

Abstract
Chromatin immunoprecipitation and sequencing (ChIP-seq) provides a static snap-shot of DNA-associated proteins which fails to reflect the dynamics of the DNA-bound proteome. Now, Catic and co-workers combine ubiquitin ChIP-seq and proteasome inhibitors to map sites of DNA-associated protein degradation on a genome-wide scale. They identify an ubiquitin ligase which targets a transcriptional repressor for destruction by the proteasome, thus activating transcription of specific genes. These findings reveal that the ubiquitin proteasome system actively regulates transcription.
See also: A Catic et al (December 2013)
Understanding mechanisms of gene expression control is perhaps the biggest challenge of the post-genomic era. Protein abundance is regulated at all levels from transcription to protein degradation (Vogel & Marcotte, 2012). Current research mainly focuses on transcriptional control by activators and repressors that bind to specific DNA sequences. These interactions can now be studied on a genome-wide scale. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) generates genomic maps of interactions between proteins and their posttranslational modifications with DNA.
It is known for some time that the ubiquitin proteasome system (UPS) also plays a role in transcription (Collins & Tansey, 2006). In fact, one of the subunits of the proteasome was originally identified on the basis of mutations that suppressed defects in the yeast transcription activator Gal4 (Swaffield et al, 1992). A growing body of evidence suggests a direct mechanistic link between the UPS and transcription. For example, activity of the UPS is required for efficient transcription of certain genes (Lipford et al, 2005). However, a comprehensive picture of DNA-associated protein turnover has until now been lacking.
Now, the lab of David Scadden use ChIP-seq to investigate DNA-associated protein turnover on a global scale (Catic et al, 2013). They transfected human and mouse cells with tagged ubiquitin which they could pull down to sequence the associated DNA. However, also histones are frequently mono-ubiquitinated, and this modification does not induce degradation. So how is it then possible to distinguish regions of degradative ubiquitination from other sites of ubiquitination? Catic et al chose a pragmatic approach and deployed the proteasome inhibitor lactacystin: An increase in ubiquitination compared to untreated cells then indicates DNA-associated protein degradation at the respective genomic location. As an added bonus, lactacystin also depletes nuclear mono-ubiquitin which reduces the background (Kim et al, 2011). They found that DNA associated protein turnover correlates with actively transcribed genes, as previously shown in yeast (Auld et al, 2006). A closer look revealed enrichment in nuclear-encoded mitochondrial genes, many of which contained binding motifs of the transcriptional enhancer CREB in their promoter regions. CREB itself however was not degraded as its binding did not change much upon proteasome inhibition. Instead, the authors identified the CREB interacting co-repressor NCoR1 as the target of ubiquitination. This ubiquitination is mediated by the E3 ligase Siah2 and leads to rapid NCoR1 degradation. Consequently, knocking down Siah2 or blocking the proteasome both affects mitochondrial function negatively.
The model emerging from these data is straightforward: To activate transcription, a ubiquitin ligase (like Siah2 in this study) targets a transcriptional repressor (here NCoR1) for degradation, thus derepressing the gene (Fig 1A). In this model, continuous degradation of the repressor is necessary for efficient transcription. Alternatively, it is also conceivable that the UPS could target enhancers and thus represses specific genes (Fig 1B). Finally, it is known that the UPS can remove “spent” transcription activators to allow binding of fresh molecules (Lipford et al, 2005). Thus, degradation of transcriptional enhancers can also increase transcription (Fig 1C).
Figure 1.
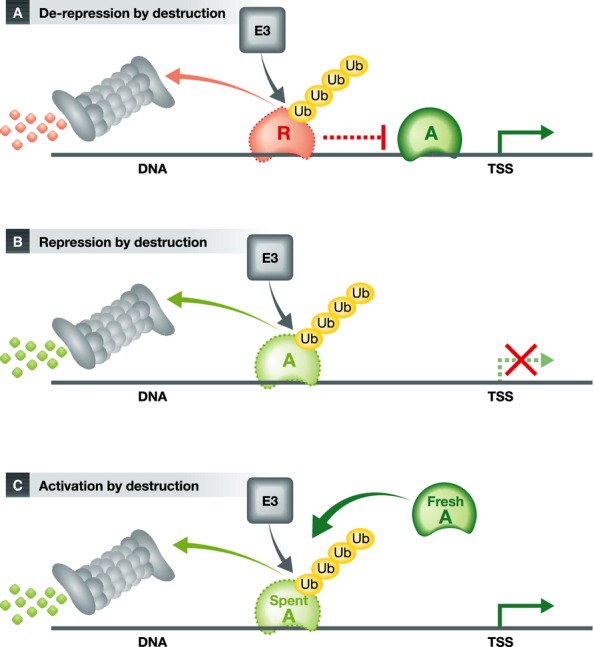
- Poly-ubiquitination by an E3 ligase (E3) and proteasomal degradation of repressors (R) can de-repress genes and promote transcription.
- A gene can also be turned off by the elimination of an essential transcription activator.
- Degradation of a “used up” activator allows a new functional one to replace it and thus increases transcription.
What are the functional implications? On one hand, targeted degradation of DNA-associated proteins adds another layer of regulation which may be required to achieve complex gene expression patterns. Accordingly, E3 ligases can be regarded as a novel class of trans-acting factors involved in transcription regulation. On the other hand, high turnover of DNA-binding proteins is also required for rapid changes in transcription. In fact, transcriptional regulators and chromatin-modifying enzymes tend to be unstable at both the protein and the mRNA level (Schwanhausser et al, 2011). To some extent, chromatin-associated protein degradation may thus simply reflect the fact that many DNA-binding proteins are generally unstable.
As is the case for many pioneering papers, the work by Catic et al opens up many questions. For example, except for NCoR1, it is not known which proteins are targeted. Proteomics could help quantifying changes in ubiquitination of individual proteins on a global scale (Kim et al, 2011; Wagner et al, 2011). Moreover, the assay developed by the authors is indirect and does not prove that proteins are indeed degraded. The use of proteasome inhibitors is a particular concern because it is expected to induce unwanted side effects. This could be addressed by using metabolic pulse labeling methods which can quantify protein turnover with less interference (Deal et al, 2010). Future studies will provide new insights into the role of the UPS in transcriptional regulation.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Auld KL, Brown CR, Casolari JM, Komili S, Silver PA. Genomic association of the proteasome demonstrates overlapping gene regulatory activity with transcription factor substrates. Mol Cell. 2006;21:861–871. doi: 10.1016/j.molcel.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Catic A, Suh CY, Hill CT, Daheron L, Henkel T, Orford KW, Dombkowski DM, Liu T, Liu XS, Scadden DT. Genome-wide map of nuclear protein degradation shows NCoR1 turnover as a key to mitochondrial gene regulation. Cell. 2013;155:1380–1395. doi: 10.1016/j.cell.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, Tansey WP. The proteasome: a utility tool for transcription? Curr Opin Genet Dev. 2006;16:197–202. doi: 10.1016/j.gde.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328:1161–1164. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipford JR, Smith GT, Chi Y, Deshaies RJ. A putative stimulatory role for activator turnover in gene expression. Nature. 2005;438:113–116. doi: 10.1038/nature04098. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Swaffield JC, Bromberg JF, Johnston SA. Alterations in a yeast protein resembling HIV Tat-binding protein relieve requirement for an acidic activation domain in GAL4. Nature. 1992;357:698–700. doi: 10.1038/357698a0. [DOI] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10:M111 013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]