

Abstract
At sites of damage DNA is resected to enable the optimal form of repair, directed by homology. But how does the cell regulate resection while coordinating with the cell cycle? In this issue of The EMBO Journal, Clerici et al define missing links between DNA resection and cell cycle arrest during repair, showing that Mec1 can inhibit resection to subsequently activate arrest by Tel1.
See also: M Clerici et al (December 2013)
Resection and cell cycle arrest enable faithful DSB repair
DNA double-stranded breaks (DSBs) are a nasty form of damage for a cell, and their repair is important, complex and deeply studied. In the best case, a DSB is repaired using a homologous substrate; less optimally by error-prone direct sealing, then by sealing to another piece of DNA, or not sealed at all. The key is that cells convert a DSB into ssDNA, a substrate repairable by homology, and cells best undergo cell cycle arrest while doing so; ssDNA and time enable faithful repair. Once repair is complete, the cell resumes division. How cells coordinate resection and arrest is the subject of a study by Clerici et al (2014). They examine, as have others, the multiple roles of upstream-acting checkpoint protein kinases Mec1 and Tel1 (the budding yeast orthologs of mammalian ATR and ATM). Models posited partially explain resection and arrest: Tel1 senses damage at DSBs and assists in initial resection (with other nucleases) to generate ssDNA, that then turns over checkpoint control to Mec1 (which is activated by the ssDNA). Activated Mec1 then phosphorylates downstream protein kinases leading to cell cycle arrest and regulation of repair proteins. What's left to learn? Plenty, when one examines the details closely as Clerici et al do! An earlier study indicated mutually exclusive activities of Mec1 and Tel1 at telomeres (Takata et al, 2004). Clerici et al also find roles for Mec1 and Tel1 at DSBs; they detail how Mec1 can limit resection and, most surprisingly, show that Tel1 acts in arrest both before and now after Mec1 activity. These results add interesting molecular details to our understanding of DSB metabolism, and pose intriguing puzzles.
To study the complexity of Mec1 and Tel1 interactions, the authors make use of the somewhat enigmatic phenomenon called ‘adaptation’; cells with a DSB that cannot be repaired first arrest in G2 (for ˜8–12 h), yet they then resume cell division with the broken chromosome (Sandell & Zakian, 1993; Toczyski et al, 1997). Clerici et al first identify a Mec1 mutant they call mec1-ad, that ‘fails to adapt’, that is cells experience a prolonged arrest with a broken chromosome. They found, as have others, that this prolonged arrest (failure to adapt) does occur through canonical checkpoint activation (i.e. downstream Rad53 checkpoint kinase is activated), suggesting study of this abnormal prolonged arrest in mec1-ad mutants may reveal aspects of normal signaling.
Impaired resection induces prolonged arrest
They then ask and answer the key question quite clearly: why do mec1-ad mutants experience a prolonged arrest? Unexpectedly they found that there is a subtle resection defect in mec1-ad mutants: while early resection from the DSB is normal, the later long-range resection is absent (early and long-range resection both occur in MEC1+ cells). Equally remarkable, they found that overexpressing a known exonuclease, Exo1, restores long-range resection and, importantly, restores the wild-type arrest phenotype (mec1-ad alleles overexpressing Exo1 arrest and then resume cell division). So resection and arrest are linked.
They go on to show how mec1-ad mutants inhibit long-range resection; by phosphorylation of two pathways known to intersect with resection (Rad9 and Rad53, and a role for H2A phosphorylation). Finally, and surprisingly, they show that the prolonged arrest in mec1-ad resection-defective cells requires Tel1 (and presumably not Mec1). The connection between resection-defects and Tel1 are deepened by additional observations; prolonged arrest in resection mutants (sgs1Δ,dna2Δ) also requires Tel1, and an earlier study showed that signaling in resection-defective sae2Δ mutants required Tel1 (Usui et al, 2001).
These results lead to the detailed model shown in Fig 1, and then to a plethora of puzzles, in our minds. In the model, MRX-Tel1 initially detects a DSB by end binding and together with other nucleases (Sae2, Exo1, Sgs1/Dna2) resects the DSB to ssDNA. Resection leads to inactivation of Tel1 and activation of Mec1 (which binds RPA on ssDNA, goes the model). Then (dramatically in mec1-ad alleles, and maybe in wild-type cells, too), resection ceases by inhibition of nucleases, some known, some not, via phosphorylation of H2A and Rad53, and recruitment of Rad9 to chromatin. When resection ceases, cells remain arrested, due to MRX-Tel1 binding to some structure, discussed below. The cessation of resection and arrest by Tel1 is dramatic in mec1-ad mutants, and one imagines they occur more subtly in wild-type (MEC1+) cells.
Figure 1.
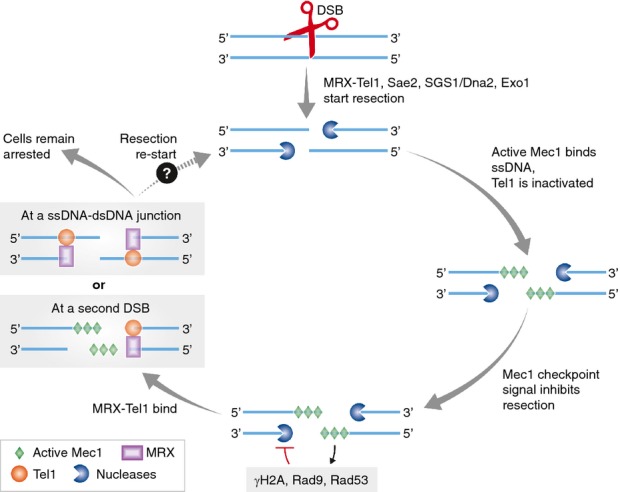
Mec1 inhibits resection to subsequently activate a Tel1-dependent cell cycle arrest.
MRX-Tel1 binds a DSB and assists other nucleases in initial resection. Continuous resection generates ssDNA and eliminates stable ssDNA-dsDNA junctions, Tel1 is thus inactive. Active Mec1 binds ssDNA and signals the checkpoint that then inhibits nuclease activity. Mec1-dependent signaling becomes inactive, perhaps by some “timer” control. When resection stops, MRX-Tel1 binds either a now-stable ssDNA-dsDNA junction or a second DSB (perhaps formed along “static” ssDNA prone to breakage) and signals a prolonged cell cycle arrest. It may be that active Tel1 re-starts resection to promote repair by homology, causing the unrepaired structure to re-enter the cycle for repair.
Interactions between Mec1 and Tel1 signaling
Now to address what seem to be deep puzzles. First, a question that remains from earlier studies and is raised here again: why does extensive resection in wild-type (MEC1+) cells not eternally activate Mec1 to cause arrest? Why can't ‘static’ ssDNA activate Mec1? And similarly, why is the cessation of resection in mec1-ad mutants, leaving static ssDNA, not sufficient to activate Mec1 and cause arrest? It seems that ssDNA is at some point not sufficient to cause arrest; perhaps arrest is under some sort of ‘timer control’ such that after up to 12 h, when DSBs should have been resolved (it takes about 1 h in a repair-proficient cell; Pellicioli et al, 2001), checkpoint arrest is overridden (adaptation) and cells resume division. (A plethora of proteins that regulate adaptation, including Cdc5, Ptc2 and Ptc3, may act on Mec1 targets to inactivate them; Toczyski et al, 1997; Leroy et al, 2003.) The nature of the ‘timer’ is, to our knowledge, unknown. (Continued resection was proposed in an earlier study as necessary for checkpoint activation, yet MEC1+ cells with active resection do not have a prolonged arrest, while mec1-ad mutants with impaired resection do; contradicting this idea.)
Second, and novel in this work; why do mec1-ad mutants require Mec1 early for arrest, and Tel1 (but not Mec1?) for the prolonged arrest later (when resection has ceased)? (It is not known if Tel1 and mec1-ad are needed for late arrest; it seems more likely that only Tel1 is required.) And what might be the cellular role for a late-acting Tel1-dependent arrest in normal cells? Perhaps when a DSB is repairable, the late Tel1-induced arrest may ensure time to complete repair or restart resection if needed; we know of no data that bear on this issue.
What structure does MRX-Tel1 bind?
A key molecular question is, what is the structure(s) of DNA to which Tel1 and MRX presumably bind to cause arrest when resection ceases? It's either a DSB or a ssDNA-dsDNA junction, one surmises. The data from Clerici et al are ambiguous on this point; ssDNA is measured out to 4 h, but not beyond, and Tel1's role in arrest begins at 12–14 h, and not sooner. So, there are two possibilities: either the ssDNA is broken near the dsDNA junction (perhaps even caused by endonuclease activity of Mre11 after binding the junction), to form a ‘late DSB’ that activates Tel1. Or, the ssDNA is not broken, and MRX-Tel1 may bind the ssDNA-dsDNA junction to cause arrest.
Which is it? A second DSB or a now-exposed ssDNA-dsDNA junction? And who cares? Clues resolving the nature of the DNA are not unambiguous, in our view. Mutants in Ku70/Ku80 have increased Mre11 binding to a DSB, and prolonged arrest via Tel1, seeming to suggest that a new DSB arises. Yet there is some evidence that Ku70/Ku80 can bind 3’ overhangs as well (Mimori & Hardin, 1986; Falzon et al, 1993) and that MRN (the mammalian MRX ortholog) binds ssDNA-dsDNA junctions (Shiotani & Zou, 2009; Duursma et al, 2013).
Implications at other DNA structures
And who cares, aside from those interested in the machinations of DSB repair? Mec1 and Tel1 regulation of resection and signaling arrest may have implications at other DNA structures, such as stalled replication forks and short telomeres. At stalled forks, Mec1 activation is proposed to require RPA coated ssDNA, Dpb11, the checkpoint clamp (Rad17-Mec3-Ddc1; 9-1-1 complex) and checkpoint clamp loader (Rad24/RFC complex). Curiously, Tel1 interactions with the 9-1-1 complex show increased MMS sensitivity that is partially rescued by overexpression of a CDC13-EST2 fusion plasmid (Piening et al, 2013). Kaochar et al (2010) found that chromosome rearrangements arising from replication errors were increased in mutants of Mec1, Dbp11 and the 9-1-1 complex as expected. Unexpectedly, rearrangements were unaffected by mutant RPA alleles and increased in tel1Δ (as well as in MRX mutants; Kaochar et al, 2010). It may be that Dpb11 and Mec1 are important for initially sensing damage and preventing replication errors but after some time, or after the replication fork passes and leaves a ssDNA gap, Tel1-MRX may be important for signaling arrest at more ‘static’ ssDNA-dsDNA junctions. (Tel1 does appear to signal at stalled forks near a DSB; [Doksani et al, 2009] though it is unknown if Tel1 signals directly from a stalled fork or from a gapped molecule generated by defective replication). Clerici's work may suggest a role for Tel1 signaling in arrest to aid replication fork recovery.
A similar interaction between Mec1, Tel1 and resection has been reported at telomeres. Takata et al found that Mec1 and Tel1 compete for telomere association during the cell cycle in a manner apparently similar to Mec1 and Tel1 binding at resected DSBs; Tel1-MRX first bind telomeres in G1 when they are double-stranded, Mec1 but also low levels of Tel1 associate during S phase and in G2 when ssDNA is present, and Tel1 and not Mec1 binds again in very late G2 when there is little ssDNA (Takata et al, 2004; Sabourin et al, 2007).
In conclusion, Clerici et al show Mec1 can inhibit resection to such an extent that Mec1 becomes inactive (via a timer?) and Tel1 becomes active (via new DSB or ssDNA-dsDNA junction?). The role of Mec1, Tel1 and resection in an actual recombination event, or at a replication fork, where DNA structures may be dynamic, may be worth contemplating.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Clerici M, Trovesi C, Galbiati A, Lucchini G, Longhese MP. Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J. 2014;33:198–216. doi: 10.1002/embj.201386041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, Bermejo R, Fiorani S, Haber JE, Foiani M. Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell. 2009;137:247–258. doi: 10.1016/j.cell.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, Cimprich KA. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell. 2013;50:116–122. doi: 10.1016/j.molcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzon M, Fewell JW, Kuff EL. EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single-to double-strand transitions in DNA. J Biol Chem. 1993;268:10546–10552. [PubMed] [Google Scholar]
- Kaochar S, Shanks L, Weinert T. Checkpoint genes and Exo1 regulate nearby inverted repeat fusions that form dicentric chromosomes in Saccharomyces cerevisiae. PNAS. 2010;107:21605–21610. doi: 10.1073/pnas.1001938107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy C, Lee SE, Vaze MB, Ochsenbien F, Guerois R, Haber JE, Marsolier-Kergoat MC. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell. 2003;11:827–835. doi: 10.1016/s1097-2765(03)00058-3. [DOI] [PubMed] [Google Scholar]
- Mimori T, Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- Piening BD, Huang D, Paulovich AG. Novel connections between DNA replication, telomere homeostasis, and the DNA damage response revealed by a genome-wide screen for TEL1/ATM interactions in Saccharomyces cerevisiae. Genetics. 2013;193:1117–1133. doi: 10.1534/genetics.113.149849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin M, Tuzon CT, Zakian VA. Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol Cell. 2007;27:550–561. doi: 10.1016/j.molcel.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Zakian VA. Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell. 1993;75:729–730. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–558. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata H, Kanoh Y, Gunge N, Shirahige K, Matsuura A. Reciprocal association of the budding yeast ATM-related proteins Tel1 and Mec1 with telomeres in vivo. Mol Cell. 2004;14:515–522. doi: 10.1016/s1097-2765(04)00262-x. [DOI] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell. 1997;90:1097–1106. doi: 10.1016/s0092-8674(00)80375-x. [DOI] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]