (AA) | 0.29/0.23 (GA) | 0.61/0.41 (GG) | 1.44 e−04/2.94 e−04 | 5.01 e−03/2.15 e−04 |
| C7493S233_CT | 0.96/0.85 (C) | 0.04/0.15 (T) | 0.94/0.77 (CC) | 0.03/0.15 (CT) | 0.03/0.08 (TT) | 0.71/0.53 (C) | 0.29/0.47 (T) | 0.59/0.31 (CC) | 0.23/0.43 (CT) | 0.18/0.26 (TT) | 1.73 e−05a/2.75 e−05a | 8.39 e−04/4.33 e−04 |
aSignificant after Bonferroni correction.
For each marker, alleles or genotypes are indicated in parenthesis below their frequencies.
4. Discussion
4.1. Large-scale marker development from NGS-generated transcriptomic resources
NGS technologies are now frequently being used to generate extensive genomic resources for non-model organisms. Although a large number of SNPs can be readily discovered from these resources, large-scale marker development and evaluation are seldom performed for such in silico predicted SNPs. In this study, we conducted the first transcriptome-wide marker development from more than 21,000 putative SNPs for a bivalve mollusc, which currently represents the largest gene-associated marker collection for the phylum Mollusca. Although stringent criteria were adopted for transcriptome assembly, the marker conversion rate remained relatively low (∼33%), but was still comparable with those reported in recent studies using the same genotyping platform.14–16 The inefficient marker conversion can largely be attributed to low passing rates in the primer and probe tests, i.e. 32.4% failed the primer test and 18% failed the probe test. This issue may be directly related to a few well-known drawbacks of NGS platforms. For example, NGS platforms usually generate a substantial higher rate of sequencing errors than traditional Sanger-based methods. In particular, the 454 sequencing platform is prone to introducing higher indel errors than other platforms, especially when stretches of homopolymeric bases are present.28–30 Sequencing errors, when occurring at positions where a primer or probe binds, may hamper the amplification efficiency and confound subsequent HRM analysis. In addition, sequencing errors are also a major source of falsely predicted SNPs. As revealed by this study, there is a trend for SNPs with higher MAF to have higher validation rates. This result suggests that SNPs with high MAF (e.g. 40–50%) are less likely to be affected by sequencing errors and thus, should be given high priority during marker development. Furthermore, NGS platforms usually produce very short reads (e.g. 35–100 bp); accurate de novo assembly of such short reads poses a significant informatics challenge.31 Assembly artefacts can arise when reads of different genomic origins are misassembled together due to high similarity of sequence context. Such artefacts may result in PCR failure when primers are targeting two genomic regions far from each other. One interesting finding is that introns were present at a substantially high frequency, even though we restricted the expected amplicon length to ∼100 bp during primer design. Approximately 14% of primer pairs produced amplicons with a larger than expected size, indicating that introns were present in the vicinity of SNPs. It should be noted that this percentage most likely underestimates the actual occurrence of introns, because very large introns would more likely result in PCR failure rather than successful amplicons of larger than expected size. The aforementioned issues cannot be easily amended without the aid of a high-quality reference genome. However, with the rapid development of sequencing technologies (i.e. much longer reads and higher accuracy) and more draft genomes being generated for non-model species, there is much hope that these issues can be resolved.
4.2. Genic versus non-genic markers in molluscan population studies
Currently, the genetic markers available for molluscs were developed predominantly from anonymous genomic sequences. It is well known that molluscan genomes are typically highly heterozygous.4,5 For example, the Pacific oyster has an average SNP density of one SNP per 60 bp in coding regions and one per 40 bp in non-coding regions.32 Such high genome heterozygosity can result in a high frequency of null alleles, i.e. alleles fail to amplify due to random mutations in primer-binding regions. Extremely high proportions of null alleles have been observed for the microsatellite markers developed in C. farreri (56%).33 Application of these markers in population genetic studies can severely distort the estimation of population structure and differentiation, parentage analysis, and assignment tests,34 and may result in misleading conclusions or inferences. In contrast, genetic markers developed from transcriptomic sequences can, to some extent, alleviate this problem, because transcribed regions are usually more conserved than non-transcribed regions. In this study, the average PCR success rate across all assayed individuals was 93%, suggesting that null alleles were indeed present at very low frequencies, as expected for gene-derived markers.
Remarkably, high population transferability was observed for the developed SNP markers, and the vast majority of which were also neutral markers. Our study, therefore, provides a valuable set of SNP markers that are suitable for population genomic studies. A major benefit of utilizing gene-associated markers is that it is possible to quickly identifying causal genes under natural selection, which usually involves identifying outlier SNP markers showing significantly increased or decreased differentiation among populations compared with neutral expectations.35 Scaling up the number of available gene-associated SNPs to thousands of markers extends the genome coverage, thereby increasing the probability of identifying outlier loci that are in tight linkage disequilibrium with loci under selection.
4.3. Cross-species transferability and comparative genomics
Gene-associated markers generally exhibit higher cross-species transferability in closely related species than markers developed from anonymous genomic sequences. However, to what extent SNP markers developed from one species are transferable to others within the Pectinidae family remains unknown. In this study, about half of the tested markers were polymorphic in at least one of the five scallop species, with the most polymorphic marker transferable to up to four species, indicating the high cross-species transferability of the developed SNP markers. However, marker transferability across the three Pectinidae subfamilies showed no correlation with their phylogenetic relationships to C. farreri. This is somewhat unexpected, suggesting that these transferable SNPs may largely represent ancestral genetic variations that have been preserved differentially among Pectinidae subfamilies. Our study also revealed that SNPs with high transferability (≥3 species) were all synonymous SNPs. This makes sense because synonymous SNPs do not alter the encoded amino acids and thus, are less likely to be removed by purifying selection. Once a polymorphism had arisen, it would be preserved for a long period of time during the evolution of the Pectinidae family.
The high cross-species transferability together with the high annotation rate (∼73%) of the developed markers hold great promise for conducting comparative genome analysis among molluscs. The recently released Pacific oyster genome represents the best assembled genome currently available for bivalve molluscs.4 It provides a valuable genomic resource not only for oyster genetic and breeding studies, but also for comparative genome analysis among molluscs. One of our ongoing projects is to construct a high-density linkage map for C. farreri based on the markers developed here. In the present study, we made an initial assessment of the usefulness of the developed markers (if they were included in a future linkage map) for comparative genome analysis between the two bivalve species. Of the annotated markers, 69% could be anchored onto the Pacific oyster genome, and 250 markers could be linked to very large oyster scaffolds that are longer than 500 kb and matched by at least five markers. Therefore, once the high-density map is constructed, these markers would help identify conserved genomic regions (i.e. synteny blocks) between the two species.
4.4. Large-scale marker association analysis of growth traits
Identification of major genes responsible for growth traits is highly desirable in scallop breeding programmes for the purpose of genetic improvement. Gene-associated markers are valuable tools for fulfilling such task, because they have great potential for quickly identifying causal genes underlying the trait of interest. The extensive marker set generated by this study provides an unprecedented opportunity to conduct a large-scale association analysis of growth traits in C. farreri. Using a selective genotyping approach, five growth-related markers were identified and confirmed in two C. farreri populations, where allele frequencies were associated with variation in growth trait. Unfortunately, four of these markers are currently uncharacterized, and therefore, their putative roles in growth regulation cannot be inferred. The marker C7493S233_CT was the only marker with annotation information (SDF4, 45 kDa calcium-binding protein). This gene belongs to the calcium-binding protein family, a large group of proteins that can regulate a variety of cellular processes including cell division, differentiation, motility, and apoptosis.36 Mutations in calcium-binding proteins have been associated with variation in growth traits, as demonstrated in Chinese cattle.37 Currently, it is largely unknown which genes or genetic loci are involved in scallop growth regulation. Our study offers the first report of candidate genes responsible for scallop growth regulation and provides markers for further genetic improvement of C. farreri in breeding programmes.
4.5. Conclusions
We developed for the first time large-scale gene-associated SNP markers for a bivalve mollusc, which currently represents the largest gene-associated marker collection in the phylum Mollusca. The properties of high polymorphism within and across populations, low frequency of null alleles, neutrality, and high cross-species transferability make these markers highly valuable for population genomic, comparative genomic, and genome-wide association studies.
Supplementary Data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was financially supported and provided by the National Natural Science Foundation of China (31130054), National Basic Research Program of China (2010CB126406), National High Technology Research and Development Program of China (2012AA10A401, 2012AA10A402, and 2012AA10A405) and Natural Science Foundation for Distinguished Young Scholars of Shandong Province (JQ201308).
Supplementary Material
Acknowledgements
We are very grateful to Xunshan Aquatic Group Corporation (Shandong Province, China) for the help in maintaining and sampling scallop materials.
Footnotes
Edited by Dr Shoji Tsuji
References
- 1.Haszprunar G., Schander C., Halanych K.M. Relationships of higher molluscan taxa. In: Ponder W., Lindberg D.R., editors. Phylogeny and Evolution of the Mollusca. Berkeley: University of California Press; 2008. pp. 19–32. [Google Scholar]
- 2.Walters E.T., Moroz L.L. Molluscanmemory of injury: evolutionary insights intochronic pain and neurological disorders. Brain Behav. Evol. 2009;74:206–18. doi: 10.1159/000258667. doi:10.1159/000258667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talmage S.C., Gobler C.J. Effects of past, present, and future ocean carbon dioxide concentrations on the growth and survival of larval shellfish. Proc. Natl. Acad. Sci. USA. 2010;107:17246–51. doi: 10.1073/pnas.0913804107. doi:10.1073/pnas.0913804107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang G., Fang X., Guo X., et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2011;490:49–54. doi: 10.1038/nature11413. doi:10.1038/nature11413. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi T., Kawashima T., Koyanagi R., et al. Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Res. 2012;19:117–30. doi: 10.1093/dnares/dss005. doi:10.1093/dnares/dss005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekblom R., Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107:1–15. doi: 10.1038/hdy.2010.152. doi:10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franchini P., van der Merwe M., Roodt-Wilding R. Transcriptome characterization of the South African Abalone Haliotis midae using sequencing-by-synthesis. BMC Res. Notes. 2011;4:59. doi: 10.1186/1756-0500-4-59. doi:10.1186/1756-0500-4-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hou R., Bao Z., Wang S., et al. Transcriptome sequencing and de novo analysis for Yesso scallop (Patinopecten yessoensis) using 454 GS Flx. PLoS One. 2011;6:e21560. doi: 10.1371/journal.pone.0021560. doi:10.1371/journal.pone.0021560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith S.A., Wilson N.G., Goetz F.E., et al. Resolving the evolutionary relationship of molluscs with phylogenomic tools. Nature. 2011;480:364–7. doi: 10.1038/nature10526. doi:10.1038/nature10526. [DOI] [PubMed] [Google Scholar]
- 10.Sadamoto H., Takahashi H., Okada T., Kenmoku H., Toyota M., Asakawa Y. De novo sequencing and transcriptome analysis of the central nervous system of mollusk Lymnaea stagnalis by deep RAN sequencing. PLoS One. 2012;7:e42546. doi: 10.1371/journal.pone.0042546. doi:10.1371/journal.pone.0042546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X., Mao Y., Huang Z., et al. Transcriptome analysis of the Octopus vulgaris central nervous system. PLoS One. 2012;7:e40320. doi: 10.1371/journal.pone.0040320. doi:10.1371/journal.pone.0040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reed G.H., Kent J.O., Wittwer C.T. High-resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics. 2007;8:597–608. doi: 10.2217/14622416.8.6.597. doi:10.2217/14622416.8.6.597. [DOI] [PubMed] [Google Scholar]
- 13.Taylor C.F. Mutation scanning using high-resolution melting. Biochem. Soc. Trans. 2009;37:433–7. doi: 10.1042/BST0370433. doi:10.1042/BST0370433. [DOI] [PubMed] [Google Scholar]
- 14.Wang S., Zhang L., Meyer E., Matz M.V. Construction of a high-resolution genetic linkage map and comparative genome analysis for the reef-building coral Acropora millepora. Genome Biol. 2009;10:R126. doi: 10.1186/gb-2009-10-11-r126. doi:10.1186/gb-2009-10-11-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ujino-Ihara T., Taguchi Y., Moriguchi Y., Tsumura Y. An efficient method for developing SNP markers based on EST data combined with high resolution melting (HRM) analysis. BMC Res. Notes. 2010;3:51. doi: 10.1186/1756-0500-3-51. doi:10.1186/1756-0500-3-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S., Hou R., Bao Z., et al. Transcriptome sequencing of Zhikong scallop (Chlamys farreri) and comparative transcriptomic analysis with Yesso scallop (Patinopecten yessoensis) PLoS One. 2013;8:e63927. doi: 10.1371/journal.pone.0063927. doi:10.1371/journal.pone.0063927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang G., Li J., Li L., Zhang L., Bao Z. Development of 44 gene-based SNP markers in Zhikong scallop, Chlamys farreri. Conserv. Genet. Resour. 2011;3:659–63. doi:10.1007/s12686-011-9427-2. [Google Scholar]
- 18.Wang X., Hu X., Li J., et al. Characterization of 38 EST-derived SNP markers in Zhikong scallop (Chlamys farreri) and their cross-species utility in Yesso scallop (Patinopecten yessoensis) Conserv. Genet. Resour. 2012;4:747–53. doi:10.1007/s12686-012-9636-3. [Google Scholar]
- 19.Huang X., Madan A. CAP3, a DNA sequence assembly program. Genome Res. 1999;9:868–77. doi: 10.1101/gr.9.9.868. doi:10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang J.F., Vosman B., Voorrips R.E., van der Linden C.G., Leunissen J.A.M. QualitySNP, a pipeline for detecting single nucleotide polymorphisms and insertions/deletions in EST data from diploid and polyploidy species. BMC Bioinformatics. 2006;7:438. doi: 10.1186/1471-2105-7-438. doi:10.1186/1471-2105-7-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhan A., Hu J., Hu X. Fine-scale population genetic structure of Zhikong scallop (Chlamys farreri), do local marine currents drive geographical differentiation? Mar. Biotechnol. 2009;11:223–35. doi: 10.1007/s10126-008-9138-1. doi:10.1007/s10126-008-9138-1. [DOI] [PubMed] [Google Scholar]
- 22.Yeh F.C., Boyle T.J.B. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg. J. Bot. 1997;129:157. [Google Scholar]
- 23.Raymond M., Rousset F. GENEPOP (Version 1.2), population genetics software for exact tests and ecumenicism. J. Hered. 1995;86:248–9. [Google Scholar]
- 24.Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., Robles M. Blast2GO, A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. doi:10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 25.Moriya Y., Itoh M., Okuda S., Yoshizawa A., Kanehisa M. KAAS, an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–185. doi: 10.1093/nar/gkm321. doi:10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldstein D.B., Weale M.E. Population genomics, linkage disequilibrium holds the key. Curr. Biol. 2001;11:R576–9. doi: 10.1016/s0960-9822(01)00348-7. doi:10.1016/S0960-9822(01)00348-7. [DOI] [PubMed] [Google Scholar]
- 27.Puslednik L., Serb J.M. Molecular phylogenetics of the Pectinidae (Mollusca, Bivalvia) and effect of increased taxon sampling and outgroup selection on tree topology. Mol. Phylogenet. Evol. 2008;48:1178–88. doi: 10.1016/j.ympev.2008.05.006. doi:10.1016/j.ympev.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Margulies M., Egholm M., Altman W.E., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huse S.M., Huber J.A., Morrison H.G., Sogin M.L., Welch D.M. Accuracy and quanlity of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. doi:10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao W., Boltz V.F., Spindler J.E., et al. Analysis of 454 sequencing error rate, error sources, and artifact recombination for detection of low-frequency drug resistance mutations in HIV-1 DNA. Retrovirology. 2013;10:18. doi: 10.1186/1742-4690-10-18. doi:10.1186/1742-4690-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin J.A., Wang Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011;12:671–82. doi: 10.1038/nrg3068. doi:10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 32.Sauvage C., Bierne N., Lapegue S., Boudry P. Single nucleotide polymorphisms and their relationship to codon usage bias in the Pacific oyster Crassostrea gigas. Gene. 2007;406:13–22. doi: 10.1016/j.gene.2007.05.011. doi:10.1016/j.gene.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 33.Zhan A., Hu J., Hu X. Construction of microsatellite-based linkage maps and identification of size-related quantitative trait loci for Zhikong scallop (Chlamys farreri) Anim. Genet. 2009;40:821–31. doi: 10.1111/j.1365-2052.2009.01920.x. doi:10.1111/j.1365-2052.2009.01920.x. [DOI] [PubMed] [Google Scholar]
- 34.Wen Y., Uchiyama K., Han W. Null alleles in microsatellite markers. Biodiversity Sci. 2013;21:117–26. [Google Scholar]
- 35.Hohenlohe P.A., Phillips P.C., Cresko W.A. Using population genomics to detect selection in natural populations, key concepts and methodological considerations. Int. J. Plant Sci. 2010;171:1059–71. doi: 10.1086/656306. doi:10.1086/656306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carafoli E., Klee C.B. Calcium as a cellular regulator. Oxford: Oxford University Press; [Google Scholar]
- 37.Li F., Chen H., Lei C.Z. Novel SNPs of the bovine NUCB2 gene and their association with growth traits in three native Chinese cattle breeds. Mol. Biol. Rep. 2009;37:541–6. doi: 10.1007/s11033-009-9732-y. doi:10.1007/s11033-009-9732-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.