

Abstract
The selective and temporal DNA methylation plays an important role in the self-renewal and differentiation of hematopoietic stem cells (HSCs), but the molecular mechanism that controls the dynamics of DNA methylation is not understood. Here, we report that the PIAS1 epigenetic pathway plays an important role in regulating HSC self-renewal and differentiation. PIAS1 is required for maintaining the quiescence of dormant HSCs and the long-term repopulating capacity of HSC. Pias1 disruption caused the abnormal expression of lineage-associated genes. Bisulfite sequencing analysis revealed the premature promoter demethylation of Gata1, a key myeloerythroid transcription factor and a PIAS1-target gene, in Pias1−/− HSCs. As a result, Pias1 disruption caused the inappropriate induction of Gata1 in HSCs and common lymphoid progenitors (CLPs). The expression of other myeloerythroid genes was also enhanced in CLPs and lineage-negative progenitors, with a concurrent repression of B cell-specific genes. Consistently, Pias1 disruption caused enhanced myeloerythroid, but reduced B lymphoid lineage differentiation. These results identify a novel role of PIAS1 in maintaining the quiescence of dormant HSCs and in the epigenetic repression of the myeloerythroid program.
Keywords: epigenetic repression, hematopoietic stem cell, lineage differentiation, protein inhibitor of activated STAT1, self-renewal
Introduction
Mouse hematopoiesis is a well characterized system to understand the regulation of hematopoietic stem cells (HSCs). HSCs are a small population of pluripotent cells in bone marrow (BM) capable of differentiating into all blood cells of the myeloerythroid and lymphoid lineages at a single cell level (Chao et al, 2008). HSCs can be identified by a combination of surface markers. Lin−Sca1+c-Kit+ (LSK) cells are enriched in HSCs. Recent studies showed that dormant HSCs (d-HSCs) within the Lin−Sca1+c-Kit+CD150+CD48−CD34− population harbor the vast majority of multipotent long-term self-renewal activity (Wilson et al, 2008). HSCs maintain the blood system through self-renewal and multi-lineage differentiation. The differentiation program of HSCs is thought to follow the hierarchy model and is governed by the coordinated expression of lineage-affiliated transcription factors (Copley et al, 2012).
Epigenetic mechanisms play essential roles in the regulation of the self-renewal and differentiation of HSCs (Attema et al, 2007; Cedar & Bergman, 2011). DNA methylation is mediated mainly by three DNA methyltransferases (DNMTs), including the de novo methyltransferases DNMT3A and DNMT3B, and the maintenance methyltransferase DNMT1 (Jaenisch & Bird, 2003). Recent studies have shown that DNMTs are critical for regulating the self-renewal and differentiation of HSCs (Tadokoro et al, 2007; Broske et al, 2009; Trowbridge et al, 2009; Challen et al, 2011). Although DNMTs do not possess sequence-specific DNA binding properties, gene-specific and temporal epigenetic changes are associated with the proper differentiation of self-renewing HSCs. The molecular mechanism that controls the methylation dynamics during HSC differentiation has not been understood.
PIAS1 (protein inhibitor of activated STAT1) is a SUMO E3 ligase that binds to chromatin to repress transcription (Shuai & Liu, 2005). Recent studies have uncovered a novel role of PIAS1 in mediating an epigenetic mechanism to restrict natural regulatory T cell (nTreg) differentiation. PIAS1 acts by recruiting DNMTs and histone modification factors such as HP1γ to promote epigenetic silencing of the Foxp3 promoter, a transcription factor crucial for nTreg differentiation (Liu et al, 2010). These findings raise an interesting question on whether the PIAS1 epigenetic gene silencing pathway plays a role in stem cell biology. Here, we demonstrated that PIAS1 is important for maintaining the quiescence of dormant HSCs, and Pias1−/− LSK cells showed profound defects in long-term competitive reconstitution assays. Furthermore, PIAS1 is essential for preventing the premature and inappropriate activation of the myeloerythroid transcription factor Gata1 through epigenetic repression. These studies identified PIAS1 as a novel epigenetic regulator of HSC self-renewal and differentiation.
Results
Altered HSCs and lineage-restricted progenitors in Pias1−/− mice
Previous studies showed that PIAS1 restricts the differentiation of CD4+Foxp3+ natural regulatory T cells (nTreg) (Liu et al, 2010). To address whether PIAS1 affects the differentiation of other lymphoid/myeloid lineages, flow cytometry analyses were performed with splenocytes and peripheral blood lymphocytes (PBL) from wild-type (WT) and Pias1−/− mice using lineage-specific markers (Fig S1). Minor increases in the percentages of myeloid cells were observed in periphery, with no differences in the cellularity of B cells, T cells, dendritic cells or erythroids. These data indicate that PIAS1 does not dramatically affect peripheral lineage differentiation in homeostasis.
Similar flow cytometry analyses were performed with bone marrow (BM) cells, where a reduction in the cell numbers as well as the percentage of B220+ B cells was observed in Pias1−/− BM (Fig 1A and Fig S2A). When the lineage-negative (Lin−) population was examined, a minor increase in the percentage of Lin− cells was observed in Pias1−/− BM, although the cell number of Lin− cells was not altered (Fig 1B and Fig S2B). Interestingly, while no significant differences were observed in common myeloid progenitor (CMP) and granulocyte monocyte progenitor (GMP) populations, a 50% decrease in common lymphoid progenitor (CLP) population was observed in Pias1−/− BM with a concurrent increase in megakaryocyte erythrocyte progenitor (MEP) as well as the myeloid-restricted Lin−Sca1−c-Kit+ (L−S−K+) cells (Fig 1C and Fig S2C). In addition, reduced Pre-B populations were observed in Pias1−/− BM (Fig 1C and Fig S2C).
Figure 1.

- Cell numbers of T cells (CD4+ or CD8+), B cells (B220+), granulocytes/monocytes (Gr1+Mac1+), dendritic cells (CD11c+) and erythroids (Ter119+) in freshly isolated bone marrow (BM) from 8 to 12 weeks old WT (Pias1+/+) and Pias1−/− littermates were assayed by flow cytometry.
- Same as in (A) except that cell numbers of Lin− cells were assayed.
- Same as in (A) except that cell numbers of myeloid-restricted Lin−Sca1−c-Kit+ (L−S−K+) populations, common myeloid progenitors (CMP), granulocyte monocyte progenitors (GMP), megakaryocyte erythrocyte progenitors (MEP), common lymphoid progenitors (CLP), Pro-B and Pre-B cells as defined in supplementary Materials and Methods were assayed.
- Same as in (A) except that cell numbers of LSK, long-term hematopoietic stem cells (LT-HSC) and short-term multi-potent progenitors (ST/MPP) as defined in supplementary Materials and Methods in total BM were assayed.
- Same as in (A) except that cell numbers of dormant hematopoietic stem cells (d-HSCs; Lin−Sca1+c-Kit+CD150+CD48−CD34−) were assayed.
Data information: Shown in each panel is a pool of 3 independent experiments (n = 9–13). Error bars represent SEM. P-values were determined by non-paired t-test. See also Fig S1 and S2.
Next, the effect of Pias1 disruption on HSCs was examined. An approximately 2-fold increase in HSC-enriched LSK cells was observed in Pias1−/− BM (Fig 1D and Fig S2D). LSK can be further divided into long-term hematopoietic stem cells (LT-HSC; Lin−Sca1+c-Kit+CD34−) and short-term multi-potent progenitors (ST/MPP; Lin−Sca1+c-Kit+CD34+). Similar increases were observed in LT-HSC, but not ST/MPP, population (Fig 1D and Fig S2D). Dormant mouse HSCs (d-HSCs) within the Lin−Sca1+c-Kit+CD150+CD48−CD34− population harbor the vast majority of multi-potent long-term self-renewal activity (Wilson et al, 2008). No significant difference was observed in the percentage or the cell number of d-HSC cells between WT and Pias1−/− BM (Fig 1E and Fig S2E).
Increased cell death of Pias1−/− lymphoid progenitors and enhanced cell proliferation of Pias1−/− dormant HSCs
To determine whether the changes in Pias1−/− progenitor populations were due to cell proliferation and/or cell death, flow cytometry analyses were performed with freshly isolated BM from WT and Pias1−/− mice using 7-AAD to reveal cell death, or Ki67 and Hoechst DNA staining to reveal cell cycle profiles. Increased cell death was observed in Pias1−/− CLP as well as Pro-B and Pre-B populations (Fig 2A), while minor difference was observed in their cell cycle properties (Fig S3). These data are consistent with the reduced B cells observed in Pias1−/− BM, and indicate that PIAS1 is important for the survival of CLP and B cell progenitors under homeostatic condition.
Figure 2.

- Cell death of indicated BM subsets from Pias1−/− mice and their WT littermates (Pias1+/+) were assayed by 7-AAD staining followed by flow cytometry.
- Cell proliferation of indicated BM subsets from WT and Pias1−/− mice was revealed by intracellular Ki67 (icKi67) and Hoechst DNA staining followed by flow cytometry. G0, icKi67−, 2N DNA content; G1, icKi67+, 2N DNA content; S+G2/M, icKi67+, > 2N DNA content.
- Same as in (B) except that d-HSCs were assayed (left). A representative cell cycle profile of WT and Pias1−/− d-HSCs was also shown (right). Numbers in top right are the percentage of cells in each cell cycle.
Data information: Shown in each panel is a pool of 3 independent experiments (n = 10–13). Error bars represent SEM. P-values were determined by non-paired t-test. See also Fig S3.
Interestingly, although no difference was observed in the cell cycle profiles of Lin−, myeloid-restricted L−S−K+, CMP, GMP, MEP and ST/MPP subpopulations, the percentage of cells in G0 phase was significantly reduced in Pias1−/− LSK cell, with a concurrent increase of the cells in G1 phase of the cell cycle (Fig 2B and Fig S3). More importantly, similar alteration in cell cycle profiles was observed in Pias1−/− LT-HSC and d-HSC, but not ST/MPP, populations as compared to that of WT controls (Fig 2B and C). These results suggest that PIAS1 is important for maintaining the quiescence of d-HSCs.
Defective long-term reconstitution capability of Pias1−/− HSCs
To directly test the functional role of PIAS1 in the regulation of HSC in vivo, competitive reconstitution assays were performed using WT or Pias1−/− BM cells (CD45.2+) as donors and the congenic WT C57SJL BM (CD45.1+) as competitors. Pias1−/− cells showed dramatic defects in reconstituting hematopoietic system of WT C57SJL recipient mice in multiple lineages, including B cells (B220+), T cells (CD3+) and myeloid cells (Mac1+) (Fig 3A and B). The defects were significant at 5 weeks, and became more profound at later time points, with Pias1−/− donor cells exhibiting over 20-fold reduction in the ability to reconstitute blood system as compared to WT cells, indicating that PIAS1 affects the functions of progenitor cells as well as long-term HSCs. Similar results were obtained with in vivo competitive reconstitution assays using FACS-sorted WT or Pias1−/− LSK cells (Fig 3C and D), confirming that the reconstitution defects of Pias1−/− BM cells lie within the LSK population. These data indicate that PIAS1 is crucial for the in vivo reconstitution activities of HSCs and their progeny.
Figure 3.
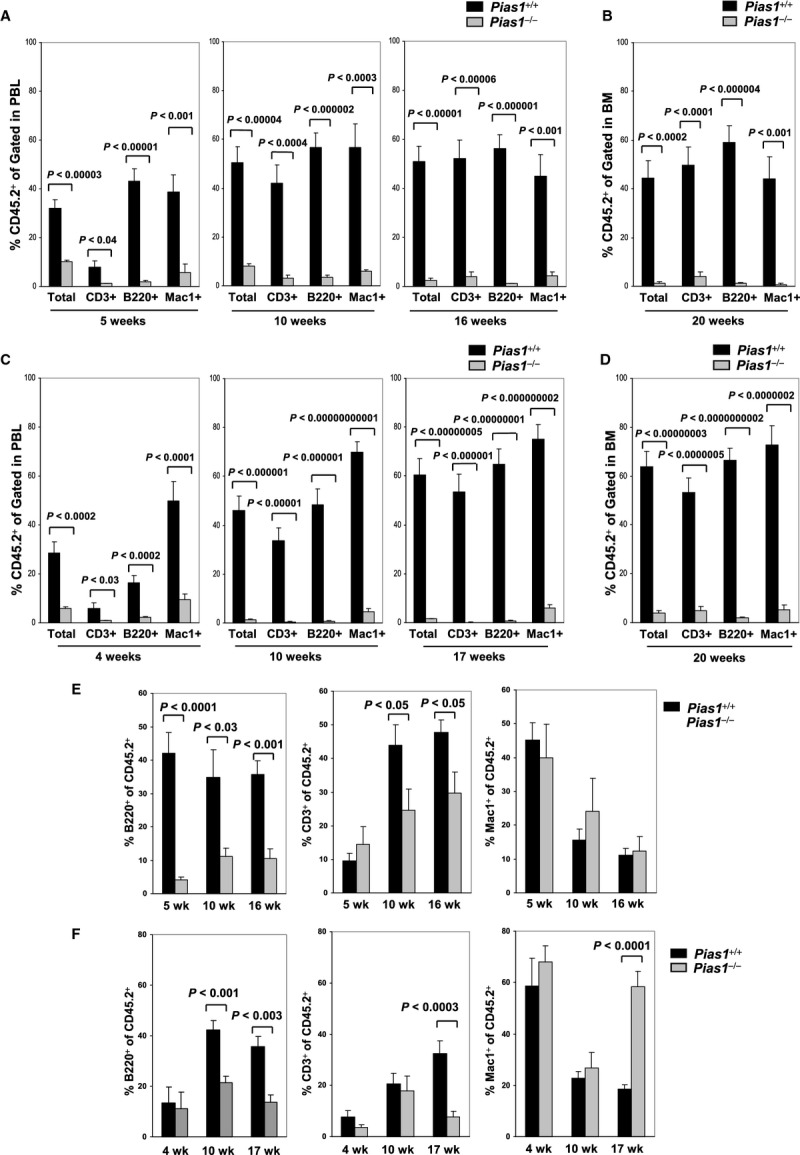
- In vivo competitive reconstitution assays. Total bone marrow cells (2 × 105) from WT or Pias1−/− littermates (CD45.2+) were mixed with 2 × 105 of WT C57SJL bone marrow cells (CD45.1+) and injected into lethally irradiated WT C57SJL mice. The percentage of T cells (CD3+), B cells (B220+) and myeloid cells (Mac1+) from donor mice in peripheral blood (PBL) were assayed by flow cytometry at 5, 10 and 16 weeks post reconstitution.
- Same as in (A) except that bone marrow (BM) cells from the recipient mice were assayed at 20 weeks post reconstitution.
- Same as in (A) except that FACS-sorted LSK cells (1000) from WT or Pias1−/− littermates were used.
- Same as in (B) except that FACS-sorted LSK cells (1000) from WT or Pias1−/− littermates were used.
- Same as in (A) except that the percentage of T cells (CD3+), B cells (B220+) and myeloid cells (Mac1+) within the donor cells (CD45.2+) in peripheral blood (PBL) were presented.
- Same as in (E) except that FACS-sorted LSK cells (1000) from WT or Pias1−/− littermates were used.
Data information: Shown is a pool of 3 independent experiments in all panels (n = 10). Error bars represent SEM. P-values were determined by non-paired t-test. See also Fig S4.
To further quantify the effect of PIAS1 on the long-term repopulation capability of HSCs, competitive limiting dilution assays were performed to determine the frequencies of functional HSCs in WT and Pias1−/− BM. No significant difference was observed in the percentage of donor-derived CD45.2+ populations in the peripheral blood 21 weeks post the primary reconstitution (WT: 57.8 ± 11%; Pias1−/−: 42.9 ± 11.5%). In contrast, by 10 weeks post secondary reconstitution, recipients of Pias1−/− BM revealed a 5-fold decrease in the competitive repopulating unit (CRU) as compared to that of WT controls (Table 1). Similar defects were observed 16 weeks post the secondary transplantation, indicating that PIAS1 is crucial for maintaining the long-term repopulation capacity of HSCs. These data strongly support for an important role of PIAS1 in the regulation of the self-renewal of HSCs in vivo.
Table 1.
Evaluation by limiting dilution analysis of competitive long-term repopulating cells (CRU) in mice transplanted with WT (Pias1+/+) or Pias1-null (Pias1−/−) bone marrow cells
| Number of cells injected into secondary recipients | CRU evaluation of primary recipientsa |
|||
|---|---|---|---|---|
| 10 weeks post transplantation |
16 weeks post transplantation |
|||
| Pias1+/+ | Pias1−/− | Pias1+/+ | Pias1−/− | |
| 2,000,000 | 3/3 (24.5 ± 9.7) | 4/4 (4.7 ± 2.7) | 3/3 (32.5 ± 11.2) | 3/4 (3.9 ± 2.5) |
| 1,000,000 | 2/2 (29.5 ± 0.1) | 1/2 (1.2 ± 1.1) | 2/2 (29.4 ± 21.2) | 1/2 (1.0 ± 0.8) |
| 300,000 | 3/3 (8.9 ± 5.3) | 0/3 (0.5 ± 0.1) | 3/3 (16.5 ± 9.9) | 0/3 (0.2 ± 0.1) |
| 100,000 | 1/5 (0.5 ± 0.7) | 0/5 (0.3 ± 0.2) | 0/5 (0.2 ± 0.3) | 0/5 (0.3 ± 0.2) |
| CRU frequency per 105 cells (range)b | 0.52 (0.20–1.34) | 0.09 (0.04–0.21) | 0.37 (0.14–0.97) | 0.06 (0.02–0.15) |
Results are expressed as number of mice repopulated with donor-derived cells (CD45.2+; >1%) over total. Numbers in parentheses represent the mean % ± s.d. of peripheral blood CD45.2+ cells found in the transplanted recipients.
CRU frequency was calculated using Extreme Limiting Dilution Analysis (ELDA) software available at http://bioinf.wehi.edu.au/software/elda/.
The effect of PIAS1 on multi-lineage differentiation
To access the role of PIAS1 in lineage differentiation in vivo, the distribution of B cells, T cells and myeloid cells within the CD45.2+ donor population at various time points was analyzed in competitive reconstitution assays. Pias1−/− BM cells showed defective B lymphocytes differentiation at both early and late time points, indicating that PIAS1 affects B cell progenitors as well as stem cells (Fig 3E). Pias1−/− BM also showed defective T cell differentiation at 10 and 16 weeks, while myeloid cells were not affected (Fig 3E). These data are consistent with the previous results that PIAS1 is important for the survival of CLP, while restricting the myeloid progenitor population. When reconstitution assays were performed for WT or Pias1−/− LSK cells, no difference was observed at 4 or 10 weeks except for the reduced B cells from Pias1−/− LSK donors at 10 weeks, further suggesting that PIAS1 is important for B cell differentiation at multiple stages (Fig 3F). In contrast, both B and T cell lineages were defective, while myeloid cell differentiation was increased from Pias1−/− LSK donors at 17 weeks. These data indicate that PIAS1 is important for balancing the long-term multi-lineage differentiation of HSC between CLP and CMP.
Increased myeloid-restricted Lin−Sca1−c-Kit+ (L−S−K+) population was observed in Pias1−/− BM (Fig 1C and Fig S2C). To address whether PIAS1 affects the differentiation of L−S−K+ cells into their progeny in vivo, short-term competitive reconstitution assays were performed with FACS-sorted L−S−K+ cells from WT or Pias1−/− littermates (Fig S4). Surprisingly, Pias1−/− L−S−K+ cells showed defects in differentiating into Mac1+ cells as compared to WT controls, indicating that PIAS1 is important for the proper differentiation of L−S−K+ cells into their progeny.
No defects in Pias1−/− BM homing and niche retention
The observed defects in the long-term reconstitution capacity of Pias1−/− HSC can be explained by several mechanisms, including the intrinsic properties of Pias1−/− cells such as self-renewal, as well as Pias1−/− cells homing to BM and niche retention. To examine whether Pias1−/− BM cells are defective in homing to BM, CFDA-SE (CFSE)-labeled WT or Pias1−/− BM cells were injected into lethally irradiated WT C57SJL recipient mice, and cells migrated to BM were determined by measuring CFSE+ cells in BM. No difference was observed in the ability of Pias1−/− cells to home to BM compared to that of WT controls (Fig 4A and B). Similar results were obtained with LT-HSC cells, suggesting that Pias1−/− LT-HSC cells are not defective in homing to BM (Fig 4C).
Figure 4.
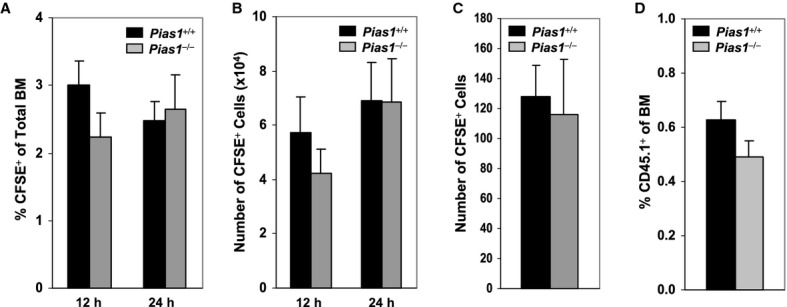
- Homing of bone marrow (BM) cells. CFDA-SE (CFSE)-labeled total BM cells (2 × 107) from WT and Pias1−/− littermates were injected into lethally irradiated WT C57SJL mice. The percentage of CFSE+ cells in BM of the recipient mice was determined by flow cytometry 12 and 24 h post injection.
- Same as in (A) except that the number of CFSE+ cells was presented.
- Same as in (A) except that CFSE-labeled long-term hematopoietic stem cells (LT-HSC; Lin−Sca1+c-Kit+CD34−) cells (2000 cells/mouse) from WT and Pias1−/− littermates were used, and the number of CFSE+ cells in BM of the recipient mice was determined 24 h post injection.
- Niche retention assays. Total BM cells (4 × 107) from WT C57SJL mice (CD45.1+; n = 12) were injected into non-irradiated WT or Pias1−/− recipient mice (CD45.2+). The percentage of CD45.1+ cells in BM of the recipient mice were assayed by flow cytometry 12 weeks post injection.
Data information: Shown in each panel is a pool of 2 independent experiments (n = 9–10). Error bars represent SEM.
HSCs are thought to reside in niches, which are cellular and molecular microenvironments that regulate stem cell quiescence, self-renewal and differentiation (Jones & Wagers, 2008; Kiel & Morrison, 2008). The ability of Pias1−/− BM cells to be retained in the niche was examined by nonablative transplant assays, where BM from WT C57SJL mice (CD45.1+) were injected into non-irradiated WT or Pias1−/− mice (Fig 4D). No difference was observed in the engraftment of WT C57SJL cells into WT or Pias1−/− mice, indicating that the HSC niche in both WT and Pias1−/− mice are stably occupied by host cells. Taken together, these results indicate that the observed defects in the long-term reconstitution capacity of Pias1−/− HSC are not due to defects in BM homing or niche retention, implying that PIAS1 is crucial for the intrinsic properties of HSCs.
No defects in the BM microenvironment of Pias1−/− mice
It is well established that BM microenvironment is important for HSC function and maintenance (Jones & Wagers, 2008; Kiel & Morrison, 2008; Ehninger & Trumpp, 2011). To test whether the impaired function of Pias1−/− HSC is due to altered BM microenvironments in Pias1−/− mice, in vivo reconstitution assays were performed by transplanting WT C57SJL BM cells into lethally irradiated WT or Pias1−/− recipient mice. Interestingly, enhanced engraftment of WT C57SJL cells into Pias1−/− mice was observed at 4 weeks and 18 weeks, while the difference was diminished at 20 weeks (Fig 5A). In addition, no differences in the distribution of mature T cells, B cells and myeloid cells within WT donor cells were observed at all time points examined (Fig 5B). These results indicate that the BM microenvironment in Pias1−/− mice is not responsible for the impaired Pias1−/− HSC functions, further suggesting that PIAS1 regulates the intrinsic properties of HSCs.
Figure 5.
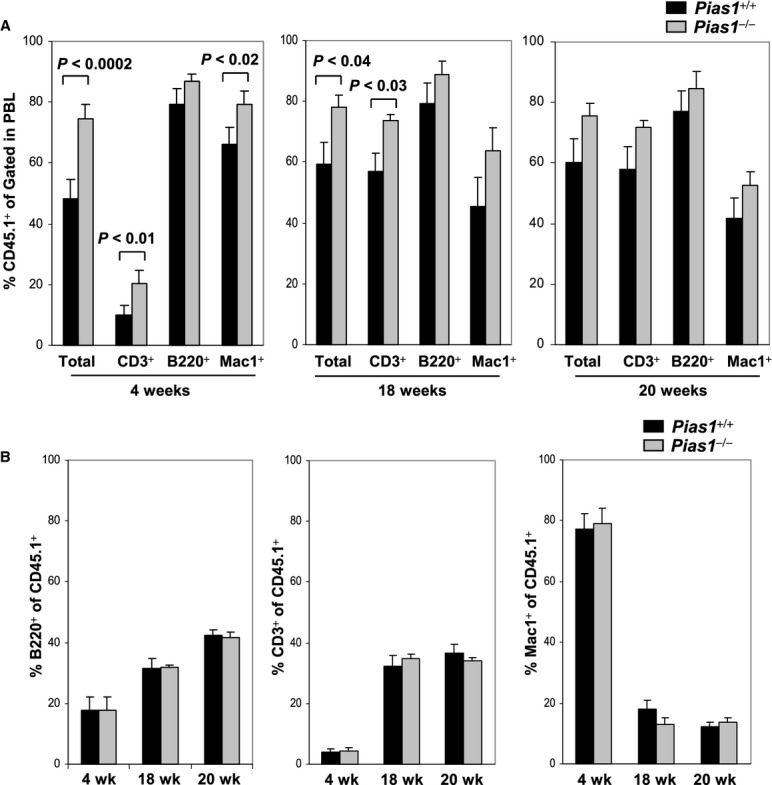
- In vivo reconstitution assays. Total BM cells (4 × 105) from WT C57SJL mice (CD45.1+) were injected into lethally irradiated WT or Pias1−/− recipient mice (CD45.2+). The percentage of T cells (CD3+), B cells (B220+) and myeloid cells (Mac1+) from donor mice in peripheral blood (PBL) were assayed by flow cytometry at 4, 18 and 20 weeks post reconstitution.
- Same as in (A) except that the percentage of T cells (CD3+), B cells (B220+) and myeloid cells (Mac1+) within the donor cells (CD45.1+) in PBL were assayed.
Data information: In (A) and (B) is shown a pool of 2 independent experiments (n = 9–10). Error bars represent SEM. P-values were determined by non-paired t-test.
Abnormal expression of lineage-specific genes in Pias1−/− cells
To test if Pias1 disruption affects the transcription of lineage-specific genes, Q-PCR assays were performed with Lin− progenitors from WT and Pias1−/− BM. While no change or slight increase was observed in the expression of myeloerythroid-specific genes, including Gata1 (GATA-binding factor 1), Gata2 (GATA-binding factor 2), Csf1r (Macrophage colony-stimulating factor 1 receptor), Mpo (Myeloperoxidase) and Cebpa (CCAAT/enhancer-binding protein alpha) (Akashi et al, 2000), transcription of genes essential for B cell differentiation, including Il7r (Interleukin-7 receptor subunit alpha), Ebf1 (Early B-cell factor 1), Pax5 (Paired box protein Pax-5) and Igll1 (Immunoglobulin lambda-like polypeptide 1) was significantly reduced (Fig 6A). In contrast, transcription of other lymphoid-associated genes, such as Ikzf1 (Ikaros family zinc finger protein 1) and T cell-specific factor Gata3 (GATA-binding factor 3), was not altered. These data are consistent with the defective B lymphoid differentiation phenotype observed in Pias1−/− BM cells, further supporting the notion that PIAS1 is important for maintaining the expression of B cell-specific genes while restricting the expression of myeloerythroid genes.
Figure 6.

- Quantitative real time polymerase chain reaction (Q-PCR) analyses were performed with total RNA from WT or Pias1−/− Lin− population. The expression of each gene in WT cells was relatively set as 1, and the results were adjusted by Hprt1.
- Same as in (A) except that common lymphoid progenitors (CLP) cells were used.
- Same as in (A) except that long-term hematopoietic stem cells (LT-HSC; Lin−Sca1+c-Kit+CD34−) cells were used.
- Same as in (A) except that CLP cells were used, and the expression of GATA1-downstream genes was quantified.
Data information: Shown in each panel is the average of 3–5 independent experiments. Error bars represent SEM. P-values were determined by non-paired t-test. See also Fig S5.
GATA1, a key transcription factor for erythropoiesis, can directly repress the transcription of Ebf1 (Iwasaki et al, 2003; Xie et al, 2004). To further investigate the role of PIAS1 in the transcriptional regulation of lineage-associated genes, Q-PCR analyses were performed with various FACS-sorted BM subpopulations from WT and Pias1−/− mice (Fig S5). Interestingly, transcription of myeloerythroid-associated genes, including Gata1, Gata2, Csf1r, Mpo and Cebpa, was dramatically increased in CLP cells, with a concurrent decrease in genes important for B cell differentiation, such as Il7r, Ebf1, Pax5 and Igll1 (Fig 6B). When HSC-enriched LT-HSC cells were examined, increased transcription of Gata1, Csf1r and Csf1r and decreases in B cell differentiation-related genes, including Il7r, Ebf1, Pax5 and Igll1, were observed (Fig 6C). In addition, transcription of these genes was not affected in CMP (Fig S5). These data indicate that PIAS1 is an important transcriptional regulator for the proper expression of lineage-affiliated genes in LT-HSC and CLP.
To test whether PIAS1 affects the transcription of GATA1-downstream genes, Q-PCR analyses were performed with FACS-sorted CLP cells from WT and Pias1−/− BM to assess the transcription of several GATA1-regulated genes, including Epor (Erythropoietin receptor), Hbb-b1 (Hemoglobin subunit beta-1) and Slc4a1 (Solute carrier family 4 member 1; an erythroid specific factor) (Fig 6D). The transcription of all 3 genes were increased in Pias1−/− CLP cells as compared to that of WT controls, suggesting that enhanced Gata1 transcription in Pias1−/− cells leads to the functional increase in GATA1-mediated gene activation.
PIAS1 suppresses Gata1 through direct epigenetic silencing
To test whether Gata1 is a direct PIAS1-target gene, chromatin immunoprecipitation (ChIP) assays were performed with WT and Pias1−/− BM cells using 2 independent PIAS1 antibodies (Fig 7A). Binding of PIAS1 to the promoter region of Gata1 was observed in WT, but not Pias1−/− BM, indicating that Gata1 is a direct target of PIAS1. ChIP assays were also performed with FACS-sorted LSK or myeloerythroid-restricted L−S−K+ cells (Fig 7B). PIAS1 also binds to the promoter region of Gata1 in these cells.
Figure 7.
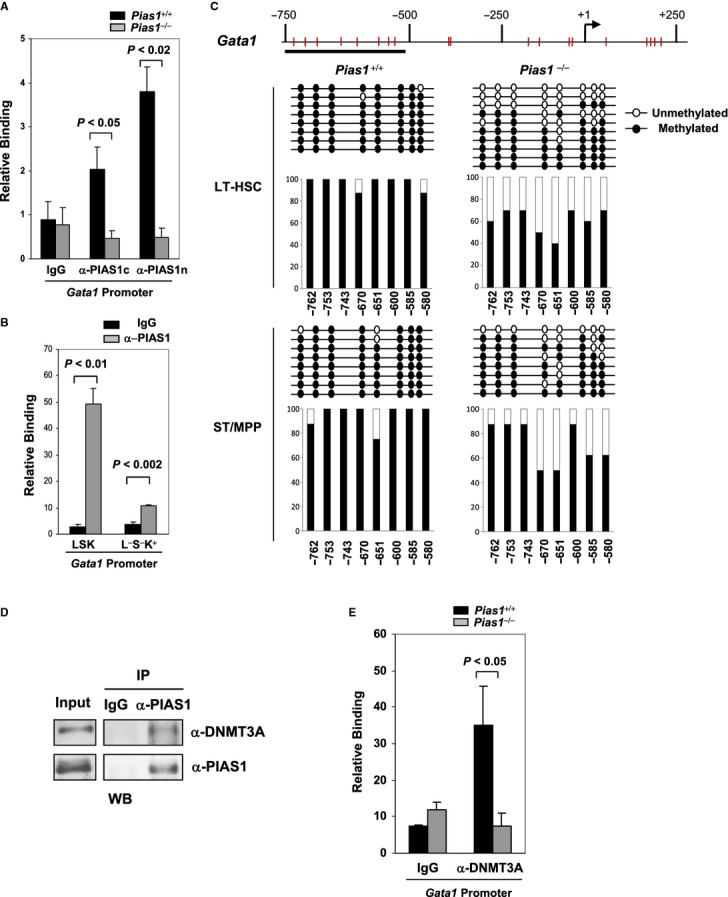
- Chromatin immunoprecipitation (ChIP) assays were performed with cell extracts from WT or Pias1−/− BM, using 2 independent antibodies against either the C-terminal (α-PIAS1c) or the N-terminal (α-PIAS1n) PIAS1, or IgG as a negative control. Bound DNA was quantified by Q-PCR with specific primers against Gata1 promoter, and normalized with the input DNA.
- Same as in (A) except that FACS-sorted LSK or Lin−Sca1−c-Kit+ (L−S−K+) cells from WT BM were used.
- Methylation analysis of the Gata1 promoter was performed by bisulfite conversion of genomic DNA from FACS sorted long-term hematopoietic stem cells (LT-HSC) and short-term multi-potent progenitors (ST/MPP) as defined in Materials and Methods from WT and Pias1−/− male mice (n = 5). The x axis represents the positions of the CpG sites relative to the transcription start site (+1); the y axis represents the percentage.
- PIAS1 interacts with DNMT3A in BM cells. Co-immunoprecipitation (Co-IP) assays were performed with cell extracts from WT BM, using anti-PIAS1 or IgG, followed by immunoblotting with anti-DNMT3A or a monoclonal anti-PIAS1.
- PIAS1 is required for the recruitment of DNMT3A to the Gata1 promoter. Same as in (A) except that anti-DNMT3A was used for ChIP assays.
Data information: Shown in each panel is a representative of 3 independent experiments (n = 4–6 for each experiment). Error bars represent SEM. P-values were determined by non-paired t-test.
PIAS1 has been shown to regulate the promoter methylation of the Foxp3 gene (Liu et al, 2010). To understand the mechanism of PIAS1-mediated transcriptional repression of Gata1, the methylation status of the Gata1 promoter was analyzed by bisulfite-sequencing of WT and Pias1−/− BM subpopulations. Several CpG sites in the Gata1 promoter were hypermethylated in WT LT-HSC and ST/MPP cells (Fig 7C). Pias1 disruption caused a significant reduction of DNA methylation in the Gata1 promoter, consistent with the enhanced transcription of Gata1 observed in Pias1−/− LT-HSC cells (Fig 6C).
It has been demonstrated that PIAS1 is important for the recruitment of DNMT3A/3B to the Foxp3 promoter (Liu et al, 2010). To test whether PIAS1 interacts with DNMT3A in BM cells in vivo, co-immunoprecipitation (Co-IP) assays were performed with cell extracts from BM using anti-PIAS1 antibody, followed by immunoblotting with anti-DNMT3A. PIAS1 can interact with DNMT3A in BM cells (Fig 7D). Furthermore, ChIP assays revealed that while DNMT3A binds to the Gata1 promoter in WT BM cells, the binding of DNMT3A to the Gata1 promoter was abolished in Pias1−/− BM (Fig 7E). These results indicate that PIAS1 is required for the recruitment of DNMT3A to the Gata1 promoter in BM, and further suggest that PIAS1 represses Gata1 transcription by maintaining DNA methylation of the Gata1 promoter in HSCs.
Discussion
PIAS1 is a SUMO E3 ligase involved in the regulation of multiple transcriptional programs (Shuai & Liu, 2005; Liu et al, 2007; Liu & Shuai, 2008). Recent studies have uncovered an important role of PIAS1 in mediating a novel epigenetic mechanism to restrict the expression of Foxp3 in natural regulatory T cells (Liu et al, 2010). In this manuscript, we explored the role of the newly identified PIAS1 epigenetic pathway in the regulation of HSCs. Our results have identified an essential role of PIAS1 in regulating the self-renewal and differentiation of HSCs (Fig 8).
Figure 8.
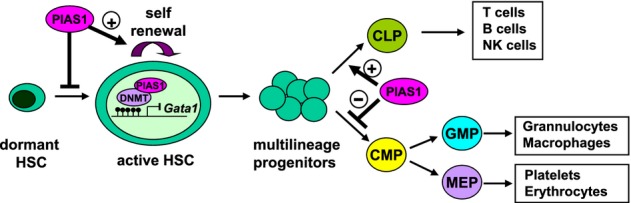
A proposed model of the essential function of PIAS1 in the regulation of self-renewal and lineage differentiation of HSCs.
PIAS1 restricts dormant cells to enter the active cell cycle, while supports the self-renewal of active HSC. PIAS1 also supports the proper differentiation of lymphoid cells, while restricting the myeloerythroid lineage.
We demonstrated that Pias1−/− BM cells as well as HSC-enriched LSK population failed to reconstitute blood system in the presence of WT competitors. No defects were observed in Pias1−/− BM microenvironment, niche retention, or homing, suggesting an intrinsic role of PIAS1 in regulating the self-renewal of HSCs. It has been documented that the Lin−Sca1+c-Kit+CD150+CD48−CD34− population is enriched in dormant HSCs (d-HSCs), and the ability of d-HSCs to remain quiescent is crucial for maintaining the capacity for lifelong replenishment of all blood cells. Pias1−/− d-HSCs showed reduced G0 population with an increase in G1 phase (non-quiescent cells). These results suggest an important role of PIAS1 in maintaining the quiescence of d-HSCs. It is likely that the increased cell proliferation of Pias1−/− d-HSCs resulted in their exhaustion and reduced long-term reconstitution capacity. The effect of Pias1 disruption on cell proliferation was only observed in HSC-enriched populations, including d-HSCs, LT-HSCs and LSK cells, but not differentiated BM progenitor subsets, such as CMP, GMP, MEP, CLP and myeloid-restricted Lin−Sca1−c-Kit+. The precise molecular mechanism responsible for PIAS1-mediated regulation on the quiescence of d-HSCs is not known. It will be very interesting to test whether the PIAS1-mediated epigenetic control mechanism is involved in this process, although this is technically challenging due to the rareness of dormant HSCs.
It has been documented that DNA methylation plays an important role in the regulation of HSC self-renewal and differentiation (Tadokoro et al, 2007; Broske et al, 2009; Trowbridge et al, 2009; Challen et al, 2011), but how DNMTs are regulated to control methylation dynamics in hematopoiesis is not known. PIAS1 interacts with DNMTs in vivo (Liu et al, 2010) (Fig 7D). Our recent studies in regulatory T cells suggest that PIAS1 can recruit DNMTs to specific gene promoters to promote DNA methylation (Liu et al, 2010), suggesting that PIAS1 may function as a cofactor of DNMTs. In this report, we showed that Gata1 is a direct target of PIAS1, and Pias1 disruption resulted in the premature demethylation of the Gata1 promoter in HSCs. Consistently, an inappropriate induction of Gata1 in HSCs and CLPs was observed. Gata1 is a key myeloerythroid transcription factor and its elevated expression can suppress the induction of crucial lymphoid genes such as Ebf1 (Iwasaki et al, 2003). Indeed, the expression of a number of B lymphoid genes was significantly repressed in Pias1-null Lin− progenitor cells (Fig 6A). As a result, defective B cell differentiation was observed in the absence of PIAS1 (Fig 1A). These results suggest that the PIAS1 epigenetic pathway plays an important role in preventing the inappropriate expression of the myeloerythroid gene program, which is essential for the balanced myeloerythroid vs lymphoid lineage differentiation.
Studies from several groups have shown that DNMTs play important roles in regulating the self-renewal and differentiation of HSCs (Tadokoro et al, 2007; Broske et al, 2009; Trowbridge et al, 2009; Challen et al, 2011). The conditional knockout of Dnmt3a impaired HSC differentiation, and Dnmt3a-null HSCs upregulated HSC multipotency genes and downregulated differentiation factors (Challen et al, 2011). The phenotype of Pias1−/− mice in regulating HSC lineage differentiation resembles that of Dnmt1 knockout mice (Broske et al, 2009). Both Pias1 disruption and DNMT1 reduction caused the premature demethylation of Gata1 promoter in HSCs, the derepression of myeloerythroid genes in Lin− progenitor cells, the reduction of CLPs, and the impaired B cell differentiation (Broske et al, 2009). These findings provide genetic evidence to support our hypothesis that PIAS1 is a key cofactor of DNMTs in promoting DNA methylation. Our results have uncovered a novel functional role of the PIAS1 epigenetic pathway in regulating the methylation dynamics in the HSC differentiation program.
Materials and Methods
Flow cytometry analysis and sorting of HSC and progenitors
Single cell suspensions were prepared from spleen, bone marrow (BM) or peripheral blood (PBL). Cells were stained with various combinations of surface markers followed by flow cytometry analysis using a FACSCalibur or FACScanX (Becton Dickinson, BD, San Jose, CA, USA). Data were analyzed using the FCS Express software (De Novo). FACS cell sorting was performed as described (Zeng et al, 2004; Wilson et al, 2008; Broske et al, 2009; Trowbridge et al, 2009; Ji et al, 2010; Mayle et al, 2012). Briefly, single cell suspensions were prepared from BM and subjected to surface staining with various markers. FACS sorting of HSC and progenitors was performed using a FACSAria (Becton Dickinson, BD). See supplementary Materials and Methods for definitions of each population.
Cell proliferation and cell death
Cell death was assessed by 7-AAD staining as described (Liu et al, 2004, 2010). Cell proliferation was assayed by intracellular anti-Ki67 and Hoechst DNA staining as described (Wilson et al, 2008). Briefly, single cell suspensions of BM were first stained with various surface markers, followed by fixation/permeabilization of the cells. Cells were then stained with anti-Ki67 and 1 μg/ml bisbenzimide (Hoechst no. 33342; Sigma, St. Louis, MO, USA), and analyzed on a LSR flow cytometer (Becton Dickinson, BD). Approximately 1 × 106 cells were collected for each sample when the cell cycle profiles of dormant HSCs (d-HSCs) were analyzed to ensure that sufficient numbers of d-HSCs were collected.
In vivo competitive reconstitution assays
In vivo competitive reconstitution experiments were performed as described (Liu et al, 2008). Briefly, bone marrow was isolated from 4 to 8 week-old Pias1−/− mice and their WT littermates (CD45.2+), or C57SJL wild-type mice (CD45.1+). Total bone marrow (2 × 105 cells) or FACS-sorted LSK cells (1000 cells) from Pias1−/− or WT mice was mixed with competitor bone marrow from C57SJL mice (2 × 105 cells), and injected intravenously via retro-orbital eye injection into 6–8-week-old C57SJL recipient mice that were lethally irradiated 24 h previously (10 Gy at a split dose). The reverse was performed in some experiments, where BM from WT SJL mice (CD45.1+; 4 × 105) were injected into lethally irradiated WT or Pias1−/− recipient mice (CD45.2+). Reconstitution of donor T cells (CD3+), B cells (B220+) and Granulocytes/monocytes (Mac1+) in peripheral blood (PBL) were assayed by flow cytometry 4–20 weeks post reconstitution.
Competitive limiting dilution assay
Competitive limiting dilution assays were performed as described (Sauvageau et al, 1995). Briefly, total bone marrow (BM) cells (3 × 105 cells) from Pias1−/− or WT mice (CD45.2+) were injected intravenously into 6–8-week-old lethally irradiated congenic WT C57SJL recipient mice (CD45.1+). Reconstitution of donor cells in peripheral blood (PBL) was assayed by flow cytometry at various time points post reconstitution. For secondary transplantation, BM cells pooled from 3 to 4 mice transplanted 21 weeks earlier with either WT or Pias1−/− cells were injected at different doses into lethally irradiated WT C57SJL recipient mice, together with a life-sparing dose of 1 × 105 competitor BM cells from C57SJL mice. The level of lymphomyeloid repopulation with donor-derived cells (CD45.2+) in these secondary recipients was evaluated by flow cytometry at 10 and 16 weeks post transplantation. Recipients with ≥ 1% donor-derived cells were considered to be repopulated by at least one competitive repopulating unit (CRU). CRU frequencies were calculated using Extreme Limiting Dilution Analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/) (Hu & Smyth, 2009).
Bone marrow in vivo homing assay
In vivo homing assays were performed as described (Adams et al, 2006; Janzen et al, 2006). Briefly, freshly isolated total bone marrow cells were labeled with 2.5 μM carboxyfluorescein diacetate succinimidyl ester (CFDA-SE; Molecular Probes, Grand Island, NY, USA) as instructed by the manufacturer. Cells were then injected intravenously via retro-orbital eye injection into 6–8 weeks old lethally irradiated WT C57SJL mice. Mice were euthanized 12 or 24 h post injection and homed cells in the BM were assayed by measuring CFDA-SE+ cells using flow cytometry. In vivo homing assays were also performed with CFSE-labeled long-term hematopoietic stem cells (LT-HSC; Lin−Sca1+c-Kit+CD34−) cells (2000 cells/mouse) FACS-sorted from WT and Pias1−/− littermates.
Niche retention assay
Niche retention assays were performed as described (Trowbridge et al, 2009). Total bone marrow cells (4 × 107) from WT C57/SJL mice (CD45.1+) were injected into non-irradiated WT or Pias1−/− recipient mice (CD45.2+). The percentage of CD45.1+ cells in bone marrow of the recipient mice were assayed by flow cytometry 12 weeks post injection.
Quantitative real time polymerase chain reaction (Q-PCR) analysis
Quantitative real-time polymerase chain reaction (Q-PCR) analyses were performed as described (Liu et al, 2004) with the following modification. A conventional PCR reaction was carried out with a mixture of primers using the following program: 1. 95°C for 3 min; 2. 95°C for 15 s, 60°C for 15 s, 72°C for 30 s for 12 cycles; 3. 72°C for 5 min. PCR products were purified by QIAquickPCR purification kit (Qiagen, Valencia, CA, USA) and used as templates for subsequent Q-PCR analyses using individual primers and SYBR Green performed on CFX96 (BioRad, Hercules, CA, USA). Results were corrected by Hprt1 (Hypoxanthine-guanine phosphoribosyltransferase 1). See supplementary Materials and Methods for primer sequences.
DNA methylation by bisulfite sequencing
Genomic DNA was purified with the ZR genomic DNA II kit (Zymo Research, Irvine, CA, USA). Methylation analysis was performed by bisulfite conversion of genomic DNA using the EZ DNA methylation-Gold kit (Zymo Research). The PCR product was cloned using the TOPO TA cloning kit (Invitrogen, Grand Island, NY, USA).
Coimmuprecipitation (Co-IP) assays
Co-IP assays were performed as described (Liu et al, 2010). Briefly, whole-cell extracts from bone marrow cells were prepared by using lysis buffer containing 1% Brij, 50 mM Tris (pH 8), 420 mM NaCl, 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 μg/ml leupeptin, 3 μg/ml aprotinin, 1 μg/ml pepstatin and 0.1 mM sodium vanadate. The mixture was incubated on ice for 20 min and centrifuged at 15 000 g for 5 min. The supernatant was adjusted to 150 mM NaCl and used for immunoprecipitation with polyclonal anti-PIAS1 antibodies (Liu et al, 1998, 2005) at 1:100 dilution or IgG, followed by immunoblotting with anti-DNMT3A (Abiocode, Agoura Hills, CA, USA) or a monoclonal anti-PIAS1 (Abiocode).
Chromatin immunoprecipitation (ChIP) and MiniChIP assays
Chromatin immunoprecipitation (ChIP) assays were performed with bone marrow (BM) cells using the ChIP Assay Kit (Upstate Biotech) as described (Liu et al, 2010). Briefly, cell extracts from WT or Pias1−/− BMs (2 × 106 per sample) were prepared and chromatin was sheared by sonication (10 s at 30% of the maximum strength for a total of six times). ChIP assays were performed using anti-PIAS1, anti-DNMT3A (Abiocode), or rabbit IgG as a negative control. Bound DNA was quantified by Q-PCR and was normalized with the input DNA. MiniChIP assays were performed essentially as described (Attema et al, 2007), using LSK and L−S−K+ cells purified by FACS sorting to a purity > 99%. For FACS sorting, a fast sort for Lin− cells was performed first to enrich Lin− cell, followed by a regular sort for LSK and L−S−K+ cells. Approximately 50 000 LSK cells were obtained from 20 WT mice and subjected to MiniChIP assays. Of 25 000 LSK cells or 50 000 L−S−K+ cells were used per sample for MiniChIP assays.
Acknowledgments
We thank Irving Garcia for technical assistance, Min Zhou for assistance with FACS data analyses and UCLA flow cytometry core facility. Supported by grants from the NIH (R01AI063286, 3R01AI063286-05S1, and R01GM085797) and the UCLA Jonsson Comprehensive Cancer Center (K.S.). B.L. was supported by a Research Scientist Development Award from the NIH (K01 AR52717). S.T. was supported by a UCLA Tumor Immunology Training Fellowship.
Author contributions
BL and KS directed the project and wrote the manuscript. BL designed and executed most of the experiments. KMY performed experiments in Figs 5 and 7, Fig S4 and Table 1. ST performed experiments in Fig 7. RM provided supports for animal experiments and also performed experiments in Fig 6 and Fig S5. CH performed experiments in Fig 6.
Conflict of interest
K.S. and B.L. are board directors of Abiocode, Inc.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, Kos CH, Pollak MR, Brown EM, Scadden DT. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439:599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Attema JL, Papathanasiou P, Forsberg EC, Xu J, Smale ST, Weissman IL. Epigenetic characterization of hematopoietic stem cell differentiation using miniChIP and bisulfite sequencing analysis. Proc Natl Acad Sci USA. 2007;104:12371–12376. doi: 10.1073/pnas.0704468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, Nerlov C, Leutz A, Andrade-Navarro MA, Jacobsen SE, Rosenbauer F. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Epigenetics of haematopoietic cell development. Nat Rev Immunol. 2011;11:478–488. doi: 10.1038/nri2991. [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, Liang S, Lu Y, Darlington GJ, Meissner A, Issa JP, Godley LA, Li W, Goodell MA. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MP, Seita J, Weissman IL. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb Symp Quant Biol. 2008;73:439–449. doi: 10.1101/sqb.2008.73.031. [DOI] [PubMed] [Google Scholar]
- Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell. 2012;10:690–697. doi: 10.1016/j.stem.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med. 2011;208:421–428. doi: 10.1084/jem.20110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Mizuno S, Wells RA, Cantor AB, Watanabe S, Akashi K. GATA-1 converts lymphoid and myelomonocytic progenitors into the megakaryocyte/erythrocyte lineages. Immunity. 2003;19:451–462. doi: 10.1016/s1074-7613(03)00242-5. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, Rossi DJ, Inlay MA, Serwold T, Karsunky H, Ho L, Daley GQ, Weissman IL, Feinberg AP. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–342. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci USA. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Mink S, Wong KA, Stein N, Getman C, Dempsey PW, Wu H, Shuai K. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat Immunol. 2004;5:891–898. doi: 10.1038/ni1104. [DOI] [PubMed] [Google Scholar]
- Liu B, Shuai K. Targeting the PIAS1 SUMO ligase pathway to control inflammation. Trends Pharmacol Sci. 2008;29:505–509. doi: 10.1016/j.tips.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Tahk S, Yee KM, Fan G, Shuai K. The ligase PIAS1 restricts natural regulatory T cell differentiation by epigenetic repression. Science. 2010;330:521–525. doi: 10.1126/science.1193787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yang R, Wong KA, Getman C, Stein N, Teitell MA, Cheng G, Wu H, Shuai K. Negative regulation of NF-kappaB signaling by PIAS1. Mol Cell Biol. 2005;25:1113–1123. doi: 10.1128/MCB.25.3.1113-1123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yang Y, Chernishof V, Loo RR, Jang H, Tahk S, Yang R, Mink S, Shultz D, Bellone CJ, Loo JA, Shuai K. Proinflammatory stimuli induce IKKalpha-mediated phosphorylation of PIAS1 to restrict inflammation and immunity. Cell. 2007;129:903–914. doi: 10.1016/j.cell.2007.03.056. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4 + CD25 + Foxp3 + regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- Mayle A, Luo M, Jeong M, Goodell MA. Flow cytometry analysis of murine hematopoietic stem cells. Cytometry A. 2012;83:27–37. doi: 10.1002/cyto.a.22093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau G, Thorsteinsdottir U, Eaves CJ, Lawrence HJ, Largman C, Lansdorp PM, Humphries RK. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 1995;9:1753–1765. doi: 10.1101/gad.9.14.1753. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med. 2007;204:715–722. doi: 10.1084/jem.20060750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 2004;23:4116–4125. doi: 10.1038/sj.emboj.7600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.