

Abstract
Metastasis is the leading cause of morbidity for lung cancer patients. Here we demonstrate that murine tumor propagating cells (TPCs) with the markers Sca1 and CD24 are enriched for metastatic potential in orthotopic transplantation assays. CD24 knockdown decreased the metastatic potential of lung cancer cell lines resembling TPCs. In lung cancer patient data sets, metastatic spread and patient survival could be stratified with a murine lung TPC gene signature. The TPC signature was enriched for genes in the Hippo signaling pathway. Knockdown of the Hippo mediators Yap1 or Taz decreased in vitro cellular migration and transplantation of metastatic disease. Furthermore, constitutively active Yap was sufficient to drive lung tumor progression in vivo. These results demonstrate functional roles for two different pathways, CD24-dependent and Yap/Taz-dependent pathways, in lung tumor propagation and metastasis. This study demonstrates the utility of TPCs for identifying molecules contributing to metastatic lung cancer, potentially enabling the therapeutic targeting of this devastating disease.
Keywords: CD24, lung cancer, metastasis, Taz, tumor propagating cells
Introduction
The average 5-year survival rate for patients diagnosed with lung cancer remains at a strikingly low 16%. The predominant contributor to this statistic is the fact that the vast majority (∼85%) of patients are diagnosed when their cancer has already progressed beyond a localized state (Siegel et al, 2013). Even for those patients who undergo resection of localized lung cancer, 72% will later develop lethal metastatic disease (Pisters et al, 2000). In the absence of novel early detection modalities, it is essential to better understand the mechanisms that control progression and metastasis in order to offer new therapeutic options for lung cancer patients.
The mucin-like cell surface adhesion molecule, CD24, has been implicated as a marker and a key regulator of advanced disease in multiple types of cancer, yet with some possibly conflicting results (Lee et al, 2011; Overdevest et al, 2011; Liu et al, 2012). High levels of CD24 detected by immunohistochemistry had a significantly increased likelihood of disease progression and cancer-related death for lung cancer patients (Kristiansen et al, 2003; Lee et al, 2010). Disseminated lung tumor cells from malignant pleural effusions also express CD24 (Yao et al, 2013). On the other hand, CD24-negative/low membrane expression was significantly correlated with poor differentiation and worse survival in lung cancers (Damelin et al, 2011). In breast cancers, the links between CD24 expression and tumor progression have also differed between studies (Al-Hajj et al, 2003; Shipitsin et al, 2007).
Tumor-propagating cells (TPCs) are tumor cells capable of serially transplanting and recapitulating the tumor phenotype and may contribute to tumor progression and metastasis, yet few studies have directly tested this idea. Notable examples have utilized cancer cell lines and xenograft models in breast cancer (Korkaya et al, 2008; Charafe-Jauffret et al, 2009, 2010; Croker et al, 2009), pancreatic (Hermann et al, 2007), and colorectal cancer (Pang et al, 2010) to link metastasis to TPCs. Murine CD24+ CD90+ TPCs play a key role in breast cancer metastasis through cell autonomous and microenvironmental requirements (Malanchi et al, 2011). However, differentiated, non-tumor propagating cancer cells can also facilitate metastasis in embryonal rhabdomyosarcoma (Ignatius et al, 2012). A CD24-expressing population (CD24+ ITGB4+ Notch-hi) was shown to be enriched for tumor propagation in a mouse model of lung cancer (Zheng et al, 2013), yet the metastatic potential of this CD24+ cell population has not been reported.
The Hippo signaling cascade, most noted for its role in organ size control and development, has been posited to play a role in cancer progression (reviewed in Pan, 2010), and emerging evidence supports this concept in lung cancer. Expression of the canonical effector of the Hippo pathway, Yes associated protein 1 (Yap1), has been correlated with poor prognosis in non-small cell lung cancer patients and was shown to modulate the tumorigenicity of human lung cancer cell lines (Wang et al, 2010). Overexpression of WW domain-containing transcription regulator protein 1 (Wwtr1, more commonly referred to as Taz), a close homolog of Yap1, was capable of transforming immortalized human bronchial epithelial cells, while knockdown of Taz in non-small cell lung cancer cell lines decreased tumorigenicity, validating Taz as an oncogene in the lung (Zhou et al, 2011).
Metastatic lung adenocarcinomas, the most common form of lung cancer in patients, can be initiated in mice harboring Lox-Stop-Lox-KrasG12D and p53flox/flox alleles (hereafter, Kras;p53-flox) upon intranasal instillation of adenovirus expressing Cre-recombinase (Jackson et al, 2005). Using this model, we have shown that the Sca1+ population of tumor cells is enriched for tumor-propagating potential (Curtis et al, 2010). In the present study, we determined that CD24a (the mouse homolog of human CD24, CD24 hereafter) is an additional lung TPC marker and a key mediator of lung cancer cell line migration and subcutaneous tumor propagation. We also show that Sca1+ CD24+ cells are enriched for metastatic potential after orthotopic delivery of tumor cells. Lung TPCs exhibit transcriptional profiles correlating with metastatic potential and poor patient prognosis. Finally, our data point to a functional role for YAP and TAZ in lung cancer progression and metastasis.
Results
CD24 expression marks cells with tumor-propagating capacity
We sought to identify additional cell surface markers of the Sca1+ Kras;p53-flox cancer cells we previously identified as lung TPCs. DAPI− CD31− CD45− Sca1+ (Sca1+ hereafter) and DAPI− CD31− CD45− Sca1− (Sca1− hereafter) cell populations from four primary Kras;p53-flox lung tumors were isolated via fluorescence activated cell sorting (FACS) as previously described (Curtis et al, 2010) and their transcriptional profiles were analyzed by Affymetrix microarray. Expression levels of cell surface markers were examined followed by quantitative real-time PCR (qPCR) and FACS analysis to identify candidate lung TPC markers. Differential expression of Sca1 (LY6A) in TPCs was confirmed (fold change in Sca1+ versus Sca1−, 30.77) in this data set. CD133, a putative marker of human lung cancer cells with increased tumorigenicity (Chen et al, 2008; Eramo et al, 2008; Levina et al, 2008; Bertolini et al, 2009), was highly expressed in Sca1+ TPCs by microarray and qPCR analysis (fold change, 23.63)(Curtis et al, 2010), yet flow cytometry for CD133 failed to show enrichment within the Sca1+ fraction (unpublished observations). Several other markers of aggressive cancer stem cells in other tissues, including CD44 and CXCR4, were not significantly differentially expressed in Sca1+ and Sca1− cells. Interestingly, CD24 was upregulated in lung TPCs (fold change, 1.8). qPCR confirmed that CD24 was overexpressed more than 3-fold in Sca1+ TPCs from Kras;p53-flox lung tumors compared to Sca1− cells (P = 0.0057) (Fig 1A). Flow cytometric analysis of Kras;p53-flox lung tumor cell preparations revealed that 87.1 ± 5.44% of Sca1+ cells were CD24+ (Fig 1B, n = 7).
Figure 1.
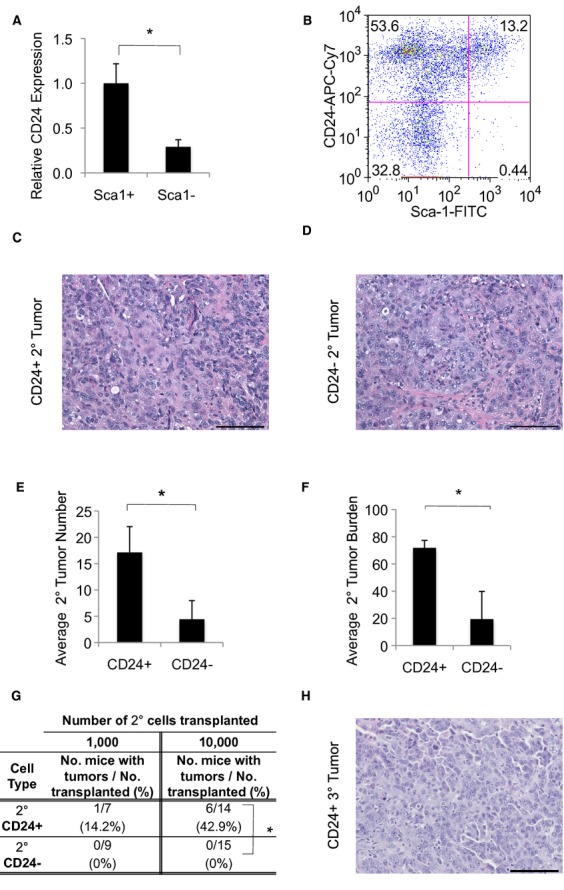
CD24 marks cells enriched for tumor-propagating ability.
A Real Time RT-PCR analysis for CD24 in sorted Sca1+ and Sca1− Kras;p53-flox lung tumor cells.
B A representative flow cytometry plot showing CD24 and Sca1 expression in Kras;p53-flox lung tumor cells. Cells shown were gated on the single, live (DAPI-negative), CD31-negative, CD45-negative population.
C, D H&E images showing a secondary Kras;p53-flox lung adenocarcinoma from the transplantation of CD24+ tumor cells (C) or CD24− tumor cells (D). 200× magnification, scale bar = 100 μm.
E, F Quantification of tumors from transplantation of 10,000 CD24+ or CD24− cells. Average tumor number per recipient mouse (E), n = 7 for each cell type transplanted). Average tumor burden in secondary mice calculated as the percentage of lung area filled with tumors (F, n = 6 for CD24+, n = 7 for CD24−).
G Table showing tertiary tumor formation ability of CD24+ and CD24− Kras;p53-flox lung tumor cells from secondary transplants. 1,000 or 10,000 CD24+ or CD24− cells were intratracheally transplanted into immunocompromised mice. The percentage of recipient mice with at least one lung tumor after 13–28 weeks is shown. *P = 0.0063, Fisher's Exact Test, two-tailed.
H H&E image showing a tertiary Kras;p53-flox lung adenocarcinoma from the transplantation of CD24+ tumor cells.
Data information: In (A, E and F): *P < 0.05. See also Supplementary Figure S1 for additional data.
We utilized our orthotopic intratracheal transplantation model (Curtis et al, 2010) to verify that CD24-expressing cells were lung TPCs. Primary Kras;p53-flox lung tumors were isolated for digestion, FACS, and transplantation. We first compared the activity of DAPI− CD31− CD45− CD24+ cells (hereafter CD24+) with the activity of DAPI− CD31− CD45− CD24− cells (CD24−). 10,000 CD24+ or CD24− cells were transplanted intratracheally into immunocompromised Foxn1nu/nu (nude) mice. Recipient mice were observed until they exhibited labored breathing, when mice were euthanized for analysis of lung tumors (22 ± 3 weeks for CD24+ cells, 25 ± 4 weeks for CD24− cells). The CD24+ and CD24− populations both propagated lung adenocarcinomas that accurately recapitulated primary Kras;p53-flox tumors (Fig 1C and D). 100% of recipient mice developed at least one lung tumor after transplantation of 10,000 CD24+ or CD24− cells (10/10 for CD24+ and 12/12 for CD24−) (Table S1). Histological analysis revealed that while CD24+ recipient mice had an average of 17 tumors, CD24− recipients had only four (P = 1.2 × 10−4), and likewise CD24+ recipients showed 72% tumor burden (lung area filled with tumor) in contrast to 19% tumor burden observed for CD24− recipients (P = 8.3 × 10−5) (Fig 1E and F).
We next tested the ability of cell populations separated by CD24 expression to serially passage tumors. Interestingly, lung tumors in recipient mice contained a CD24+ population regardless of whether CD24+ or CD24− cells were transplanted despite ∼95% cell purity at the time of tumor cell isolation (Supplementary Fig S1A and B). 1,000 or 10,000 CD24+ or CD24− cells from secondary tumors were FACS-sorted and transplanted orthotopically to test for serial tumor formation ability. Recipients of CD24− cells from secondary tumors did not develop lung tumors (0/9 and 0/15 for 1,000 and 10,000 cells, respectively). In contrast, CD24+ cells from secondary tumors could form tertiary tumors. CD24+ cells from secondary tumors yielded tumors in 1/7 mice after transplantation of 1,000 cells and in 6/14 mice after transplantation of 10,000 cells, P = 0.0063 (Fig 1G and H, Supplementary Fig S1C and D, Supplementary Table S2). These data supported our hypothesis that CD24 is a marker of lung cancer cells with tumor-propagating capacity.
We next used the combination of Sca1 and CD24 to further delineate Kras;p53-flox TPCs. Limiting dilution transplantation experiments were used to compare TPC activity in the Sca1+ CD24+ , Sca1−CD24+ , Sca1+ CD24− and Sca1−CD24− fractions. Recipients of Sca1+ CD24+ and Sca1−CD24+ tumor cells had higher rates of tumor formation than Sca1−CD24− and Sca1+ CD24− recipients, further showing CD24 is a TPC marker (Sca1−CD24+ vs Sca1−CD24−, P = 0.0116, Sca1−CD24+ versus Sca1+ CD24−, P = 0.0334) (Fig 2A). Similar to our results using CD24 alone, Sca1+ CD24+ and Sca1−CD24+ cells serially propagated tumors (2/5 and 3/4 mice respectively), while Sca1−CD24− could not (0/5) (Fig 2B). Importantly, cytospin and qPCR showed the four Sca1/CD24-sorted populations had similar epithelial cell content (Supplementary Fig S2A,B,C and D). Thus, Sca1+ CD24+ and Sca1−CD24+ cells are enriched for Kras;p53-flox TPCs.
Figure 2.
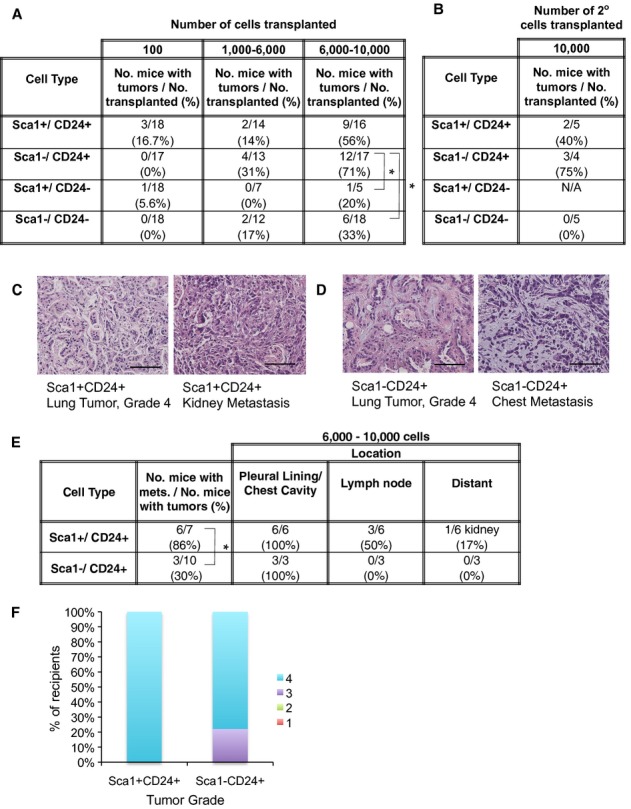
- Table showing tumor propagation ability of Sca1+ CD24+ , Sca1−CD24+ , Sca1+ CD24− and Sca1−CD24− Kras;p53-flox lung tumor cells. 100–10,000 cells were intratracheally transplanted into immunocompromised mice. The percentage of recipient mice with at least one lung tumor after 17–33 weeks is shown.
- Table showing tertiary tumor formation ability of Sca1+ CD24+ , Sca1−CD24+ , and Sca1−CD24− Kras;p53-flox lung tumor cells from secondary transplants: 10,000 CD24+ or CD24− cells were intratracheally transplanted into immunocompromised mice. The percentage of recipient mice with at least one lung tumor after 20–29 weeks is shown.
- H&E images showing a lung tumor (left) and kidney metastasis (right) from the transplantation of Sca1+ CD24+ tumor cells. 200× magnification, scale bars = 100 μm.
- H&E images showing a lung tumor (left) and chest cavity metastasis (right) from the transplantation of Sca1−CD24+ tumor cells. 200× magnification, scale bars = 100 μm.
- Table of metastatic incidence in secondary mice from transplantation of 6,000–10,000 Sca1+ CD24+ , Sca1−CD24+ , Sca1+ CD24− and Sca1−CD24− Kras;p53-flox lung tumor cells. The number and (percentage) of recipient mice with at least one metastatic lesion are shown in the first column. Location columns show the number and (percentage) of recipient mice with metastases in the noted site.
- The percentage of recipient mice bearing lung tumors from transplantation of Sca1+ CD24+ (n = 7) or Sca1−CD24+ (n = 9) cells with the indicated tumor grade (Scale 1–4) is shown. Grade 1 or 2 lesions were not observed in recipient mice.
Sca1+ CD24+ status correlates with metastatic potential
Orthotopically transplanted Kras;p53-flox tumor cell populations also produced metastases in recipient mice, allowing us to also test the role of TPCs in metastasis. First, a cohort of CD24+ and CD24− cell recipients were euthanized for collection of tissues from common metastatic sites for lung cancer, including connective tissue in the pleural cavity, lymph nodes, heart, liver, spleen and adrenal glands. To more fairly compare metastatic capacity, we analyzed CD24− cell recipients 26–29 weeks after transplantation, when they had primary tumors of similar size and grade as tumors in CD24+ cell recipients analyzed at 22 weeks (Supplementary Fig S1G and H). It is possible that larger tumors are more likely to give rise to metastases than smaller tumors. Therefore, we only assessed metastasis in recipient mice bearing lung tumors of similar size. With these parameters, there was no significant difference in metastasis in recipients of CD24+ cells versus CD24− cells (Supplementary Fig S1E). CD24+ cell recipients developed metastatic lesions in the chest wall, local lymph nodes, and the heart (Supplementary Fig S1E and F). Similarly, mice that received CD24− cells developed small chest cavity lesions and local lymph node metastases yet did not have metastatic lesions in the heart. Thus, CD24 status alone is not sufficient as a metastatic marker.
Having identified the Sca1+ CD24+ and Sca1−CD24+ populations as TPC-enriched, we tested their relative metastatic capability. Recipients of Sca1+ CD24+ or Sca1−CD24+ populations developed high-grade lung tumors and metastases (Fig 2C and D). When we compared mice bearing lung tumors of equal size and comparable grades that arose from either of the TPC populations, recipients of Sca1+ CD24+ cells (6/7) were significantly more likely to have metastases than the Sca1− CD24+ recipients (3/10) (P = 0.0498, Fig 2E and F, Supplementary Fig S2E). Thus, at least two TPC populations can be identified with Sca1 and CD24 staining, and the Sca1+ CD24+ phenotype is most highly associated with metastatic activity.
CD24 plays a functional role in metastatic activity
To further probe the role of CD24 in the metastatic spread of lung cancer, we generated a series of cell lines from the Kras;p53-flox lung adenocarcinoma model. The CK1750 and SC241 cell lines exhibit many properties of metastatic lung tumor cells. Firstly, CK1750 cells robustly formed adenocarcinomas when transplanted intratracheally (10/12 recipients formed tumors when 1,000–10,000 cells were injected) (Fig 3A). Furthermore, these recipient animals developed metastases in multiple sites including the chest wall (5/10), lymph nodes (4/10), adrenal glands (2/10), and liver (1/10), with approximately 50% of recipients exhibiting metastasis to one or more sites (Fig 3A). Nearly all CK1750 and SC241 cells expressed CD24 and Sca1 (Fig 3B). Thus, the cell lines functionally and phenotypically resembled the most metastatic Kras;p53-flox lung TPCs.
Figure 3.

- H&E images of a lung tumor (left) and adrenal gland metastasis (right) from intratracheal transplantation from the Kras;p53-flox CK1750 cell line. Images are 200× magnification, scale bars = 100 μM.
- FACS plot from CK1750 cells showing expression of CD24 and Sca1. Cells shown were gated on the single, live (DAPI-negative), CD31-negative, CD45-negative population.
- Analysis of cellular migration from in vitro transwell migration assays, determined by scoring cells migrating towards serum-containing (10% FBS) media with CK1750 CD24 knockdown cells (shCD24-Low-1 & Low-5, n = 3) or control CK1750 shGFP cells (shGFP, n = 6). Results shown are the average of three independent experiments.
- Average number of lung metastases after tail vein injection of CK1750 shGFP cells (circles), CK1750 shCD24-Low-1 cells (squares), or CK1750 shCD24-Low-5 cells (triangles). Lungs were analyzed for metastases 2 weeks after injection of 500,000 cells. Each symbol represents an individual mouse analyzed.
- Table showing the number of recipient mice with lung tumor formation after intratracheal injection with 100,000 shGFP cells or shCD24-Low-1 cells out of the total injected mice (percentage).
Data information: *P < 0.05. See also Supplementary Figure S3 for additional data.
Utilizing two different shRNA constructs against CD24 (shCD24-1 targets the 3′ UTR, while shCD24-5 targets the coding region) we diminished CD24 expression to ∼20–30% of normal transcript levels compared to shGFP control cells after selection for CK1750 and SC241 cells expressing the lowest levels of CD24 by FACS (termed “Low”) (Supplementary Fig S3A). Importantly, knockdown of CD24 in the CK1750 cell line did not affect the growth of the cells in standard cell culture conditions (Supplementary Fig S3B). However, in transwell in vitro migration assays, the CD24 knockdown lines showed markedly less migration compared with control shGFP cells (Fig 3C); the shCD24-Low-1 and shCD24-Low-5 lines showed a 3-fold and 4-fold reductions in migration, respectively (P = 0.033 & P = 0.015). CD24 knockdown in SC241 cells showed similar results; the shCD24-Low-1 and shCD24-Low-5 lines also showed 3-fold and 4-fold reductions in migration, respectively (P = 0.013 & P = 0.0024) (Supplementary Fig S3C). To assess metastatic capacity in vivo, shGFP and shCD24 cells were injected into the tail vein of nude mice. Transplantation of the shCD24-Low-1 and shCD24-Low-5 lines resulted in a significantly lower number of lung metastases (P = 0.7 × 10−4 and P = 0.027) and smaller tumor burden for shCD24-Low-1 transplants (Fig 3D, Supplementary Fig S3D and E). However, average tumor size was not significantly different between the shGFP control and shCD24 groups, suggesting engraftment rather than proliferation is affected by knockdown of CD24 in this tail vein assay (Supplementary Fig S3F). Intratracheal injections showed that shCD24 cells were devoid of TPC activity compared with shGFP controls; shCD24-Low-1 cells produced lung tumors after IT administration in 0/5 recipients whereas shGFP cells yielded tumors in 4/6 recipients (P = 0.04, Fig 3E). While CD24 knockdown cells were capable of subcutaneous tumor formation, the shCD24-Low-1 line showed a longer latency to first palpable tumor (P = 0.04) and yielded smaller tumors 3 weeks post-transplant (P = 0.001) (Supplementary Fig S3G). These findings strongly suggest that CD24 plays a functional role in TPC activity and in promoting lung cancer metastasis.
Murine lung TPCs exhibit clinically relevant, metastatic gene expression profiles
To better understand the connections between TPCs and metastatic potential, we next took an unbiased gene expression profiling approach. Microarray data from the Kras;p53-flox TPCs were further interrogated by bioinformatics. 278 differentially expressed, upregulated probes corresponding to 250 unique genes (log2 fold-change > 1.53 and P-value ≤ 0.02) were used in this analysis (Fig 4A and Supplementary Data). Gene set enrichment analysis (GSEA) on the expression data revealed that gene sets involved in processes such as cytoskeletal rearrangement, cell motility/migration, and invasion, all of which are associated with metastatic capacity, were significantly overrepresented in TPCs (Fig 4B). Sca1+ and Sca1− populations had equally low levels of vimentin (Vim) expression and high levels of E-cadherin (Cdh1) (Supplementary Fig S4A and B), confirming that enrichment for these gene sets was not a result of stromal contamination. These results supported the idea that lung TPCs are involved in metastatic spread of Kras;p53-flox lung tumors, and that this activity is not related to an epithelial-to-mesenchymal transition as observed in other tumor types (Cordenonsi et al, 2011).
Figure 4.

Lung tumor-propagating cells exhibit clinically relevant gene expression profiles that suggest metastatic potential.
A Heatmap showing the 250 genes significantly upregulated in Sca1+ TPCs compared to Sca1− non-TPCs from Kras;p53-flox lung tumors as assessed by Affymetrix Mouse 430 2.0 gene expression microarray. Red indicates upregulated expression, green indicates downregulated expression.
B Table of the top five most significantly enriched gene sets (based on GSEA enrichment and False Discovery Rate (FDR) q-value less than 0.25) in the TPC cell population. Hippo conserved signature genes found in the indicated gene set are listed in the last column.
C The Director's Challenge collection of lung cancer data sets was interrogated for expression of a gene signature upregulated in murine TPCs. Higher expression of the TPC signature (red) versus lower expression of the TPC signature (blue) was compared for patient metastasis (left) and overall survival (right).
D–G Expression of Yap1 (D), Taz (Wwtr1) (E), Nf2 (F) and Cyr61 (G) by qPCR in sorted TPC or non-TPC cells from Kras;p53-flox lung tumors. n = 8 samples each.
H The Director's Challenge collection of lung cancer data sets was interrogated for expression of the Hippo conserved signature. Higher expression of the Hippo signature (red) versus lower expression of the Hippo signature (blue) was compared for patient metastasis (left) and overall survival (right).
Data information: *P < 0.05. See also Supplementary Figure S4 for additional data.
As we had found increasing evidence that TPCs contributed to metastasis, we sought to determine a relationship between murine TPCs and lung adenocarcinoma patient data. We generated a gene signature consisting of 126 human orthologs corresponding to the significantly upregulated genes in Kras;p53-flox TPCs (Supplementary Data). The TPC gene signature was examined in the Director's Challenge cohort of lung adenocarcinoma patients and was found to be significantly associated with decreased overall survival (P = 0.0006) and with metastasis (P = 0.049) (Fig 4C). This indicated that our results using murine models to study tumor propagation and metastasis are applicable to human lung cancer.
Yap/Taz in metastatic lung TPCs
We next used the gene expression analyses to identify molecules other than CD24 that may be crucial for lung tumor propagation and metastatic potential. The gene sets enriched in lung TPCs, including Actin Filament Based Processes, Actin Cytoskeleton Organization and Biogenesis, and Regulation of Cell Migration, included members of the Hippo signaling cascade (Fig 4B). Furthermore, GSEA with a Yap/Taz Gene Signature (Cordenonsi et al, 2011) revealed that Sca1+ TPCs were significantly enriched for this gene set; 25 out of 52 genes in the Yap/Taz gene set exhibited core enrichment in lung TPCs (Supplementary Fig S4C).
We validated expression differences in Hippo pathway components in lung TPCs. Yap1 was not significantly differentially expressed between the Sca1+ and Sca1− cells by Real Time RT-PCR analysis (Fig 4D, P = 0.10). However, Wwtr1 (Taz), which has been shown to be important in lung development (Mitani et al, 2009), was over 2-fold overexpressed in the Sca1+ TPCs compared to Sca1− cells (Fig 4E, P = 0.0087). Importantly, the potent negative regulator of the Hippo pathway, Nf2, was greater than 15-fold down-regulated in the Sca1+ cell population (Fig 4F, P = 0.0001), while a known downstream target of activated Hippo signaling, cysteine-rich angiogenic inducer 61 (Cyr61), was over 2-fold up-regulated in the Sca1+ cell population (Fig 4G, P = 0.021). In the Director's Challenge cohort of lung adenocarcinoma patients, the Yap/Taz signature was significantly associated with worse overall survival (P = 0.00001) and with metastasis (P = 0.009) (Fig 4H). These data show that Yap/Taz is associated with TPCs and human lung cancer progression.
We next examined Yap1/Taz protein levels in tissue sections from murine lung cancer models, since differences in protein levels and localization of the Hippo transcriptional controllers may be more important than overall expression levels detected at the RNA level. Yap1/Taz activity is tightly controlled at the protein level: phosphorylated Yap1/Taz is sequestered in the cytoplasm and inactive, but dephosphorylated Yap1/Taz can translocate into the nucleus to activate target gene transcription. Using an antibody that recognizes both Yap1 and Taz proteins, we performed immunostaining of tumors from the non-metastatic Kras model (Jackson et al, 2001) and the Kras;p53-flox metastatic tumors. Interestingly, Yap1/Taz expression in Kras tumors was very weak and almost exclusively cytoplasmic (Fig 5A, n = 4). In stark contrast, in Kras;p53-flox tumors (n = 4) ∼30–50% of tumor cells expressed nuclear Yap1/Taz, and in lymph node metastases from Kras;p53-flox mice, almost all cells expressed nuclear Yap1/Taz (n = 3) (Fig 5A). These results supported the concept that Yap1/Taz expression and activity is correlated with more aggressive disease in lung cancer.
Figure 5.

- Representative Yap1/Taz IHC (brown staining) in a Kras primary lung tumor (top), Kras;p53-flox tumor (middle), and Kras;p53-flox lymph node metastasis (bottom). 400× magnification, with zoom as shown, approximately 75 μm.
- Percentage of mice of each genotype with tumors of grade 0–4 scored from paraffin sections. Grade shown is the highest tumor grade present in each mouse. Grade 0 indicates no tumors present.
- Number of lung tumors per mouse was scored from paraffin sections and averaged for each genotype (n = 15 Kras mice, 14 Kras;Yap mice).
- Tumor burden was measured as the percentage of lung filled with tumors. Mice were euthanized 7 months after Cre or earlier if symptoms of lung tumors were apparent (n = 15 Kras mice, 14 Kras;Yap mice). NS, not significant.
- Percentage of Ki67+ cells per 200× field. NS, not significant.
- The average number of migratory cells observed in transwell migration assays, determined by the number of cells migrating towards serum-containing (10% FBS) media in control, Yap1 or Taz knockdown CK1750 cells. Results shown are the average of three independent experiments.
- Average number of lung metastases after tail vein injection of CK1750 shGFP cells (circles), CK1750 shYap1 cells (squares, up triangles), or CK1750 shTaz cells (down triangles, diamonds). Lungs were analyzed for metastases 2 weeks after injection of 100,000 cells. Each symbol represents an individual mouse analyzed. Data shown are the combination of two independent experiments (n = 6).
Data information: *P < 0.05, one-tailed t-test. See also Supplementary Figure S5 for additional data.
To determine if Yap activity is sufficient to affect lung tumor progression, we used a doxycycline-inducible, constitutively active allele of Yap1 in combination with oncogenic Kras in vivo. In tetO-YapS127A mice, a mutated version of Yap1 exhibits enhanced nuclear localization of Yap1 and constitutive signaling activity (Schlegelmilch et al, 2011). In a binary genetic approach, we activated Yap1 activity in lung tumor cells by treating Kras; Rosa26-LSL-rtTA; tetO-YapS127A mice with Adeno-Cre to initiate lung tumorigenesis, followed by treatment with doxycycline to activate Yap signaling concomitant with tumor initiation. Mice were euthanized when they showed signs of distress, 7 months after Adeno-Cre and doxycycline administration. Histological analysis showed that tumors of Kras; LSL-rtTA; tetO-YapS127A mice were significantly higher grade than those in Kras controls (Cochran-Armitage test, P = 0.04, Fig 5B). Kras; LSL-rtTA; tetO-YapS127A mice had significantly more tumors than Kras controls (Fig 5C), but no difference in overall tumor burden, average tumor size, or Ki67 staining index (Fig 5D and E, Supplementary Fig S5A,B and C). These results indicated that Yap activation is sufficient to promote lung tumor progression in vivo.
We interrogated the role of Yap/Taz in metastatic capacity using in vitro migration and in vivo tail vein assays in the Kras;p53-flox cell lines after knockdown of Yap1/Taz. Two different shRNAs were used to reduce Yap1 or Taz levels to 40–80% of control levels (Supplementary Fig S5D and E). A significant reduction (1.5-3-fold compared to shGFP) in migration was observed in the shYap1 or shTaz cells exhibiting knockdown (P < 0.05) compared with the negative control shGFP (Fig 5F). To examine whether YAP knockdown in the human lung cancer cell line A549 would yield similar results, we interrogated the ability of two different doxycycline-inducible YAP shRNAs to affect cell migration. Dox-induced YAP knockdown resulted in significantly fewer migrated cells using both hairpins (P = 0.023 and P = 0.020); migration was over 2-fold decreased in A549 YAP knockdown cells (Supplementary Fig S5F). Finally, tail vein injection was performed to assess the impact of knockdown on metastatic capacity in vivo. Lung metastases were significantly less frequent in recipients of shTaz cell lines compared to shGFP or shYap cells (shYap1108 P = 0.061, shYap1824 P = 0.18, shTaz771 P = 0.029, shTaz1616 P = 0.0029), and recipients of shTaz1616 cell lines in particular showed very few or no metastases or tumor burden (Fig 5G, Supplementary Fig S5G and H). Average tumor size was also smaller in shTaz recipients (Supplementary Fig S5I). These data indicated that Yap/Taz is necessary and sufficient to drive key steps in advanced lung tumorigenesis, including progression, migration, and metastasis.
To investigate the mechanism of Yap/Taz activity and to discover potential Taz targets in lung cancer cells, gene expression microarray analysis was performed. Three paired shGFP or shTaz Kras;p53-flox murine lung tumor cell lines (CK1750, SC241, and Tmet)(Supplementary Fig S6A) were compared; differentially expressed genes are listed in Supplementary Data. Gene Set Enrichment Analysis of the shTaz vs shGFP data was used to identify gene sets with negative enrichment scores, indicating cellular processes possibly impaired by Taz knockdown (Supplementary Fig S6B). The Yap/Taz gene signature (Cordenonsi et al, 2011) and genes differentially expressed in the Taz knockout mouse lungs (Mitani et al, 2009) were significantly enriched in the shTaz data set, validating our approach. qPCR confirmed differential expression of Cyr61, a known Yap/Taz target, and Ltbp4, a mediator of the TGF-β pathway and possible Yap/Taz target, in SC241 shGFP and shTaz cells (Supplementary Fig S6C), further validating our gene expression data set. Published data sets connected to migration and metastasis (Lung Cancer Metastasis, Nguyen et al, 2009; Breast Cancer Invasion, Patsialou et al, 2012; Breast Cancer Migration, Gunawardane et al, 2005; Bladder Cancer Migration, Wu et al, 2008) were also enriched, validating the metastatic capacity associated with Yap/Taz activity.
Gene sets of interest that were significantly enriched in shTaz cells revealed several pathways and activities that may be critical for Yap/Taz function in lung cancer progression and metastasis. Several gene sets associated with NF Kappa B signaling were significantly enriched: I KappaB Kinase NF KappaB Cascade, Regulation of I KappaB Kinase NF KappaB Cascade, and Positive Regulation of I KappaB Kinase NF KappaB Cascade (Supplementary Data). Expression of Bcl2, an NF-KB target that inhibits apoptosis, was validated for significantly decreased expression in shTaz cells (Supplementary Fig S6E). Gene sets corresponding to actin cytoskeletal processes were also highly decreased after Taz knockdown, including Actin Cytoskeleton, Cytoskeletal Protein Binding, Actin Binding, Actin Filament Binding, and Actin Filament Based Processes.
Discussion
Our results provide novel insight into the cells and pathways that drive the progression and metastasis of lung adenocarcinoma using TPCs as a discovery platform. TPCs expressing Sca1 and CD24 were enriched for metastatic activity after orthotopic transplantation. CD24 knockdown impaired metastatic processes including migration and ectopic tumor growth. Our findings also implicate a role for the Hippo signaling mediators Yap and Taz in lung TPCs and the progression and metastasis of lung cancer. Importantly, expression of TPC and Yap/Taz gene signatures predicted metastasis and patient survival, demonstrating the relevance of our findings in mouse models to human disease. These results delineate new ways to uncover clinically relevant mechanisms in advanced lung cancer.
Our data provide new understanding of the links between tumor-propagating cells and metastasis, an idea that has been postulated for at least a decade with limited experimental backing (reviewed in Adhikari et al, 2011). The significant correlation of our TPC gene expression signature with both metastasis and patient survival adds new support for this proposition. Lung TPCs also exhibited downregulation of Nkx2.1, a mechanism known to correspond with increased metastatic potential (Curtis et al, 2010; Winslow et al, 2011). We show that several TPC populations can produce metastatic lesions, albeit with differing degrees of activity; Sca1+ CD24+ cells were more metastatic than Sca1−CD24+ cells. Differing from our work, tumor cell barcoding studies in colon cancer showed that only one TPC fraction contributed to metastases (Dieter et al, 2011). Therefore, in some tumor types one unique metastasis-propagating cell population may exist, whereas our data suggest a gradient of metastatic capacity exists within lung tumor cell populations.
CD24 appears to be required for lung TPCs to propagate tumors and possibly to drive a metastatic cascade. CD24 expression levels were similar in lung tumors arising in recipient mice after transplantation of CD24+ or CD24− cells; it is possible that activation of CD24 expression is critical during a particular stage of tumorigenesis or metastatic progression. Furthermore, results in bladder (Overdevest et al, 2011) and liver (Lee et al, 2011) cancer, in combination with the present study, make a strong argument for CD24 being a marker and functional mediator of metastatic potential in multiple tumor types. As proliferation and apoptosis were not significantly altered in CD24 knockdown cells, the mechanism by which CD24 affects tumorigenesis and metastasis remains to be determined.
We demonstrate for the first time that Yap/Taz are active in lung-tumor propagating cells with metastatic potential, and that Yap activation affects lung tumor progression in a mouse model in vivo. Interestingly, the Yap/Taz gene signature predicted lung cancer patient metastasis and survival. Taz expression was also found to be significantly associated with poor differentiation, poor survival, and metastasis in patients with non-small cell lung cancer (Xie et al, 2012). Previously, Yap1 and Taz have both been implicated in different processes of lung development and disease (see also Materials and Methods). Taz interacts with the master transcription factor Nkx2.1 during mouse lung development to promote alveolarization (Mitani et al, 2009). These and our own observations of elevated Taz expression in TPCs and decreased lung metastases from tail vein injections after knockdown of Taz tempt us to speculate that Taz, rather than Yap1, is primarily utilized by lung tumors to promote metastasis. Supporting this idea, Taz may regulate the cancer stem cell phenotype in breast cancers (Cordenonsi et al, 2011). In our studies, constitutive mutant Yap was sufficient to significantly impact Kras-driven lung tumor progression, yet metastasis did not develop in Kras;Yap mice. Notably, the presence of more high-grade tumors in Kras;Yap mice required euthanasia and may have precluded our ability to assess later metastatic progression. It is also possible that further activation of Yap1 through mutation at more than one site is necessary to drive metastases. It remains possible that both Yap1 and Taz play important and complementary roles in lung TPC function and metastatic potential.
Yap/Taz may control lung cancer cell migration and metastasis through a number of mechanisms implicated in this study. In tail vein assays, the average tumor size was significantly less in recipients of shTaz cells vs shGFP cells, pointing to the potential importance of Taz regulation of proliferation in advanced lung tumors. Supporting this idea, we found many gene sets enriched for cell growth in shTaz cells. GSEA also suggested a role for Taz regulation of the NF-KappaB pathway, which has been shown to be required for Kras;p53-flox lung tumorigenesis (Meylan et al, 2009; Xue et al, 2011). Further studies are needed to which, if any, of the cellular activities identified in our expression analyses are critical for Yap/Taz contribution to aggressive lung cancer.
It will be important to understand the cellular markers and mechanistic drivers of metastatic cancer cells in each tumor type of interest. In bladder cancer, CD24+ cells rely on the STAT3/NANOG network to promote metastasis (Lee et al, 2011), whereas CD24+ cells utilize the SRC-family kinase YES in metastatic melanoma (Liu et al, 2012). Additionally, we show here that Yap/Taz activity may mediate lung cancer metastasis in lung TPCs. It remains to be determined if there is a direct interaction between CD24 and Yap1/Taz regulation in lung tumorigenesis, since we found that these molecules were all required for lung cancer cell metastatic behavior. Interestingly, Hippo signaling has been implicated in regulation of expression of CD44, another surface marker of tissue-specific TPCs such as in breast cancer (Xu et al, 2010; Cordenonsi et al, 2011). Although there may be overlap between the markers and pathways responsible for metastasis in different tumor types, many combinations are likely utilized by aggressive cancer cells. Synergistic investigations into the phenotype of cells that are responsible for metastatic spread of lung cancer and the pathways that mediate this progression should ultimately lead to new ways to target the most functionally important and deadly lung cancer cells.
Materials and Methods
Generation of cell lines and in vitro assays
CK1750 and SC241 cells were generated by culturing tumor cells from a Kras;p53-flox donor mouse in DMEM + 10% FBS. Tmet cells were obtained from Monte Winslow (Winslow et al, 2011). These cells were infected with lentiviral constructs from The RNAi Consortium, pLKO.1 based system. The two hairpins directed at CD24a were TRCN0000077028 (shCD24a-1) and TRCN0000077032 (shCD24a-5), the two hairpins directed at Yap1 were TRCN0000095866 (Yap1108) and TRCN0000095864 (Yap1824), the two hairpins directed at Taz were TRCN0000095949 (Taz1616) and TRCN0000095951 (Taz771), with a non-targeting hairpin directed against GFP (Sigma SHC005) as a control. Transduced cells were selected with 3 μg/ml puromycin for 1–2 weeks. Knockdown was confirmed by quantitative RT-PCR and flow cytometry. To generate shCD24-Low-1 and shcD24-Low-5 cell lines, sorts for the lowest CD24-expressing cells were completed. All lines were maintained in DMEM + 10%FBS. 24-well format cell migration assays (Corning) were performed as per the manufacturer's instructions. Briefly, cells were serum-starved overnight, harvested with Accutase (Millipore) and counted. Cells were plated in serum-free conditions (30,000 cells per well) on the top chambers and the bottom chambers contained either serum-free DMEM or DMEM + 10% FBS. After a 24 h incubation, non-migratory cells were removed using a cotton swab, migratory cells were stained with DAPI for imaging and nuclei were counted using ImageJ. All conditions were completed with three to four replicates and averaged.
Mice and tissues
Lox-Stop-Lox-KrasG12D;p53fl/fl and Lox-Stop-Lox-KrasG12D mice (Jackson et al, 2001, 2005) were maintained in virus-free conditions on 129 SvJae background. Lox-Stop-Lox-KrasG12D;Lox-Stop-Lox-rtTA;tetO-YapS127A mice were maintained in virus-free conditions on a mixed 129/C57Bl6 background. All mouse experiments were approved by the BCH Animal Care and Use Committee and by the Dana-Farber Cancer Institute Institutional Animal Care and Use Committee, both accredited by AAALAC, and were performed in accordance with relevant institutional and national guidelines and regulations. Lung tissue preparation was as described (Curtis et al, 2010) and sections were analyzed for tumors by at least two investigators including a pathologist with expertise in murine lung cancer. Tumor burden, the percentage of lung filled with tumor, was calculated using ImageJ to measure total lung and tumor area from H&E stained paraffin sections from each mouse. Tumor size was measured using ImageJ to quantify tumor area from paraffin sections and the average area of all lung tumors per mouse was calculated. For metastases, heart, liver, spleen, kidneys, lymph nodes, and chest wall were also analyzed. Immunohistochemistry was performed as described (Schlegelmilch et al, 2011) with anti-Yap1 (1:50, Cell Signaling #4912) or anti-Ki67 (1:10,000, Novocastra) antibodies and developed using Vectastain Elite ABC kit (Vector Labs). Immunofluorescent staining for pro-SPC was performed with anti-pro-SPC (1:500, Chemicon). Imaging was performed with a Nikon 90i camera and NIS-Elements software and processed with NIS-Elements and Adobe Photoshop.
FACS analysis and sorting
Mice were euthanized with avertin overdose and lungs were dissected and examined grossly for tumor formation. Tumors were dissected from the lungs of primary mice and tumor tissue was prepared as described (Curtis et al, 2010). Briefly, tumors were isolated, minced, digested rotating for 1 h at 37°C with 2 mg/ml Collagenase/Dispase (Roche) and then filtered twice (100 μm, then 40 μm) after a 5-minute incubation with 0.025 mg/ml DNase. Cell lines were trypsinized and then filtered (40 μm). Single-cell suspensions were stained using Sca1-FITC, –PE, or APC-Cy7, CD45-APC, CD31-APC, CD24-Biotin (with Streptavidin-APC/Cy7) or –FITC (BD Pharmingen) with 7AAD (Molecular Probes) or DAPI (Sigma) staining to visualize dead cells. All antibodies were incubated for 15-20 minutes at 1:100 dilutions for primaries and 1:200 for secondaries. Cell sorting was performed with a Beckman Coulter/Cytomation MoFlo or a BD FACS Aria, and data were analyzed with FlowJo software (Tree Star, Inc.).
Tumor transplants
Intratracheal transplants were performed as described (Curtis et al, 2010). Mice were monitored for signs of lung tumor onset and euthanized for gross and histological analysis upon signs of distress. Cells for tail vein injections were resuspended in PBS and mice were sacrificed 2 weeks after injection for analysis of lung metastasis. Cells for subcutaneous injections were resuspended in a 1:1 mixture of PBS and matrigel (BD Biosciences), and subcutaneous tumor volume was measured (volume = length × width × depth).
Quantitative RT-PCR gene expression analysis
RNA from sorted cell populations was extracted from 8–12 tumors using the Stratagene Absolutely RNA Nanoprep Kit. cDNA was made using the SuperScript III kit (Invitrogen) and analyzed using TaqMan Assays (Applied Biosystems) for Acta2 (Mm00725412_s1), Bcl2 (Mm00477631_m1), Cd24a (Mm00782538_sH), Cdh1 (Mm01247357_m1), Cyr61 (Mm00487498_m1), Ltbp4 (Mm00723631_m1), Nf2 (Mm00477771_m1), Vim (Mm01333430_m1), Wwtr1 (Mm01289583_m1), Yap1 (Mm01143263_m1), or YAP1 (Hs00902712_g1), with a BioRad iQ5 iCycler or StepOnePlus™ Real-Time PCR System (Applied Biosystems) and software as per the manufacturer's recommendations. Mouse Actb (B-actin, 4352341E) or Gapdh (4352339E) was used as an endogenous control for normalization.
Microarray analysis
For Sca1+/Sca1− arrays, four primary tumors from Kras;p53-flox mice were dissociated and sorted into CD31−/CD45−/Sca1− and CD31−/CD45−/Sca1+ populations. RNA was isolated as above and subsequently amplified using the WT-Ovation Pico kit (NuGEN). The amplified cDNA was fragmented and biotin labeled using the FL-Ovation Biotin V2 kit (NuGEN). The Children's Hospital Boston Molecular Genetics Core Facility performed the hybridization and data acquisition using an Affymetrix Mouse Genome 430 2.0 expression array. Array normalization, expression value calculation and clustering analysis were performed using DNA-Chip Analyzer (http://www.dchip.org, Schadt et al, 2001). The Invariant Set Normalization method was used to normalize arrays at probe cell level to make them comparable, and the model-based method was used for probe-selection and computing expression values. These expression levels were attached with standard errors as measurement accuracy, which were subsequently used to compute 90% confidence intervals of fold changes between the Sca1+ and Sca1− groups. The lower confidence bounds of fold changes were conservative estimate of the real fold changes. Genes with more than 2-fold increased expression in Sca1+ cells relative to Sca1− cells and an associated P-value of 0.02 were selected for further study (Sca1+ up gene signature, Supplemental Data). The Affymetrix probe match tool was used for matching the Sca1+ up gene list to human U133A probes (Supplemental Data). Gene set enrichment analysis (GSEA) was performed by the Center for Computational Cancer Biology at the Dana Farber Cancer Institute with the pre-ranked implementation of the GSEA software package (Subramanian et al, 2005) using the moderated t-statistic from limma to determine rank order (Smyth, 2004).
shGFP/shTaz Arrays were performed in the Boston Children's Hospital Molecular Genetics Core facility on Affymetrix mouse Gene2.0ST slides. Array quality was assessed using the R/Bioconductor package (http://www.bioconductor.org/). Raw CEL files from U133A Affymetrix arrays were processed using the robust multi-array average (RMA) algorithm (Irizarry et al, 2003). To identify genes correlating with the phenotypic groups, we used limma (Smyth, 2004) to fit a statistical linear model to the data and then tested for differential gene expression in the two groups: shTaz (SC241, CK1750 and Tmet) vs shGFP (SC241, CK1750 and Tmet). GSEA (Subramanian et al, 2005) was performed using the javaGSEA desktop application on pre-ranked gene lists constructed from RMA normalized expression data.
Human tumor data Kaplan-Meier analysis
Raw gene expression data from human lung adenocarcinoma samples were obtained from publications (Director's Challenge, 2008; Chitale et al, 2009, http://cbio.mskcc.org/Public/lung_array_data/). Among these datasets, 3 are annotated for survival post biopsy while 1 dataset is annotated for both survival and metastatic spread of disease. Probes intensities from the Affymetrix U133A platform used in these studies were normalized and modeled using dChip software. Kaplan-Meier survival analyses were implemented after the samples were hierarchically clustered using centroid linkage into two risk groups using either the human orthologues of the Sca1+ up gene set or the Yap/Taz conserved gene set (Cordenonsi et al, 2011). Differences of the survival risk between the two risk groups were assessed using the Mantel-Haenszel log rank test. The larger area between the two risk groups and its associated smaller P-value from the Mantel-Haenszel log rank test implicate a better classification model. For the dataset for which metastatic spread was annotated, both survival and metastatic spread of the two risk groups were queried.
Statistical analysis
Except where indicated, Student's t-test was used to compare measurements between two conditions. For tumor cell transplants, data organized in tables was analyzed using Fisher's exact test. GraphPad Prism (GraphPad Software, Inc.), Excel and Mstat (McArdle Laboratory for Cancer Research, University of Wisconsin) were used for graphing and statistical analyses. Unless noted otherwise, pooled data is represented by the mean and standard deviation. P-values less than 0.05 with a one-tailed distribution were considered significant.
Acknowledgments
We thank the DFCI, BIDMC, and CHB HemOnc FACS facilities, M. Correll and S. Duan of the DFCI Center for Computational Cancer Biology, and members of the Kim Lab for technical assistance and discussions, R. Bronson for histopathology, L. Zon, C. Cepko, S. Armstrong, and W. Hahn for helpful discussions. This work was supported by DoD, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate Fellowships, 32 CFR 168a (to S.J.C. and A.N.L.), PF-09-121-01-DDC and PF-12-151-01-DMC from the American Cancer Society (to K.W.S. and C.M.F., respectively), the Ladies Auxilliary to the Veterans of Foreign Wars (to C.M.F.), Dana Farber Harvard Cancer Center Lung Cancer SPORE grant P50 CA090578, R01 AG2400401, R01 CA122794, R01 CA140594 (to K–K.W.), the V Foundation for Cancer Research, American Cancer Society Research Scholar Grant #RSG-08-082-01-MGO, the Freeman Trust, the Harvard Stem Cell Institute, NIH/NHLBI U01 HL100402 and R01 HL090136 and The Lung Cancer Alliance (to C.F.K.).
Author contributions
ANL, SJC, SPR, MM, DEW, DTM, KWS, and JB performed experiments and analyzed results. CMF performed bioinformatic analyses. ANL, AMB and JB managed mouse colony. ZEW, DJW, KKW, and FDC generated mice and/or reagents. ANL, SJC, DEW and CFK designed experiments. ANL and CFK wrote the article.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
for this article is available online: http://emboj.embopress.org
References
- Adhikari AS, Agarwal N, Iwakuma T. Metastatic potential of tumor-initiating cells in solid tumors. Front Biosci. 2011;16:1927–1938. doi: 10.2741/3831. [DOI] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S, Roz E, Caserini R, Lo Vullo S, Camerini T, Mariani L, Delia D, Calabro E, Pastorino U, Sozzi G. Highly tumorigenic lung cancer CD133 + cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, Houvenaeghel G, Extra JM, Bertucci F, Jacquemier J, Xerri L, Dontu G, Stassi G, Xiao Y, Barsky SH, Birnbaum D, Viens P, Wicha MS. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16:45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, Hung SC, Chang YL, Tsai ML, Lee YY, Ku HH, Chiou SH. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE. 2008;3:e2637. doi: 10.1371/journal.pone.0002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitale D, Gong Y, Taylor BS, Broderick S, Brennan C, Somwar R, Golas B, Wang L, Motoi N, Szoke J, Reinersman JM, Major J, Sander C, Seshan VE, Zakowski MF, Rusch V, Pao W, Gerald W, Ladanyi M. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;31:2773–2783. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, Basso G, Bicciato S, Piccolo S. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell. 2011;147:759–772. doi: 10.1016/j.cell.2011.09.048. [DOI] [PubMed] [Google Scholar]
- Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, Allan AL. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009;13:2236–2252. doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis SJ, Sinkevicius KW, Li D, Lau AN, Roach RR, Zamponi R, Woolfenden AE, Kirsch DG, Wong KK, Kim CF. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell. 2010;7:127–133. doi: 10.1016/j.stem.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damelin M, Geles KG, Follettie MT, Yuan P, Baxter M, Golas J, DiJoseph JF, Karnoub M, Huang S, Diesl V, Behrens C, Choe SE, Rios C, Gruzas J, Sridharan L, Dougher M, Kunz A, Hamann PR, Evans D, Armellino D, et al. Delineation of a cellular hierarchy in lung cancer reveals an oncofetal antigen expressed on tumor-initiating cells. Cancer Res. 2011;71:4236–4246. doi: 10.1158/0008-5472.CAN-10-3919. [DOI] [PubMed] [Google Scholar]
- Dieter SM, Ball CR, Hoffman CM, Nowrouzi A, Herbst F, Zavidij O, Abel U, Arens A, Weichert W, Brand K, Koch M, Weitz J, Schmidt M, Kalle von C, Glimm H. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell. 2011;9:357–365. doi: 10.1016/j.stem.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Director's Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma. Shedden K, Taylor JM, Enkemann SA, Tsao M, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giodano TJ, Misek DE, Chang AC, Zhu CQ, Strumpf D, Hanash S, Shepherd FA, Ding K, Seymour L, Naoki K, Pennell N, Weir B, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–827. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Virgilio Di A, Conticello C, Ruco L, Peschle C, Maria De R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- Gunawardane RN, Sgroi DC, Wrobel CN, Koh E, Daley GQ, Brugge JS. Novel role for PDEF in epithelial cell migration and invasion. Cancer Res. 2005;65:11572–11580. doi: 10.1158/0008-5472.CAN-05-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Ignatius MS, Chen E, Elpeck NM, Fuller AZ, Tenente IM, Clagg R, Liu S, Blackburn JS, Linardic CM, Rosenberg AE, Nielsen PG, Mempel TR, Langenau DM. In Vivo imaging of tumor-propagating cells, regional tumor heterogeneity, and dynamic cell movements in embryonal rhabdomyosarcoma. Cancer Cell. 2012;21:680–693. doi: 10.1016/j.ccr.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkaya H, Paulso A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27:6120–6130. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen G, Schluns K, Yongwei Y, Denkert C, Dietel M, Petersen I. CD24 is an independent prognostic marker of survival in nonsmall cell lung cancer patients. Br J Cancer. 2003;88:231–236. doi: 10.1038/sj.bjc.6600702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Choe G, Jheon S, Sung SW, Lee CT, Chung JH. CD24, a novel cancer biomarker, predicting disease-free survival of non-small cell lung carcinomas: a retrospective study of prognostic factor analysis from the viewpoint of forthcoming (seventh) new TNM classification. J Thorac Oncol. 2010;5:649–657. doi: 10.1097/JTO.0b013e3181d5e554. [DOI] [PubMed] [Google Scholar]
- Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9:50–63. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS ONE. 2008;3:e3077. doi: 10.1371/journal.pone.0003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Monahan KB, Pfefferle AD, Shimamura T, Sorrentiono J, Chan K, Roadcap DW, Ollila DW, Thomas NE, Castrillon DH, Miller CM, Perour CM, Wong KK, Bear JE, Sharpless NE. LKB1/STK11 inactivation leads to expansion of a pro-metastatic tumor sub-population in melanoma. Cancer Cell. 2012;21:751–764. doi: 10.1016/j.ccr.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-KappaB signaling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani A, Nagase T, Fukuchi K, Aburatani H, Makita R, Kurihara H. Transcriptional coactivator with PDZ-binding motif is essential for normal alveolarization in mice. Am J Respir Crit Care Med. 2009;180:326–338. doi: 10.1164/rccm.200812-1827OC. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, Gerald WL, Massague J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62. doi: 10.1016/j.cell.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overdevest JB, Thomas S, Kristiansen G, Hansel DE, Smith SC, Theodorescu D. CD24 offers a therapeutic target for control of bladder cancer metastasis based on a requirement for lung colonization. Cancer Res. 2011;71:3802–3811. doi: 10.1158/0008-5472.CAN-11-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK, Ng L, Cheung LW, Lan XR, Lan HY, Tan VP, Yau TC, Poon RT, Wong BC. A subpopulation of CD26 + cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell. 2010;6:603–615. doi: 10.1016/j.stem.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Patsialou A, Wang Y, Lin J, Whitney K, Goswami S, Kenny PA, Condeelis JS. Selective gene-expression profiling of migratory tumor cells in vivo predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012;14:R129. doi: 10.1186/bcr3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisters KM, Ginsberg RJ, Giroux DJ, Putnam JB, Kris MG, Johnson DH, Roberts JR, Mault J, Crowley JJ, Bunn PA. Induction chemotherapy before surgery for early-stage lung cancer: A novel approach. Bimodality Lung Oncology Team. J Thorac Cardiovasc Surg. 2000;119:429–439. doi: 10.1016/s0022-5223(00)70120-6. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Li C, Ellis B, Wong WH. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. J Cell Biochem Suppl. 2001;37:120–125. doi: 10.1002/jcb.10073. [DOI] [PubMed] [Google Scholar]
- Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell. 2011;144:782–795. doi: 10.1016/j.cell.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dong Q, Zhang Q, Li Z, Wang E, Qiu X. Overexpression of yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010;101:1279–1285. doi: 10.1111/j.1349-7006.2010.01511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, Yoon S, Crowley D, Bronson RT, Chiang DY, Meyerson M, Jacks T. Suppression of lung adenocarcinoma progression by Nk2–1. Nature. 2011;473:101–104. doi: 10.1038/nature09881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Siadaty MS, Berens ME, Hampton GM, Theodorescu D. Overlapping gene expression profiles of cell migration and tumor invasion in human bladder cancer identify metallothionein 1E and nicotinamide N-methyltransferase as novel regulators of cell migration. Oncogene. 2008;27:6679–6689. doi: 10.1038/onc.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Zhang L, He CS, Hou JH, Lin SX, Hu ZH, Xu F, Zhao HY. Prognostic significance of TAZ expression in resected non-small cell lung cancer. J Thorac Oncol. 2012;7:799–807. doi: 10.1097/JTO.0b013e318248240b. [DOI] [PubMed] [Google Scholar]
- Xu Y, Stamenkovic I, Yu Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010;70:2455–2464. doi: 10.1158/0008-5472.CAN-09-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Meylan E, Oliver TG, Feldser DM, Winslow MM, Bronson R, Jacks T. Response and resistance to NF-kB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. 2011;1:236–247. doi: 10.1158/2159-8290.CD-11-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Labelle M, Lamb CR, Dugan JM, Williamson CA, Spencer DR, Christ KR, Keating RO, Lee WD, Paradis GA, Begum S, Hynes RO, Wittrup KD. Determination of 35 cell surface antigen levels in malignant pleural effusions identifies CD24 as a marker of disseminated tumor cells. Int J Cancer. 2013;133:2925–2933. doi: 10.1002/ijc.28312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Cruz de la CC, Sayles LC, Alleyne-Chin C, Vaka D, Knaak TD, Bigos M, Xu Y, Hoang CD, Shrager JB, Fehling HJ, French D, Forrest W, Jiang Z, Carano RA, Barck KH, Jackson EL, Sweet-Cordero EA. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch 3 for self-renewal. Cancer Cell. 2013;24:59–74. doi: 10.1016/j.ccr.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hao Y, Liu N, Raptis L, Tsao MS, Yang X. TAZ is a novel oncogene in non-small cell lung cancer. Oncogene. 2011;30:2181–2186. doi: 10.1038/onc.2010.606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.