

Abstract
Senescence is a form of cellular aging that limits the proliferative capacity of cells. Senescence can be triggered by different stress stimuli, such as DNA damage or oncogene activation. Two recent articles published in Cell have uncovered an unexpected role for cellular senescence during development, as a process that contributes to remodeling and patterning of the embryo. These findings are exciting and have important implications for the understanding of normal developmental and the evolutionary origin of senescence.
See also: Muñoz-Espín et al (Cell, November 2013) and M Storer et al (Cell, November 2013)
Nearly half a century ago, Leonard Hayflick described ‘the limited in vitro lifetime of human diploid cell strains’ (Hayflick, 1965). He observed that human cells cultured in vitro stopped proliferating after a limited number of divisions, yet cells remained alive and metabolically active, although irreversibly arrested. This phenomenon was termed cellular senescence. Since then, cellular senescence has been implicated in aging, tissue repair and cancer (Rodier & Campisi, 2011). However, until now the prevailing view was that senescence occurs only under pathological conditions, and no role in normal development had been described.
Now, two recent articles published in Cell by the Serrano and the Keyes groups drastically change this view by describing a role for cellular senescence during development (Muñoz-Espín et al, 2013; Storer et al, 2013). Using different markers for senescence, both groups observed widespread appearance of senescent cells in distinct patterns and specific stages of developing vertebrate embryos. Although most of the experiments were performed in mice, similar patterns of senescence were seen in chick (Storer et al, 2013) and human embryos (Muñoz-Espín et al, 2013), suggesting that this is a conserved feature of vertebrate embryonic development. The two studies focused on different regions of the embryo: Storer et al investigated the developing limb and the neural tube, while Muñoz-Espín et al concentrated on the mesonephros and the endolymphatic sac of the inner ear. Yet both studies come to similar conclusions. In both cases, developmentally-programmed senescence is mediated by p21, but seems to be independent of DNA damage and other typical mediators of senescence, including p53. Senescence was compromised in p21 mutant mice, but the resulting developmental defects were rather minor, presumably because of compensatory mechanisms. While both studies largely agreed there were also some differences. The Serrano group explored the mechanism underlying programmed senescence and found that TGFβ/SMAD and PI3K/FOXO were required for developmental senescence, both in the mesonephros and in the endolymphatic sac. On the other hand, the Keyes group uncovered a role of pERK-mediated signaling from the stroma for inducing senescence in the apical ectodermal ridge of the limb bud.
One particularly interesting aspect that both studies pointed out is the connection between senescence and apoptosis. Apoptosis is an active form of cell death that, very much like senescence, plays an important role for tumor suppression and is also activated in response to stress and tissue damage (Fuchs & Steller, 2011). However, a role of apoptosis as a sculpting force during development has long been recognized, and apoptotic cells are rapidly cleared by phagocytes. Interestingly, Muñoz-Espín et al now demonstrate that removal of senescent cells by macrophages is instrumental for the regression of the Wolffian duct in females and to regulate the balance between different cell populations in the endolymphatic sac. And there are other common features between senescent and apoptotic cells. Senescent cells can release various factors into the environment and this capacity, commonly known as SASP (senescence-associated secretory phenotype), can have opposing effects (Rodier & Campisi, 2011). In some cases, senescent cells induce proliferation in neighboring cells and promote tumor growth (Krtolica et al, 2001). In other cases senescent cells can induce non-autonomous senescence to achieve the opposite outcome (Acosta et al, 2013). A similar complexity in secreted signals is seen for apoptotic cells: doomed cells can produce mitogenic signals and thereby stimulate proliferation in their environment to promote wound repair and tumor growth (Perez-Garijo et al, 2004; Ryoo et al, 2004). On the other hand, apoptotic cells can also release pro-death factors to promote non-autonomous cell killing at a distance (Perez-Garijo et al, 2013). In this regard, Storer et al show that senescent cells also play important instructive roles during development, since p21 mutant mice have decreased FGF expression in the apical ectodermal ridge, which in turn leads to reduced proliferation in the underlying mesenquima. This indicates that both apoptosis and developmentally programmed senescence play important roles in the removal of unwanted structures and in patterning (Fig 1).
Figure 1.
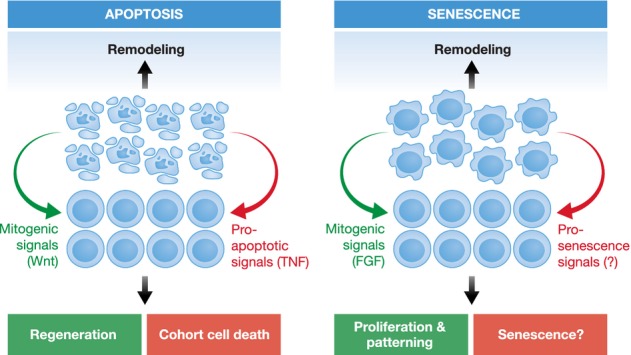
Apoptosis and senescence during development.
There are many similarities in the role of apoptosis and senescence during development. In both cases, cells are recognized and eliminated by marcrophages, and both processes are used to remodel and sculpt tissues and organs. Classic examples for apoptosis-driven sculpting include the removal of interdigital membranes in vertebrates, elimination of superfluous sex organs, and the loss of juvenile tissues during metamorphosis. Likewise, senescence has now been shown to play an important role in the regression of female Wolffian ducts. Moreover, signaling from both apoptotic and senescent cells can serve as an instructing force for patterning. For example, the production of mitogenic signals by apoptotic cells can contribute to wound healing and regeneration, while the secretion of pro-apoptotic signals like TNFα helps to orchestrate cohort cell death. In a similar way, FGF produced by senescent cells is instrumental for proliferation and patterning of the mesenchyme underlying the apical ectodermal ridge. Senescent cells are also capable of inducing non-autonomous senescence, but it remains to be determined if this signaling contributes to coordinate senescence during development.
While these observations point to obvious connections between senescence and apoptosis, the underlying mechanism remains obscure. In some cases senescence can lead to apoptosis, while in others apoptosis appears to be a compensatory mechanism that takes over when senescence is impaired. At this time, it is not clear how and when a senescent cell decides to undergo apoptosis. Likewise, we do not understand how a cell that fails to become senescent undergoes apoptosis. If both phenomena partially compensate for each other, this would help explain the relatively mild phenotypes of mice impaired in either apoptosis or senescence. Future studies with mice deficient for both senescence and apoptosis are likely to provide novel and significant insights in this regard.
Another important implication of the new work is for the very basic question of why senescence arose during evolution. Senescence is not an unavoidable consequence of old age since only some cells senesce whereas others remain ‘forever young’. Given the potential role of cellular senescence in aging, insights in this area may also relate to why organisms age. Until now, the most plausible explanation for the benefit of senescence was based on its tumor-suppressive role. The recent studies provide an additional and perhaps even more compelling reason: cellular senescence may have originated in evolution as a crucial part of normal development, and perhaps damage-induced senescence was later adopted as a mechanism to eliminate potentially dangerous cells.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Perez-Garijo A, Fuchs Y, Steller H. Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. Elife. 2013;2:e01004. doi: 10.7554/eLife.01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–5598. doi: 10.1242/dev.01432. [DOI] [PubMed] [Google Scholar]
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]