

Abstract
Calcium phosphate precipitation is a convenient and economical method for transfection of cultured cells. With optimization, it is possible to use this method on hard-to-transfect cells like primary neurons. Here we describe our detailed protocol for calcium phosphate transfection of hippocampal neurons cocultured with astroglial cells.
Keywords: Neuroscience, Issue 81, Primary hippocampal neuron, calcium phosphate transfection, Coculture, astroglial cells, DNA
Introduction
Primary neurons are one of the hardest cell types to transfect as they are postmitotic and are very sensitive to micro-environmental changes. There are four commonly used types of methods for expression of exogenous genes and short-hairpin RNAs (shRNAs) in these cells1. Each has its own advantages and disadvantages. For example, electroporation is usually performed on freshly isolated neurons2, as cells must be transferred into cuvettes for transfection. Virus infection can usually achieve very high efficiency3, but is more labor-intensive and risky for operators. Many lipid-mediated transfection reagents are available commercially, with varying degrees of success in neurons and different levels of cytotoxicity.
Calcium phosphate transfection represents a convenient and economical method for introducing foreign genes into neurons. The method was first used to introduce adenovirus DNA into mammalian cells by Graham and Van Der Eb (1973)4. Transfection was performed by mixing calcium chloride with recombinant DNA in a phosphate buffer. This allows the formation of DNA/calcium phosphate precipitates which, when gradually dropped onto a monolayer of cells, adhere to the cell surface, are taken up by endocytosis and finally enter the nucleus5. This process would lead to the expression of introduced foreign genes in the target cell. Typical efficiencies of calcium phosphate transfection range between 0.5-5%6-8. However, with careful optimization and consistent execution of the experimental protocol, it is possible to reach a transfection efficiency of almost 50%. Here we describe our detailed protocol for calcium phosphate transfection of primary hippocampal neurons, which are cocultured with astroglial cells in a sandwich format9.
Protocol
1. Preparing Rat Astrocyte Culture for Conditioned Media and Astrocyte-neuron Cocultures.
Prepare dissection buffer (BSS, see Table 1 for recipe) and store at 4 °C until ready for use.
Anesthetize neonatal rat pups (P0-P2) with isoflurane in a 500 ml beaker.
When pups are immobile, spray with 70% ethanol and decapitate.
- Remove the brain.
- Hold the head firmly with a pair of Dumont #5 forceps, and use fine scissors to make a midline incision through the skin and skull.
- Expose the brain by reflecting the skull to the sides, and remove the brain into a dish containing cold BSS.
Separate the cerebral hemispheres from the diencephalon and the brain stem under a dissecting scope. Carefully remove all meninges by stabilizing the tissue with one pair of forceps and gently pulling away the meninges with another pair of forceps.
Collect all hemispheres in one 60 mm dish. Using small scissors, mince tissue as finely as possible. Then transfer the minced tissue to a 50 ml conical tube.
Add 1.5 ml of 1% DNase I and 1.5 ml of 2.5% trypsin and 12 ml of BSS (total volume of 15ml) to brains. Incubate in a shaking water bath for 15 min at 37 °C. To ensure good mixing, tube is swirled by hand every 5 min.
Transfer supernatant through a 70 μm cell strainer over a new 50 ml conical tube. Add 3 ml FBS to this supernatant.
To the remaining pieces, add 13.5 ml of BSS and 1.5 ml 2.5% trypsin and incubate in the shaking waterbath for another 15 min.
Transfer the remaining supernatant through the cell strainer and combine with the supernatant from Step 1.8.
Centrifuge at 1,000 RPM for 5 min to pellet the cells.
Resuspend pellets in 5 ml of glial media. Supernatant can be centrifuged again to pellet the remaining cells.
Count cells using a hemocytometer.
Plate cells at a density of 1 x 107 per 150 cm2 flask.
Change media to fresh glial media the day after plating. Afterwards, feed cells twice a week with glial media.
When cells have reached >80% confluency (approximately 10 days after plating), freeze cells down in 90% horse serum and 10% DMSO and keep stocks in liquid nitrogen. Cells are frozen at 2 x 106 per vial. Approximately 5 vials can be frozen from each flask.
About 10-14 days before setting up the hippocampal culture, glial cells are plated from the frozen stocks. We usually plate five 6-well plates, which is enough to support the growth of 90 coverslips of hippocampal neurons (three 15 mm round coverslips per well). We also plate an additional eight to ten 60 mm dishes of glial cells, which is used for conditioning N2.1 media for transfection. The day before neuronal culture, cells should be fed with NB27 media. The astrocytes should ideally be >90% confluent at this point. We find that freezing greatly reduces the number of microglia in the astroglial cultures. Since increased neurotoxicity is observed when microglia is present in the astroglial cultures, we always use astroglial culture prepared from frozen stocks for coculture with neurons.
2. Neuronal Culture
Clean glass coverslips by first rinsing in milliQ water 2x. They are then soaked in concentrated nitric acid for 24 hr, and rinsed with milliQ water for at least five times over a total of 2 hr. Coverslips are dried and sterilized by baking in an oven at 225 °C for 6 hr.
Transfer coverslips to 60 mm dishes after sterilization. Place four dots of sterile paraffin near the outer edge of each coverslip to keep neurons separate from glial cells during coculture.
Coat coverslips with 1 mg/ml poly-L-lysine in 0.1 M borate buffer overnight in 37 °C incubator.
The next day, rinse coverslips twice in sterile milliQ water for at least 15 min each. They are then incubated overnight in plating media in 37 °C incubator.
Euthanize pregnant rat (E18) by isoflurane anesthesia followed by pneumothorax, and euthanize embryonic rats by decapitation. Remove brains and dissect out the cerebral hemispheres as described above.
Dissect out the hippocampi from the cerebral hemisphere. Place all hippocampi into a 15 ml conical tube, and digest with 0.25% trypsin (0.5 ml 2.5% Trypsin, 4.5 ml BSS) at 37 ℃ for 15 min.
Digested hippocampi are rinsed in BSS 3x for 5 min each. They are then triturated with a glass Pasteur pipette, and the cell density is determined by using a hemocytometer. Neurons are then seeded at a density of 2 x 105 cells per 60 mm dish.
About 2-4 hr after plating, transfer coverslips with attached neurons to the dishes containing astroglial cells, with neurons facing down towards the glia. At DIV3, add cytosine arabinoside to the cells at a final concentration of 5 μM to stop glia proliferation.
3. Calcium Transfection
Condition N2.1 media on glia the day before transfection.
On the day of the transfection, take conditioned N2.1 media off of glia and transfer into a new dish. Transfer coverslips with neurons into the conditioned N2.1 media. Let equilibrate in incubator for 10-30 min.
To one set of sterile tubes, combine 1-4 μg of DNA, 12.5 μl of 2 M CaCl2, and sterile H2O for a total volume of 100 μl. To a second set of tubes, add 100 μl of 2x HBS.
Add ⅛ volume of 2x HBS (12.5 μl) at a time to the tube containing the CaCl2/DNA mixture, vortexing for a few seconds each time. Allow the tubes to sit for 15 min.
After 15 min incubation, add the transfection mixture dropwise to the cells. Incubate for 1-1.5 hr. A layer of sand-like precipitates should be visible under the microscope using a 10X objective.
After incubation, rinse coverslips twice with warm HBS wash buffer.
Return coverslips to original dishes with glia, add kynurenic acid to a final concentration of 0.5 mM.
Transfected neurons can be imaged live or processed for immunocytochemistry as early as the next day. Neurons can survive up to three weeks after transfection.
Representative Results
When the different parameters of transfection are optimized and carefully controlled from experiment to experiment, it is possible to obtain transfection efficiencies of up to 50%. Figure 1 shows a field of neurons that are transfected with GFP on DIV4. The field contains a total of 28 neurons, among which 16 were transfected. This represents an efficiency of over 50%. A sampling of other fields on the same coverslip shows the overall efficiency is around 50% (data not shown). With the astroglial coculture system, neurons remain healthy and develop normally after transfection. Figure 2 shows a hippocampal neuron at DIV15, 10 days after transfection with a PSD-95-GFP construct. The neuron has developed numerous mature mushroom-shaped dendritic spines, which indicates the neurons are healthy. The high transfection efficiency and minimal toxicity of this protocol makes it a valuable tool for studying gene functions in primary hippocampal neurons. Finally, this protocol can potentially be adapted to transfect cultures of other types of neurons in the brain, as well as other hard-to-transfect cell types.
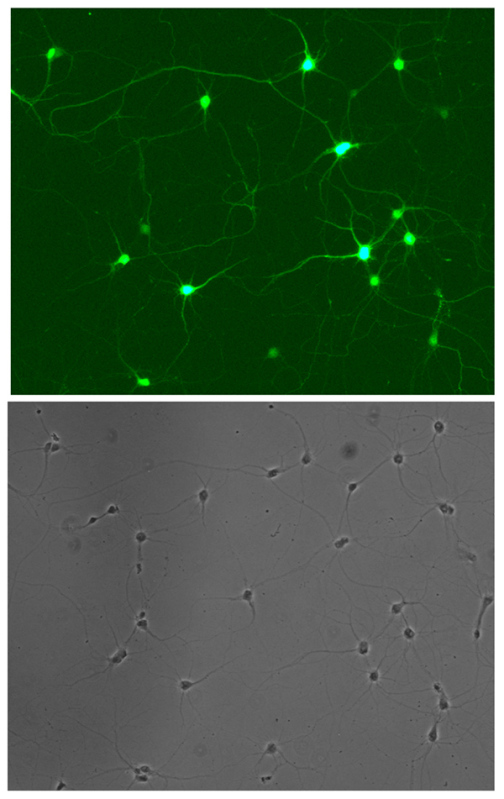 Figure 1. Hippocampal neurons transfected with GFP at DIV4, showing high transfection efficiency. 16 out of 28 neurons in the field are positive. Click here to view larger image.
Figure 1. Hippocampal neurons transfected with GFP at DIV4, showing high transfection efficiency. 16 out of 28 neurons in the field are positive. Click here to view larger image.
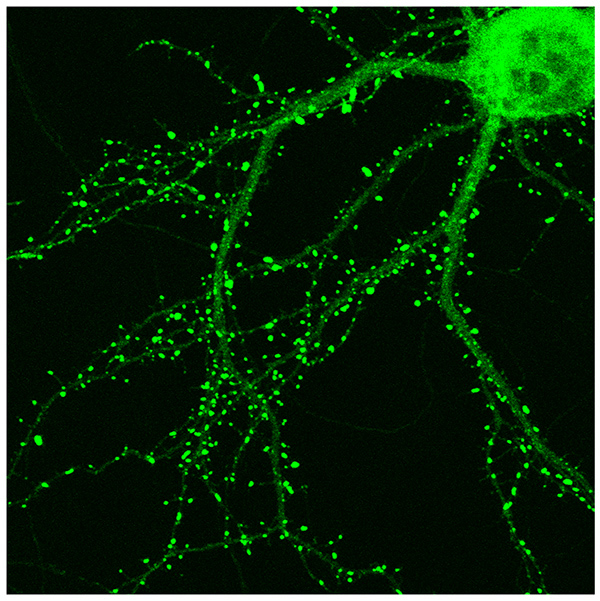 Figure 2. Hippocampal neuron transfected with PSD-95-GFP at DIV15, showing numerous mature mushroom-shaped dendritic spines.Click here to view larger image.
Figure 2. Hippocampal neuron transfected with PSD-95-GFP at DIV15, showing numerous mature mushroom-shaped dendritic spines.Click here to view larger image.
Table 1. Buffer/Media Recipes.
| Reagent | Components |
| Glial Media | 425 ml MEM 5 ml GlutaMAX 5 ml sodium pyruvate, 100 mM 15 ml 20% glucose 25 ml FBS (final 5%) 25 ml calf serum (final 5%) 5 ml Penicillin/Streptomycin |
| 0.1M Borate Buffer | 2.48 g boric acid 3.9 g sodium tetraborate (Borax) 800 ml milliQ H2O pH with NaOH to 8.5 filter sterilize |
| Plating Media | 425 ml MEM 5 ml GlutaMAX 5 ml sodium pyruvate, 100 mM 15 ml 20% glucose 50 ml FBS |
| NB27 Media | 485 ml Neurobasal Media 5 ml GlutaMAX 2 ml B27 supplement (50x) |
| Dissection Buffer (BSS) | 50 ml 10x HBSS 5 ml 1 M HEPES, pH 7.3 5 ml Penicillin/Streptomycin 440 ml H2O |
| N2.1 Media | 425 ml MEM 5 ml GlutaMAX 5 ml sodium pyruvate, 100 mM 15 ml 20% glucose 50 ml ovalbumin, 1% in MEM 5 ml N2 supplement |
| 2x HEPES Buffered Saline (HBS), sterile | 274 mM NaCl 9.5 mM KCl 15 mM glucose 42 mM HEPES 1.4 mM Na2HPO4 Prepare batches of 3 different pH (7.05, 7.10, 7.15) |
| Wash Buffer (HEPES Buffered Saline) | 50 ml 10x HBSS 5 ml 1 M HEPES, pH 7.3 445 ml H2O |
| 50 mM Kynurenic acid | Dissolve 0.473 g of kynurenic acid in 1x PBS. Add drops of NaOH to get it into solution. Then use HCl to adjust pH back to 7.0 |
Table 2. Time line for culture preparation.
| Day | Action |
| Before starting | Prepare astrocyte culture and freeze cells down. One batch of astrocyte culture can usually support several months of hippocampal cultures. |
| Week 1 Monday | Thaw astrocytes. |
| Week 1 Tuesday | Feed astrocytes. |
| Week 1 Friday | Feed astrocytes. |
| Week 2 Monday | Feed astrocytes. Rinse coverslips in H2O, then soak in nitric acid for 24 hr. |
| Week 2 Tuesday | Rinse coverslips 5x. Dry sterilize coverslips. |
| Week 2 Wednesday | Place paraffin dots on coverslips. Coat coverslips with poly-L-lysine. |
| Week 2 Thursday | Rinse coated coverslips in sterile H2O, then incubate coverslips in plating media overnight at 37 °C. Change media on astrocytes to NB27. |
| Week 2 Friday | Hippocampal dissection and plating. |
Discussion
There are several key parameters that need to be carefully controlled for consistently successful transfections10,11. The most critical parameter for calcium phosphate transfection is the pH value of 2x HBS, which in our hands usually varies between 7.10-7.15. We recommend making three batches of stocks with pH values in 0.05 increments to account for the difference between pH meters. Alternatively, the Clontech mammalian transfection kit provides 2x HBS that consistently yields good efficiency. Keep in mind that the pH of the solution will change over time, even when stored in the freezer. We generally keep aliquots of 2x HBS at 4 °C for up to one month, and at -20 °C for up to one year.
The second parameter is the pH of the culture medium. To keep this consistent, N2.1 medium are routinely conditioned on astroglial cultures overnight but not more than 24 hr. In addition, we try to use astroglial cultures that are similar in confluency. The use of glia-conditioned media also helps reduce toxicity to neurons during transfection. Conditioned media are equilibrated in the incubator before transfection to keep the pH consistent.
The size and density of the calcium phosphate precipitates is key to successful transfection. Small precipitates would lead to low transfection efficiency, while large, clumpy precipitates usually lead to cytotoxicity10. To ensure consistent calcium phosphate precipitate formation, we use intermittent vortexing when mixing the DNA/CaCl2 and 2x HBS11. Another commonly used method for mixing is through blowing air bubbles using a Pasteur pipette12. We have found the intermittent vortexing to yield more consistent precipitate formation, especially for small volumes of transfection mixture.
Other variables to consider include the quality of the DNA, and the age of the neurons. We find that the transfection start working consistently when neurons reach DIV2; however, the efficiency starts to decline when neurons are beyond DIV10. Finally, the health of the neuronal cultures themselves represents another important parameter. We find the use of the coculture system to be the best way to generate healthy primary hippocampal cultures. If the neurons are not healthy, they will not survive long after transfection. With the coculture system, low density neurons can survive for up to 3 weeks after transfection, which facilitates long term studies.
Disclosures
No conflicts of interested declared.
Acknowledgments
This work is supported by NIH grant NS065183 and start-up funds from the Rutgers Robert Wood Johnson Medical School.
References
- Karra D, Dahm R. Transfection techniques for neuronal cells. J. Neurosci. 2010;30:6171–6177. doi: 10.1523/JNEUROSCI.0183-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitelhofer M, et al. High-efficiency transfection of mammalian neurons via nucleofection. Nat. Protoc. 2007;2:1692–1704. doi: 10.1038/nprot.2007.226. [DOI] [PubMed] [Google Scholar]
- Janas J, Skowronski J, Van Aelst L. Lentiviral delivery of RNAi in hippocampal neurons. Method Enzymol. 2006;406:593–605. doi: 10.1016/S0076-6879(06)06046-0. [DOI] [PubMed] [Google Scholar]
- Graham FL, vander Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Craig AM. Transfecting cultured neurons. In: Banker GaGK., editor. Culturing Nerve Cells. 2nd edition. Cambridge, MA: MIT Press; 1998. [Google Scholar]
- Alavian KN, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Modulation of synaptic function by VAC14, a protein that regulates the phosphoinositides PI(3,5)P(2) and PI(5)P. EMBO J. 2012;31:3442–3456. doi: 10.1038/emboj.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Ghosh A, Greenberg ME. Calcium phosphate transfection of DNA into neurons in primary culture. Curr. Protoc. Neurosci. 2001;Chapter 3:Unit 3 11. doi: 10.1002/0471142301.ns0311s03. [DOI] [PubMed] [Google Scholar]
- Goslin KAH, Banker G. Rat Hippocampal Neurons in Low-Density Culture. In: Banker GaGK., editor. Culturing Nerve Cells. Cambridge, MA: MIT Press; 1998. [Google Scholar]
- Kohrmann M, et al. Fast, convenient, and effective method to transiently transfect primary hippocampal neurons. J. Neurosci. Res. 1999;58:831–835. [PubMed] [Google Scholar]
- Jiang M, Chen G. High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat. Protoc. 2006;1:695–700. doi: 10.1038/nprot.2006.86. [DOI] [PubMed] [Google Scholar]
- Goetze B, Grunewald B, Baldassa S, Kiebler M. Chemically controlled formation of a DNA/calcium phosphate coprecipitate: application for transfection of mature hippocampal neurons. J. Neurobiol. 2004;60:517–525. doi: 10.1002/neu.20073. [DOI] [PubMed] [Google Scholar]