

Abstract
TMEM106B variants are genetically associated with frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP), and are considered a major risk factor for this disease. As TMEM106B may be involved in other pathologies such as Alzheimer's disease (AD) and amyotrophic lateral sclerosis (ALS), uncovering its cellular functions has become a priority. In this issue of The EMBO Journal, Schwenk et al (2014) combine loss-of-function experiments, live imaging and proteomics to unveil the physiological roles played by TMEM106B and its binding partner MAP6 in lysosomal function and transport.
See also: BM Schwenk et al (March 2014)
FTLD comprises a group of clinically and genetically diverse pathologies characterised by atrophy of the frontal and temporal brain lobes. This family of disorders represents one of the major forms of pre-senile dementia, coming third in incidence after AD and Parkinson's disease (PD) (Rademakers et al, 2012). In addition to dementia, FTLD patients display a range of clinical symptoms including language disabilities and behavioural disorders. Much of the gross atrophy observed in FTLD could also lead to cognitive impairment and might result from synapse loss, dendritic atrophy and neuronal death, often accentuated in superficial layers of the brain.
Approximately 50% of FTLD patients exhibit nuclear and cytoplasmic inclusions of ubiquitinated TDP-43 (TAR-DNA binding protein 43), which are very similar to those found in ALS patients (Vass et al, 2011). Mutations in several genes including progranulin (GRN), microtubule-associated protein Tau (MAPT), as well as a hexanucleotide repeat expansion in the non-coding-region of C9orf72 (chromosome 9, open reading frame 72), have been associated to FTLD-TDP (Rademakers et al, 2012), yet the functional links between these mutations and the onset of disease are unclear. Similarly, although TMEM106B has been described as a major risk factor for FTLD-TDP in several genome-wide association studies (Van Deerlin et al, 2010; Finch et al, 2011; van der Zee et al, 2011), our understanding of its physiological role remains very limited, with the exception of its membrane topology and its subcellular localisation to late endosomes and lysosomes (Lang et al, 2012).
In this issue of The EMBO Journal, Schwenk et al (2014) investigated the physiological function of TMEM106B, providing direct evidence for its role in lysosomal transport. Furthermore, the authors uncovered a new function of TMEM106B in dendritic development and trafficking, which is in line with the reported role of late endosome dynamics in dendrite morphogenesis (Puram & Bonni, 2013).
TMEM106B mostly displayed a somatodendritic distribution in primary neurons, and was found associated with late endosomes and lysosomes (Chen-Plotkin et al, 2012; Lang et al, 2012; Brady et al, 2013). The clustering of LAMP2-positive organelles in the perinuclear region induced by TMEM106B downregulation suggests that this protein is important for lysosomal trafficking and/or positioning. Interestingly, TMEM106B knockdown in hippocampal neurons induced an overt change in dendritic morphology. In these cells, dendrites exhibited blunted arborisations with significantly reduced complexity at all developmental stages, suggesting an additional function of TMEM106B in dendritic maintenance (Fig 1). This hypothesis is supported by the dramatic changes in spine morphology and reduction of pre-and post-synaptic markers observed in TMEM106B-depleted neurons, leading potentially to synaptic loss.
Figure 1.
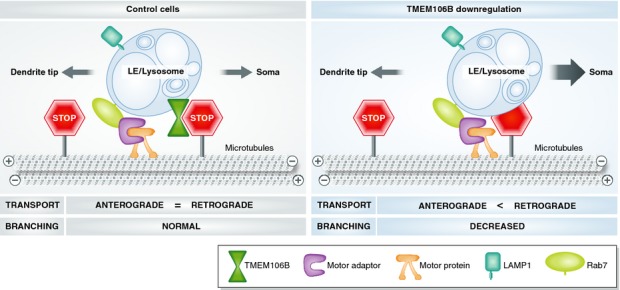
TMEM106B-MAP6 (STOP) complex controls late endosomes/lysosomes transport in dendrites.
In control cells, the interaction between TMEM106B and MAP6 inhibits retrograde transport of LE/lysosomes along dendrites and is required for branching (left panel). Upon TMEM106B downregulation, LE/lysosomes no longer bind to MAP6. As a consequence, their retrograde transport is facilitated and dendritic branching impaired (right panel).
Biochemical and imaging data indicate an interaction between the cytosolic N-terminal domain of TMEM106B and the C-terminal domain of MAP6 (microtubule-associated protein 6), also known as STOP (stable tubule-only polypeptide). Interestingly, the over-expression of MAP6 phenocopied TMEM106B knockdown, whereas MAP6 downregulation or MAP6/TMEM106B double knockdown restored dendritic arborisation. Destabilisation of microtubules by nocodazole partially rescued this phenotype. These results indicate that these two proteins are likely to function in the same microtubule-dependent pathway. Live-imaging showed that TMEM106B knockdown or MAP6 overexpression induced an increase in the number of mobile Rab7A-positive compartments and altered their dynamics by facilitating microtubule-dependent retrograde transport over anterograde transport (Fig 1). Interestingly, MAP6 downregulation or nocodazole treatment restored the anterograde versus retrograde transport balance.
Altogether, these results by Schwenk et al suggest that late endosome and lysosome dynamics are important for dendritic branching and neuronal development. Moreover, these findings strengthened the possibility that deficits in endo-lysosomal transport and/or fusion may be involved in the development of FTLD-TDP, as suggested by pathogenic mutations previously identified in components of the endosomal sorting complex required for transport (ESCRT), such as CHMP2B, and VCP (valosin-containing protein) (Rademakers et al, 2012).
This study also opens the way to new investigations to better understand the possible links between progranulin function, the endocytic pathway and neurodegeneration. Indeed, given the genetic relationship between GRN variants and FTLD-TDP, and the colocalisation of progranulin with TMEM106B in late endosomal compartments, it has been suggested that TMEM106B might modulate the level of progranulin in these organelles (Chen-Plotkin et al, 2012). The variable clinical symptoms of FTLD caused by GRN mutations as well as its low penetrance support the hypothesis that alterations in TMEM106B expression and/or function might modify disease severity in these patients (Van Deerlin et al, 2007). In spite of the preferential localisation of TMEM106B in the somatodendritic compartment, this work revealed an inverse correlation between TMEM106B knockdown and axonal length in developing neurons. However, no molecular mechanism at the basis of this finding has been provided.
The majority of transport defects associated with animal models of neurodegeneration, such as ALS, AD and Huntington's disease, have been identified in axons rather then in dendrites. Indeed, these abnormalities, also called “transportopathies” involve mostly defects in anterograde and/or retrograde transport of organelles and proteins along the axon. Although the causal links between axonal transport deficits and neuronal death are not completely understood, several neurodegenerative disorders are caused by mutations in genes encoding essential components of the vesicular transport machinery, such as motor proteins, Rab GTPases and their regulators, and microtubule-associated proteins (Perlson et al, 2010).
The elegant data provided by Schwenk et al provide further characterisation of TMEM106B, and importantly, functional evidence on its molecular role in neurons. These new findings highlight how deficits in late endosomal and lysosomal trafficking may lead to neurodegeneration and suggest the possibility that cellular assays based on the dynamics of these organelles could be used as a new strategy for drug discovery.
References
- Brady OA, Zheng Y, Murphy K, Huang M, Hu F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet. 2013;22:685–695. doi: 10.1093/hmg/dds475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, Busch JI, Akle S, Grossman M, Deerlin Van V, Trojanowski JQ, Lee VM. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. 2012;32:11213–11227. doi: 10.1523/JNEUROSCI.0521-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, Crook J, Finger E, Hantanpaa KJ, Karydas AM, Sengdy P, Gonzalez J, Seeley WW, Johnson N, Beach TG, Mesulam M. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, Haass C. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem. 2012;287:19355–19365. doi: 10.1074/jbc.M112.365098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Maday S, Fu MM, Moughamian AJ, Holzbaur EL. Retrograde axonal transport: pathways to cell death? Trends Neurosci. 2010;33:335–344. doi: 10.1016/j.tins.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Bonni A. Cell-intrinsic drivers of dendrite morphogenesis. Development. 2013;140:4657–4671. doi: 10.1242/dev.087676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8:423–434. doi: 10.1038/nrneurol.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K, Lichtenthaler SF, Hoogenraad CC, Capell A, Haass C, Edbauer D. The FTLD risk factor TMEM106B and MAP6 control dendtrici trafficking of lysosomes. EMBO J. 2014;33:450–467. doi: 10.1002/embj.201385857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerlin Van VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, Swieten van JC, Murrell JR, Ghetti B, Spina S, Grafman J. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerlin Van VM, Wood EM, Moore P, Yuan W, Forman MS, Clark CM, Neumann M, Kwong LK, Trojanowski JQ, Lee VM, Grossman M. Clinical, genetic, and pathologic characteristics of patients with frontotemporal dementia and progranulin mutations. Arch Neurol. 2007;64:1148–1153. doi: 10.1001/archneur.64.8.1148. [DOI] [PubMed] [Google Scholar]
- Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, Elman L, McCluskey L, Lee VM, Deerlin Van VM, Trojanowski JQ, Chen-Plotkin AS. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121:373–380. doi: 10.1007/s00401-010-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee van der J, Langenhove van T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, Santens P, Broeck van den M, Joris G, Brys J, Mattheijssens M, Peeters K, Cras P, Deyn De PP, Cruts M, Broeckhoven van C. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134:808–815. doi: 10.1093/brain/awr007. [DOI] [PMC free article] [PubMed] [Google Scholar]