

Abstract
Lysosomal Ca2+ homeostasis is implicated in disease and controls many lysosomal functions. A key in understanding lysosomal Ca2+ signaling was the discovery of the two-pore channels (TPCs) and their potential activation by NAADP. Recent work concluded that the TPCs function as a PI(3,5)P2 activated channels regulated by mTORC1, but not by NAADP. Here, we identified Mg2+ and the MAPKs, JNK and P38 as novel regulators of TPC2. Cytoplasmic Mg2+ specifically inhibited TPC2 outward current, whereas lysosomal Mg2+ partially inhibited both outward and inward currents in a lysosomal lumen pH-dependent manner. Under controlled Mg2+, TPC2 is readily activated by NAADP with channel properties identical to those in response to PI(3,5)P2. Moreover, TPC2 is robustly regulated by P38 and JNK. Notably, NAADP-mediated Ca2+ release in intact cells is regulated by Mg2+, PI(3,5)P2, and P38/JNK kinases, thus paralleling regulation of TPC2 currents. Our data affirm a key role for TPC2 in NAADP-mediated Ca2+ signaling and link this pathway to Mg2+ homeostasis and MAP kinases, pointing to roles for lysosomal Ca2+ in cell growth, inflammation and cancer.
Keywords: channel, lysosome, Mg2+, NAADP, TPC2
Introduction
The pleiotropic effects of Ca2+ dictates highly compartmentalized and regulated cellular Ca2+ pools. Ca2+ is stored and released by several organelles, including the ER, mitochondria (Kiselyov et al, 2006; Clapham, 2007), and most notably the acidic endolysosomes (Galione & Ruas, 2005; Patel et al, 2010; Morgan et al, 2011; Patel & Muallem, 2011). In addition to regulating several endolysosomal functions, such as apoptosis (Kroemer et al, 2010), trafficking (Saftig & Klumperman, 2009), energy metabolism (Jewell et al, 2013) and fusion/fission events (Kiselyov et al, 2012), endolysosomal Ca2+ serves as a trigger that sensitizes Ca2+ release by neighboring IP3 (Morgan et al, 2013) and ryanodine (Kinnear et al, 2004) receptors, possibly at membrane contact sites (Kilpatrick et al, 2013). Thus, understanding endolysosomal Ca2+ homeostasis is central to understanding endolysosomal function and cellular Ca2+ homeostasis.
At present, lysosomal Ca2+ homeostasis is poorly understood. Although it is established that lysosomal Ca2+ uptake depends on the lysosomal H+ gradient, the transporter(s) coupling H+ and Ca2+ transport is not known. The mechanism of Ca2+ release from the lysosomes is being studied more extensively since the seminal discoveries of the potent second messenger NAADP (Lee & Aarhus, 1995) and its ability to release Ca2+ from lysosomes (Churchill et al, 2002). Subsequently, the action of NAADP has been demonstrated in virtually all cell types examined and shown to mediate several lysosomal-related functions (Guse & Lee, 2008).
The identity of the channel activated by NAADP remains controversial. The major Ca2+-permeable channels localized to the lysosomes are TRPML1 (Kiselyov et al, 2005; Pryor et al, 2006) and two pore channel TPC2 (Brailoiu et al, 2009; Calcraft et al, 2009). Although when activated pharmacologically, TRPML1 releases Ca2+ from an acidic compartment (Shen et al, 2012) consistent with its identity as the NAADP-activated channel (Zhang et al, 2011), other studies have ruled out its role in NAADP signaling (Yamaguchi et al, 2011). A breakthrough was made with the discovery of Two Pore channels (TPCs) as the more likely NAADP targets (Brailoiu et al, 2009; Calcraft et al, 2009; Zong et al, 2009). This is based on: (i) loss of response to NAADP in cells in which TPC expression was knocked-down with RNA interference (Brailoiu et al, 2009; Calcraft et al, 2009; Dionisio et al, 2011) and cells expressing channel-dead dominant-negative (DN) TPCs (Brailoiu et al, 2009; Pereira et al, 2011), (ii) a large increase in Ca2+ release and higher apparent affinity for NAADP in cells transfected with TPC1 or TPC2 (Brailoiu et al, 2009, 2010; Pereira et al, 2011; Yamaguchi et al, 2011), (iii) activation of ion currents by NAADP in bilayers reconstituted with TPC1 (Rybalchenko et al, 2012), TPC2 (Pitt et al, 2010; Schieder et al, 2010) and plasma membrane (PM) patches obtained from cells expressing PM-targeted TPC2 (Brailoiu et al, 2010) and (iv) most notably, elimination of the response to NAADP in cells from TPC2 knockout mice (Calcraft et al, 2009; Tugba Durlu-Kandilci et al, 2010).
Nevertheless, two recent studies challenged a role of the TPCs in NAADP-mediated Ca2+ release. The first study reported that TPCs functions as a Na+-selective channels that are activated by the endolysosomal phospholipid phosphatidylinositol-3,5-bisphosphate (PI(3,5)P2) (Wang et al, 2012). Recording TPC2 channel activity in lysosomes enlarged by treatment with vacuolin or TPC2 targeted to the PM, the authors failed to activate the channel by NAADP. In addition, in cells from a TPC1 and TPC2 double knockout mouse line, Ca2+ release by NAADP remained intact, while the PI(3,5)P2-activated Na+ current was inhibited. In the second study, it was reported that the PI(3,5)P2-activated TPCs currents are inhibited by the mTORC1 kinase complex (Cang et al, 2013).
The unexpected finding that the TPCs may not function in the NAADP response in spite of the numerous studies linking TPCs to NAADP, prompted us to re-examine the properties and regulation of TPC2. We show that TPC2 is activated by PI(3,5)P2 and is Na+-permeable. We discovered profound regulation of TPC2 by Mg2+ ions whereby cytosolic Mg2+ selectively inhibits TPC2 outward current while lysosomal Mg2+ modestly inhibits both the outward and inward currents. In the absence of Mg2+, NAADP readily activates TPC2, with properties similar to PI(3,5)P2. Further, we report novel regulation of TPC2 by P38 and JNK kinases. Notably, changes in PI(3,5)P2, Mg2+ and the activity of JNK and P38 kinases function modified NAADP-mediated Ca2+ release similar to their effect on TPC2 current. These findings help to resolve conflicting views regarding activation of TPCs and its role in lysosomal Ca2+ signaling and reveal novel regulatory modes of major relevance to lysosome function.
Results
Regulation of TPC2 by Mg2+
TPC2 was reported to function as a PI(3,5)P2-activated channel when in lysosomes enlarged by vacuolin treatment to facilitate electrophysiological recording (Wang et al, 2012). We adopted the same approach to characterize the properties of TPC2. Fig 1A shows the effect of vacuolin on the subcellular distribution of TPC2-GFP expressed in COS7 cells. Mechanical release of the enlarged lysosomes and recording of whole lysosomal current using symmetrical Na+ solutions confirmed activation of a TPC2-mediated Na+ current by PI(3,5)P2 (Fig 1B). During the course of our studies, we noted that TPC2 is strongly inhibited by cytoplasmic Mg2+ (Mg2+cyt). Intriguingly, Mg2+cyt predominantly inhibited outward currents (Na+ flowing from the cytoplasm into the lysosome, see Bertl et al, 1992 for definition) (Fig 1B and C). At 2 mM, Mg2+cyt inhibited the PI(3,5)P2-activated outward current by 74 ± 11% and the inward current by 23 ± 7% (n = 6). To independently confirm these results we re-routed TPC2 to the PM by mutation of a lysosomal, targeting sequence in TPC2L11L12/AA (Fig 1D). In cells expressing high levels of the mutant channel, it was present either predominantly in the plasma membrane (PM) or in both the PM and attached large vesicular structures. Patches excised from the two cell types showed the same Na+ current, although the current from the cells containing the vesicular structures was higher. Mg2+cyt preferentially inhibited PI(3,5)P2-mediated TPC2 outward currents in PM patches (Fig 1E and F). Inhibition was concentration-dependent (Fig 1G) and was fitted well by a single exponential equation with a Hill coefficient of 0.8 and an apparent affinity for Mg2+cyt of 0.13 mM (Fig 1H), well within the physiological range of free Mg2+cyt of 0.2–0.5 mM (Gunther, 2006).
Figure 1.
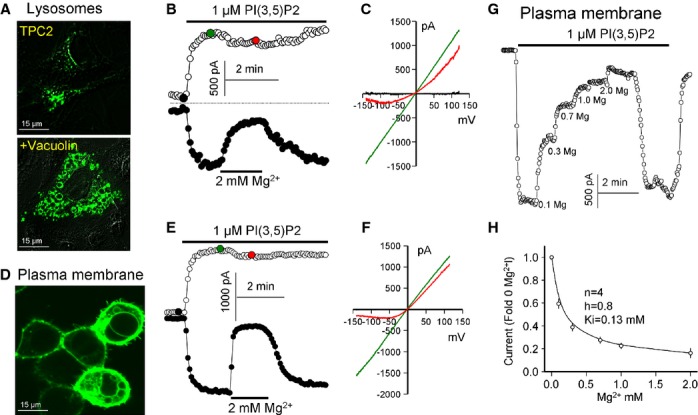
Regulation of PI(3,5)P2-activated TPC2 current by Mg2+cyt.
A The images show localization of TPC2-GFP expressed in COS-7 cells with the lower cell treated with 1 μM vacuolin for 1 h.
B, C TPC2 current measured in enlarged lysosome extracted from COS-7 cell treated with vacuolin. At the time indicated by the bar, the lysosome was exposed to 1 μM of water-soluble PI(3,5)P2 analogue, which activated TPC2. After maximal TPC2 current activation, the lysosome was exposed to 2 mM Mg2+cyt. (C) shows the I/V at the time indicated by the filled circles in (B). The mean ± s.e.m. is given in the text.
D An image of HEK cells transfected with the plasma membrane (PM)-targeted GFP-TPC2L11L12/AA mutant. Note the presence of TPC2-expressing vesicles attached to the PM in the cells expressing high level of TPC2. These cells were used for most experiments with excised PM patches since patches from these cells responded to NAADP.
E, F Excised PM patch expressing TPC2 was activated by 1 μM PI(3,5)P2 and inhibited by 2 mM Mg2+cyt. (F) shows the I/Vs at the times indicated in (E) by the filled circles.
G TPC2 in a PM patch was activated by 1 μM PI(3,5)P2. After maximal current activation the bath (cytoplasmic face) was perfused with solutions containing the indicated concentrations of Mg2+.
H Mg2+cyt dependence of TPC2 inhibition. The results were fitted by a Hill equation and the fitted parameters are listed in the figure.
To further investigate the effects of Mg2+, we examined whether TPC2 is regulated by luminal Mg2+ (Mg2+lys). At 2 mM Mg2+lys, both outward and inward PI(3,5)P2-mediated currents in excised PM patches were reduced equally by about 50% (Fig 2A–C). Mg2+cyt still specifically inhibited the residual outward current (Fig 2A–C). Since the lysosomal lumen is acidic, we also tested the effect of lysosomal pH (pHlys) on the inhibition by Mg2+lys and Mg2+cyt. Reducing pHlys to 6.5 had no effect on the extent of the current (Fig 2A and D), the shape of the I/V curves (Supplementary Fig S1) or the Na+/K+ selectivity (Fig 2D). However, under these conditions, inhibition by Mg2+lys was completely relieved whereas inhibition by Mg2+cyt was unaffected (Fig 2E and F). Together, data in Figs 1 and 2 reveal a novel role for Mg2+ in regulating TPC2 activity.
Figure 2.
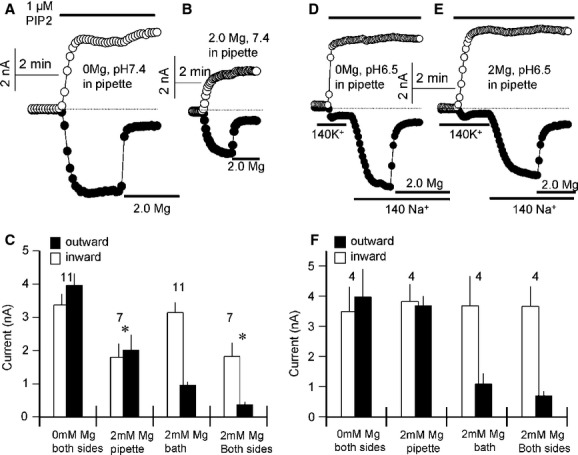
Regulation of PI(3,5)P2-activated TPC2 current by Mg2+lys and pHlys.
All experiments were with excised patches from HEK cells expressing TPC2.
A, B, D, E The pipette solution (equivalent to lysosomal lumen) was Mg2+-free, pH 7.4 in (A), contained 2 mM Mg2+, pH 7.4 in (B), was Mg2+-free, pH 6.5 in (D) and contained 2 mM Mg2+, pH 6.5 in (E). In all experiments TPC2 was activated by 1 μM PI(3,5)P2 and after maximal activation of the current the patches were exposed to a bath solution containing 2 mM Mg2+ to assay inhibition by Mg2+cyt. In (D, E) the experiments started with bath solution containing 140 mM K+ that was then replaced with solutions containing 140 mM Na+.
C Summary of the indicated number of experiments similar to those in (A, B).
F Summary of the indicated number of experiments similar to those in (D, E) * denotes P < 0.05 for the outward current relative to 0 mM Mg2+, pH 7.4 pipette conditions as in (A).
TPC2 is regulated by NAADP in a Mg2+-sensitive manner
In light of conflicting reports regarding the sensitivity of TPCs to NAADP and the inhibitory effects of Mg2+ on TPC activity reported here, we re-examined the effects of NAADP on TPC2 activity. Importantly, we demonstrate using three experimental paradigms that NAADP activates a current similar to PI(3,5)P2.
First, in 42 out of 44 lysosome recordings from six independent transfections, NAADP (100 nM) evoked a Na+ current (Fig 3A). A higher concentration of NAADP (1 μM), also evoked a current but in 8 out of 20 experiments the responses were transient (Supplementary Fig S3A), perhaps indicative of desensitization (Aarhus et al, 1996; Genazzani et al, 1996). When the current was sustained, channel activity was lost upon washout of NAADP (Supplementary Fig S3B). Second, NAADP-mediated channel activity could also be recorded in PM patches (Fig 3D). The success rate was lower (146/318) and activation by NAADP was observed mostly in cells expressing high levels of TPC2L11L12/AA (Fig 1D). The excised patches were used to determine the dependence of the current on NAADP concentration. Variability of the current (Fig 3C, left panel) precluded accurate determination, although when normalized to the current activated by PI(3,5)P2 in the same patches, the current could be activated by as low as 3 nM NAADP (Fig 3C, right panel), consistent with the high affinity for activation of Ca2+ release by NAADP (Galione & Ruas, 2005; Patel et al, 2010). The difference between the currents activated by the indicated NAADP concentrations did not reach statistical significance and for 3/10 nM, 3/1 μM, and 10/1 μM it was between P = 0.09–0.13. The magnitude of the currents however were only approximately 10% of those evoked by PI(3,5)P2 (Fig 3C).Third, infusion of NAADP (1 μM) evoked a Na+ current in whole cell recording (Fig 3F and G) similar to PI(3,5)P2 (Fig 3D and E).
Figure 3.
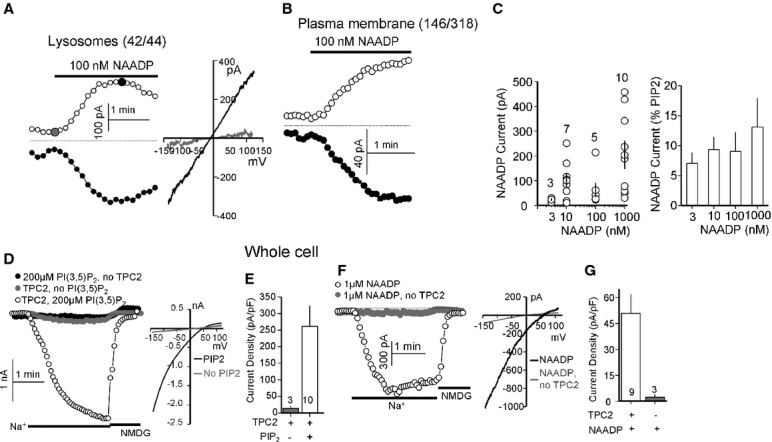
Activation of TPC2 by NAADP
A Lysosomes expressing TPC2 were stimulated with 100 nM NAADP. The I/V at the times indicated by the closed circles. Activation by NAADP was observed in 42 out of 44 lysosomes.
B PM patches expressing TPC2 were stimulated with 100 nM NAADP. Current activation was observed in 146/318 patches.
C Excised PM patches were used to determine the dose response for activation of TPC2 by NAADP. The individual currents are shown in the scatter plot (left panel) and the averaged currents normalized to the current activated by PI(3,5)P2 is shown in the columns (right panel).
D, E The whole-cell Na+ current was measured in non-transfected HEK cells infused with 200 μM PI(3,5)P2 (•) or HEK cells expressing TPC2 and infused with buffer ( ) or 200 μM PI(3,5)P2 (○). (E) shows the mean ± s.e.m. of the indicated number of experiments similar to those in (D).
) or 200 μM PI(3,5)P2 (○). (E) shows the mean ± s.e.m. of the indicated number of experiments similar to those in (D).
F, G The whole-cell Na+ current was measured upon infusion of either non-transfected () or TPC2-expressing (○) cells with 1 μM NAADP. (G) shows the mean ± s.e.m. of the indicated number of experiments similar to those in (F).
Importantly, NAADP-evoked currents were regulated by Mg2+ independently of the experimental method used. Thus, the outward lysosomal current was inhibited by Mg2+cyt (100%, n = 4; Fig 4A) and selective for Na+ over K+ (Fig 4B and C). NAADP-mediated currents in excised PM patches were also blocked by Mg2+cyt with an apparent affinity of 60 μM (Fig 4D and E). However, this value is less reliable than that obtained with PI(3,5)P2 (Fig 1) due to the small and variable current. Finally, whole-cell currents in response to both NAADP and PI(3,5)P2 were also Mg2+-sensitive (Supplementary Fig S2). Intracellular Mg2+ (equivalent to Mg2+cyt) inhibited the outward current by more than 98% and the inward current by about 20% (Supplementary Fig S2A and B). Extracellular Mg2+ (equivalent to Mg2+lys) inhibited both currents by about 20% (Supplementary Fig S2C and D). We noted that in these recordings, the outward current was smaller than the inward current. The reason for this is not clear at the present time but may reflect channel regulation that is not observed in excised patches or isolated lysosomes. Nevertheless, regulation by Mg2+ is clearly evident. Taken together, these findings show that under defined ionic conditions, TPC2 can be readily activated by both NAADP and PI(3,5)P2.
Figure 4.
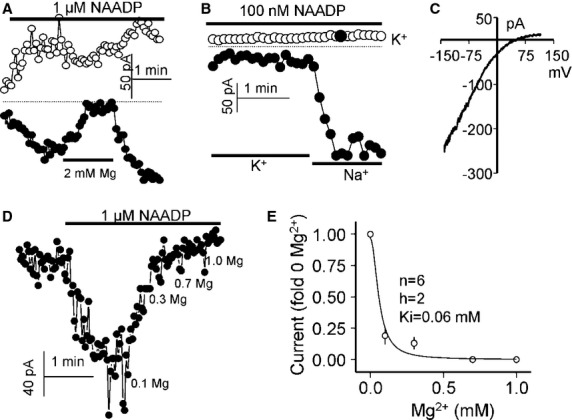
Selectivity and Mg2+cyt dependence of the NAADP-activated TPC2 current.
A Lysosomes expressing TPC2 were stimulated with 1 μM NAADP and then exposed to 2 mM Mg2+, which completely inhibited the outward and partially the inward currents.
B, C TPC2 current in endolysosomes activated by NAADP is highly Na+/K+-selective. (C) shows the I/Vs at the time indicated by the closed circle in the upper symbols.
D, E Shows the Mg2+cyt dependence of the NAADP-activated TPC2 current in endolysosomes. The results in (E) were fitted with a Hill equation and the parameters are listed in the Figure.
TPC2 is regulated by multiple protein kinases
A recent study showed that the mTORC1 kinase complex inhibits TPC2 activity (Cang et al, 2013). In accord, we found that in lysosomes expressing TPC2 and PM patches expressing TPC2AA, MgATP induced a time-dependent inhibition of PI(3,5)P2 currents (Supplementary Fig S4). This effect was slowed by chemical (rapamycin) and molecular (overexpression of DN mTORC1) inhibition of mTORC1 but unaffected by overexpression of wild-type mTORC1 (Supplementary Fig S4). The relatively modest effect of mTORC1 manipulation on the inhibitory effects of MgATP prompted us to explore the role of other protein kinases in regulating TPC2 activity. We focused on MAP kinases given their established roles in regulating lysosomal functions such as apoptosis, autophagy and energy metabolism (Wagner & Nebreda, 2009; Settembre et al, 2013). In an initial screen using MAP kinase inhibitors at concentrations five times the reported IC50, blockade of MAP2K and MEKs had no effect of the rate or extent of TPC2 inhibition by MgATP. Inhibition of the MgATP response was observed with the inhibitors of JNK and P38 and was thus characterized in detail.
The effect of the kinases was evaluated both in lysosomes and PM patches. The JNK inhibitor SP600125 (SP60) markedly delayed inhibition of TPC2 currents by MgATP in lysosomes (Fig 5A) and PM patches (Fig 5C). A summary of pooled data quantifying the extent and time constants for inhibition by MgATP under the various conditions is provided in Fig 5B and D. Importantly, expression of DN-JNK strongly suppressed inhibition whereas expressing wild-type JNK had the opposite effect (Fig 5A and C). In PM patches from cells overexpressing high levels of JNK, PI(3,5)P2-mediated currents in the absence of MgATP were substantially lower than in control cells presumably reflecting phosphorylation of TPC2 in intact cells. Consistent with this notion, pre-treating cells with 0.4 μM SP60 prior to recording of PI(3,5)P2-mediated currents reversed the inhibitory effects of JNK overexpression (Supplementary Fig S5A and B). These data reveal a novel role for JNK in regulating TPC2.
Figure 5.
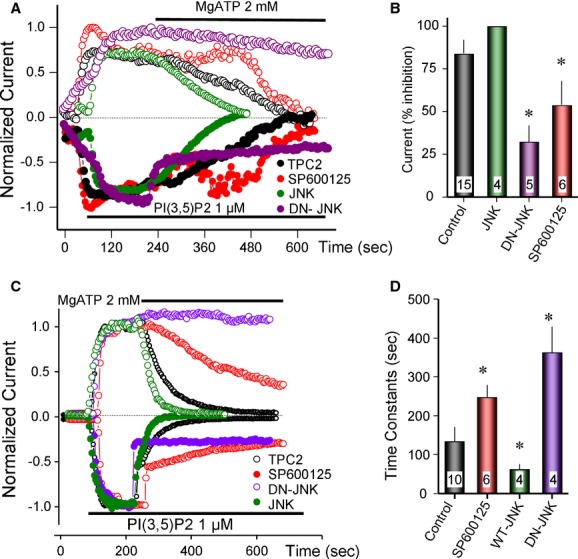
Regulation of TPC2 by JNK.
A, B All experiments are with TPC2 in lysosomes. The effect of JNK and DN-JNK was tested by transfecting the cells for 24 h with 1 μg/ml TPC2 and the kinases. The JNK inhibitor SP600125 was used at 0.4 μM (five times the Ki) and lysosomes were treated with the inhibitor for about 1–3 min before exposure to MgATP. The current was activated by 1 μM PI(3,5)P2 and after maximal current was stable, the lysosomes were exposed to 2 mM MgATP. The outward current (filled symbols) was partially inhibited by the free Mg2+ in the MgATP mixture. The summary of current inhibition by MgATP is given in (B) and the results are given as the mean ±s.e.m. of the listed number of experiments. * denotes P < 0.05 relative to control. Note the inhibition of the MgATP effect by SP600125 and DN-JNK and its acceleration by JNK.
C, D Experiments similar to those in (A, B) using TPC2 in excised PM patches. The effect of MgATP was determined by calculating the time constants for inhibition by MgATP. The summary of current inhibition is given in (D) and the results are given as the mean ± s.e.m. of the listed number of experiments. * denotes P < 0.05 relative to control. Note the increased time constant by SP600125 and DN-JNK and reduced time constant by JNK.
Similar experiments manipulating P38 activity are shown in Fig 6. As with JNK, chemical inhibition of P38 with 1.5 μM SB202190 (SB20) or overexpression of DN-P38 suppressed inhibition of PI(3,5)P2 currents by MgATP. When TPC2 was targeted to the PM, expression of P38 strongly inhibited the current in the absence of MgATP even when it was transfected at low levels (0.2 μg/ml). The inhibition was reversed by inhibition of P38 with SB20 (Supplementary Fig S5B). In experiments in which the current was inhibited by only about 50% by P38, it accelerated the inhibition of TPC2 by MgATP (normalized green trace in Fig 6C, and summary in D). Together, the results in Figs 5 and 6 and Supplementary Fig S4 indicate that TPC2 function is regulated by multiple protein kinases.
Figure 6.
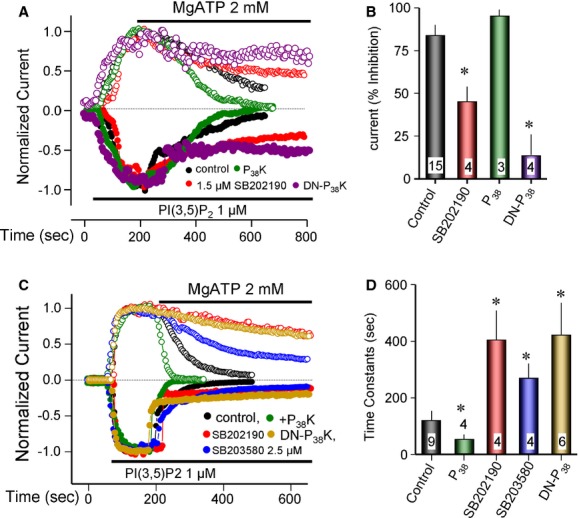
Regulation of TPC2 by the P38 MAP kinase.
A, B All experiments are with TPC2 in lysosomes. The transfection with P38 and DN-P38 and all experimental protocols are exactly as described in Fig 5 for JNK, except that the P38 inhibitor SB202190 was used at 1.5 μM (five times the Ki) and transfection was with 0.2 μg/ml P38 cDNA. After maximal current activation by 1 μM PI(3,5)P2 lysosomes were treated with 2 mM MgATP. The summary of current inhibition by MgATP is given in (B) and the results are given as the mean ± s.e.m. of the listed number of experiments. * denotes P < 0.05 relative to control. Note the inhibition of the MgATP effect by SB202190 and DN-P38 and its acceleration by P38.
C, D The experiments are with TPC2 in excised PM patches. Expression of P38 markedly reduced the basal current. In four experiments there was a residual current, the extent of which is shown for one cell in the green trace of Supplementary Fig S5B. (C) shows the normalized currents. Inhibition of the residual current by MgATP was accelerated by P38. The DN-P38 strongly inhibited the effect of MgATP. Inhibition of the MgATP effect was also observed with two P38 inhibitors that act by different mechanisms, SB202190 and SB203580. The summary of current inhibition by MgATP is given in (D) and the results are given as the mean ± s.e.m. of the listed number of experiments. * denotes P < 0.05 relative to control.
Regulation of the NAADP-mediated Ca2+ signals by PI(3,5)P2, Mg2+ and protein kinases
To determine the physiological significance of the present findings in intact cells and determine whether TPC2 currents and Ca2+ mobilizing activity of NAADP are related, we examined the effect of manipulating PI(3,5)P2, Mg2+, JNK and P38 levels on NAADP-mediated Ca2+ release. Fig 7A and B show that inhibition of PI(3,5)P2 synthesis by YM201636 (an inhibitor of the organellar PI3P kinase PIKfyve) inhibited endogenous NAADP-mediated Ca2+ signals in SKBR3 cells (Fig 7A). YM201636 also inhibited NAADP responses in cells transfected with TPC2 (Fig 7B). Conversely, increasing endolysosomal PI(3,5)P2 by expression of PIKfyve (Ho et al, 2012) increased the extent and duration of endogenous NAADP-evoked Ca2+ signals (Fig 7A). Increasing cytoplasmic Mg2+ to between about 0.3–1 mM dose-dependently inhibited the response to NAADP (Fig 7C). Notably, expression of the JNK and P38 kinases also inhibited, while expression of dominant-negative JNK or P38 increased the extent and duration of the NAADP-mediated Ca2+ release (Fig 7D). Similarly, treating the cells with the JNK and P38 kinase inhibitors augmented the NAADP response (Supplementary Fig S6). Hence, the TPC2 current and NAADP-mediated Ca2+ release are similarly regulated by PI(3,5)P2, Mg2+ and protein kinases.
Figure 7.
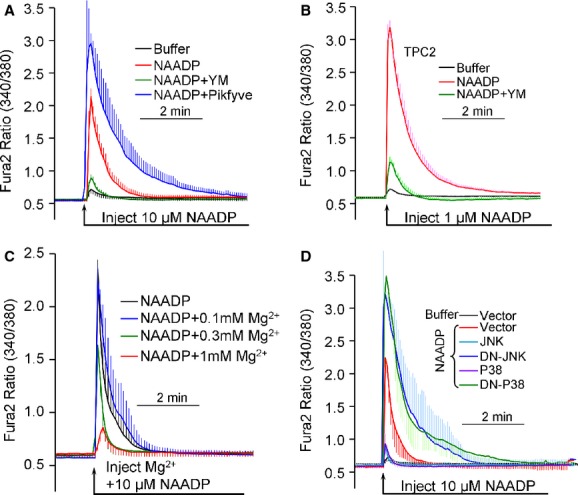
- SKBR3 cells loaded with Fura2 were injected with intracellular-like buffer (black trace) or buffer containing 10 μM NAADP to result in a final concentration of about 100 nM (red trace). The cells were treated with 1 μM YM201636 for 2 h to deplete intracellular PI(3,5)P2 (green trace) or transfected with Pikfyve for 24 h to increase intracellular PI(3,5)P2 (blue trace) before injection with NAADP.
- Experiments as in (A), except that the SKBR3 cells were transfected with TPC2 and the injected buffer contained 1 μM NAADP, resulting in a final concentration of about 10 nM.
- Experiments were as in (A), except that the cells were injected with solution containing 10 μM NAADP and either 0 (control, black trace), 30 (blue trace), 100 (green trace) or 300 (red trace) mM MgCl2 to yield a final concentrations of about 100 nM NAADP and 0.1, 0.3 or 1 mM free Mg2+, respectively.
- SKBR3 cells were transfected with empty vector (black and red traces), JNK (Turquoise trace), DN-JNK (blue trace), P38 (purple trace) or DN-P38 (green trace) for 24 h before injection with buffer (black trace) of 10 μM NAADP (all other traces). In all experiments the traces are the mean ± s.e.m of 6–8 injections.
Discussion
In addition to their canonical function in protein turnover, lysosomes play additional key roles in processes ranging from cellular energetics to cell death (Settembre et al, 2013). Lysosomal Ca2+ homeostasis is increasingly implicated in lysosomal function and dysfunction (Galione & Ruas, 2005; Patel et al, 2010; Patel & Muallem, 2011). In particular there has been much interest in the Ca2+ mobilizing messenger NAADP which releases Ca2+ from these acidic Ca2+ stores in a TPC-dependent manner (Brailoiu et al, 2009; Calcraft et al, 2009). Although multiple lines of evidence link NAADP-mediated Ca2+ release and the TPCs, the molecular mechanism underlying lysosomal Ca2+ release has proven controversial (Wang et al, 2012; Cang et al, 2013) and we know little of how this event is regulated.
Several discoveries in the present study, in particular inhibition of TPC2 by Mg2+ and multiple protein kinases, provide new insight into regulation of TPC2. We confirmed activation of TPC2 by PI(3,5)P2 and its permeability to Na+ (Fig 1). Notably, we found that in the absence of Mg2+, TPC2 is readily activated by NAADP. The properties of the current activated by PI(3,5)P2 and NAADP were highly similar. Thus, activation of the Na+ current by NAADP could be observed only in lysosomes and PM patches obtained from cells transfected with TPC2 (Fig 3), and the current was partially inhibited by Mg2+lys and strongly by Mg2+cyt (Figs 1, 2 and 4, Supplementary Fig S2). The current activated by NAADP and PI(3,5)P2 is selective for Na+ over K+ (Fig 4). In this respect, when reconstituted into bilayers, the TPC2 current activated by NAADP conducted K+ (Pitt et al, 2010) or even showed high Ca2+ selectivity (Schieder et al, 2010). This suggests that the lipid environment may affect channel selectivity. Interestingly, activation by NAADP was more consistent when the current was recorded in lysosomes compared to plasma membrane patches (Fig 3). This observation may relate to the finding that NAADP likely binds to small proteins that associate with TPCs (Lin-Moshier et al, 2011; Walseth et al, 2011). Redirecting TPC2 to the PM may therefore partially dissociate it from these regulatory proteins thus accounting for variability.
Another feature of our study is convergent regulation of TPC2 currents and NAADP-evoked Ca2+ release in intact cells by PI(3,5)P2, Mg2+ and kinases. These data provide fresh evidence to support a role for TPCs in NAADP action. Interestingly, the response to NAADP in intact cells was augmented by increasing cellular PI(3,5)P2 and inhibited by depletion of PI(3,5)P2 (Fig 7). Hence, although PI(3,5)P2 can fully activate TPC2 when applied to the cytoplasmic face of the channel under defined recording conditions, it is possible that in intact cells PI(3,5)P2 has a permissive effect promoting activation of the channel by NAADP. Changes in endolysosomal PI(3,5)P2 levels may thus modulate the size of the NAADP response. This topic needs further examination.
Considering the similarities in the regulation of NAADP-mediated Ca2+ release and the NAADP-activated TPC2 current, at present however, it is not easy to explain loss of TPCs current but retention of the NAADP-evoked Ca2+ responses in cells from TPC1/2 double knockout mice (Wang et al, 2012). Wang et al deleted only a portion of the N termini of the TPCs. Whether this resulted in complete deletion of the channels or simply redirected them to either the PM or the ER, given the presence of targeting information in these regions (Brailoiu et al, 2010; Churamani et al, 2013), remains to be established. In addition, Wang et al used a high concentration of NAADP-AM to characterize NAADP-evoked Ca2+ signals. Under these conditions, this compound may cause non-specific Ca2+ release from stores (Lu et al, 2013). In this respect, it is of note that responses to NAADP were eliminated in cells from an independent TPC2 knockout mouse line (Calcraft et al, 2009; Tugba Durlu-Kandilci et al, 2010).
Recent work suggests that lysosomes maintain high Na+ levels (Wang et al, 2012) and that activation of TPC2 by PI(3,5)P2 hyperpolarize the lysosomes (Cang et al, 2013), raising the question of the role of NAADP and TPC2 in Ca2+ release. Lysosomal hyperpolarization by NAADP is not likely to cause Ca2+ release since hyperpolarization should inhibit the release. TPC2 itself may mediate the Ca2+ release. Several studies have reported that TPCs are permeable to Ca2+ (Brailoiu et al, 2010; Pitt et al, 2010; Rybalchenko et al, 2012), although the selectivity for Ca2+ relative to Na+ appears to be low (∼1:10) (Wang et al, 2012). However, the situation with TPC2 may be similar to the NMDA receptors, where both Na+ and modest Ca2+ fluxes are physiologically relevant (Verkhratsky & Kirchhoff, 2007). In addition, we showed previously that NAADP could cause Ca2+ influx in cells expressing PM-targeted TPC2 in the face of 140 mM extracellular Na+ (Brailoiu et al, 2010). The preliminary results in Supplementary Fig S7 show that PI(3,5)P2 is also able to mediate Ca2+ influx through TPC2 and this influx is potentiated when extracellular Na+ is lowered. Significantly, Ca2+ influx by activated TPC2 is comparable or higher than Ca2+ influx by PM-targeted TRPML1 which may release Ca2+ from the lysosomes (Dong et al, 2010). Hence, even limited permeability of TPC2 to Ca2+ may be sufficient to evoke global Ca2+ signals in an intact cell setting given intimate functional coupling between TPCs and ER Ca2+ channels. This is particularly relevant if such coupling occurs at microdomains (Kilpatrick et al, 2013). Secondary activation of other channels and/or transporters upon lysosomal hyperpolarization in response to NAADP however cannot be ruled out at this stage.
A key finding of the present work described in Figs 4 and Supplementary Fig S2 is the novel and complex regulation of TPC2 by Mg2+. Both, Mg2+lys and Mg2+cyt affect TPC2 activity. The equal inhibition of the outward and inward currents by Mg2+lys suggests a general channel block that might be due to pore blockade, similar to the block of NMDA receptors by Mg2+ (Verkhratsky & Kirchhoff, 2007). Luminal lysosomal H+ relieve inhibition of both inward and outward currents by Mg2+lys, probably by dissociating Mg2+. These experiments also showed that channel conductance and selectivity appears to be unaffected by luminal pH, at least as low as pH 6.5 (Fig 2 and Supplementary Fig S1).
Arguably more interesting is regulation of TPC2 by Mg2+cyt that selectively inhibits outward Na+ currents. The inhibition occurs with an apparent affinity of 0.06–0.13 mM. This is within the cytoplasmic free Mg2+ concentration which is 0.19–0.5 mM (Gunther, 2006). Hence small changes in Mg2+cyt markedly affect the activity of TPC2 and thus the lysosomal membrane potential. Decrease in Mg2+cyt will depolarize the lysosomal membrane potential to affect the transport of all electrogenic coupled and uncoupled transporters, including facilitation of Ca2+ release from the lysosomes. Thus, our findings indicate that a change in Mg2+cyt is a key regulator of lysosomal function. Several forms of cell stimulation that increase Ca2+ or cAMP cause an increase or a decrease in cytoplasmic Mg2+ that are associated with changes in anabolic and catabolic functions and cell growth (Gunther, 2006). The finding of regulation of TPC2 by Mg2+cyt may provide the link between the associated regulation of lysosomal and cellular functions.
The present work revealed that TPC2 is regulated by multiple protein kinases, thus implicating TPC2 in diverse cellular functions. The reported inhibition of TPC2 by mTORC1 linked TPC2 to energy metabolism (Cang et al, 2013). Although we could confirm inhibition of TPC2 by mTORC1, the inhibition was modest (Supplementary Fig S2), leading us to identify prominent regulation by JNK and P38 (Figs 5 and 6). These kinases play central roles in cell proliferation, differentiation and cancer. JNK and P38 are structurally and enzymatically similar (Johnson & Lapadat, 2002) that may account for their similar regulatory effects on TPC2, although they have largely different cellular functions. The most prominent function of P38 linked to lysosomes is its role in inflammation and associated autophagy (Cuenda & Rousseau, 2007). Many cell stressors that induce apoptosis do so by acting on P38 (Wagner & Nebreda, 2009). The main function of the JNKs is in cell proliferation through transcriptional activation (Jaeschke et al, 2006). In addition, JNKs also regulate autophagy through phosphorylation of BCL-2 to release it from Beclin 1 to control Beclin 1 transcription (Mehrpour et al, 2010). It is thus possible that lysosomal P38 and JNK inhibit TPC2 to control lysosomal membrane potential and Ca2+ release (see Fig 7) to inhibit autophagy. Indeed, NAADP/TPC2 has recently been shown to regulate autophagy (Pereira et al, 2011; Lu et al, 2013).
In summary, the present work revealed novel regulatory modalities of the endolysosomal channel TPC2 by Mg2+ and multiple protein kinases. These forms of regulation should have profound effect on the function of the lysosome. The protein kinases appear to inhibit the function of TPC2 on a time scale of many minutes and thus can set the steady-state function of the lysosomes in response to inputs related to inflammatory cell stressors (P38), transcriptional demand (JNK), induction of apoptosis (P38 and JNK) or nutrient supply (mTORC1). Regulation by Mg2+ is on a sub-seconds time scale and is most suited to control the lysosomal membrane potential and thus its ionic fluxes and content. Further studies are required to determine how each of the regulatory modalities comes into play during various cellular demands for lysosomal function.
Materials and Methods
Reagents and solutions
The GFP-TPC2 clone was described elsewhere (Yamaguchi et al, 2011) and GFP-TPC2-L11L12/AA mutant was generated by site directed mutagenesis. Several clones were obtained from Addgene (cat no.) mTORC1 (26603), DN-mTORC1 (26605), JNK (19731) DN-JNK (19730), P38 (20355), DN-P38 (20356). The Pikfyve clone was from Open Biosystems (MHS1010-202696678). The MAP kinase inhibitors kit was from Tocris. The PI(3,5)P2 kinase inhibitor YM201636 and PI(3,5)P2 were from Cayman Chemical. NAADP and vacuolin were from Sigma. The PI(3,5)P2 carrier and lipid soluble PI(3,5)P2 kit were from Echelon Biosciences. For recording lysosomal current the standard pipette (luminal) solution contained (mM) 140 NaCl, 5 KCl, 10 HEPES (pH 7.4 with NaOH) and the standard bath (cytoplasmic) solution contained 140 NaCl, 5 KCl or 140 KCl, 10 HEPES (pH 7.4 with NaOH or KOH). As needed, Mg2+ was added to either or to both solutions. The same solutions were used to record current in excised plasma membrane (PM) patches, except that where indicated the pipette (luminal) solution was adjusted to pH 7.4 or 6.5. The whole-cell current was recorded with pipette solution containing (mM) either 140 Na+-Gluconate or Cs+-MeSO4, 10 NaCl or CsCl, 10 HEPES (pH 7.4 with NaOH or CsOH), 0.39 Ca2+, 1 EGTA (final free Ca2+ of 100 nM) and with or without 1 Mg2+ and with or without 200 μM PI(3,5)P2 or 1 μM NAADP. The bath solution contained 140 Na+-Gluconate or NMDG+-Gluconate, 5 KCl, 10 HEPES (pH 7.4 with NaOH or Tris base).
Cells
HEK293T and COS-7 cells were maintained in DMEM and 10% fetal bovine serum supplemented with penicillin (100 units/ml)/streptomycin (100 μg/ml), and the cells were maintained in a water-saturated 5% CO2, 95% air atmosphere. Cells were transiently transfected with GFP-TPC2/GFP-TPC2-L11L12/AA with and without the kinases using Lipofectamine 2000 and were used 24–36 h post-transfection. In experiments with cells transfected with P38 or high level of JNK kinases that strongly inhibited the basal TPC2 activity, the cells were also treated with the respective kinase inhibitors for 2 h before current measurement to demonstrate reversal of the inhibition. Lysosomes were isolated from COS-7 cells transfected with GFP-TPC2 and the indicated kinases. Lysosomes were liberated mechanically and patched within 1–2 min of release. Plasma membrane patches were excised from HEK cells transfected with GFP-TPC2 and the kinases. SKBR3 were maintained in McCoy's 5A modified media and 10% fetal bovine serum. Cells were transiently transfected using Turbofectin 8. Cells were used 48–72 h after transfection.
Current recording
Whole lysosomal current recordings were performed using a modified patch clamp method (Dong et al, 2010). Transfected COS-7 cells were released and replated on Petri dishes and incubated with culture media for 1 h to allow attachment. Then the cells were treated with 1 μM vacuolin-1, for 1–2 h. Patch pipettes had a resistance of 5–8 MΩ when filled with the pipette solution. Enlarged lysosomes were released by quickly pressing patch pipettes against the cell and pulling back. The enlarged lysosomes were identified by monitoring GFP fluorescence. After formation of a GΩ seal, capacitance transients were compensated by Axopatch 200B patch clamp amplifier. For breaking into the lysosomes, quick voltage steps of positive 350 mV were applied. The whole lysosomal configuration was confirmed by reappearance of the capacitance transient. The current was recorded by 400-ms rapid alterations of membrane potential (RAMP) from −150 to +120 mV from a holding potential of 0 mV at 4 s intervals. The whole-lysosomal currents were filtered at 1 kHz with an internal four-pole Bessel filter, sampled at 5 kHz, and stored directly to a hard drive by Digidata 1322. Whole-cell current was recorded from transfected HEK293T cells using standard technique. Patch pipettes were heat-polished and had a resistance of 3–5 MΩ when filled with the pipette solution. Current was measured with an Axopatch 200B patch clamp amplifier that was controlled by pCLAMP9 software to allow the delivery of voltage-step protocols with concomitant digitization. The currents were sampled at 5 kHz and filtered at 1 kHz. Currents were obtained by voltage ramps with command voltage from −150 to +120 mV over 400 or 200 ms every 4 s from a holding potential of 0 mV. Inside-out patches were excised from HEK cells with pipettes having resistance of 3–5 MΩ. After formation of a GΩ seals patches were pulled quickly away from the cell. The current was recorded by 400-ms RAMPs from −150 to +120 mV from a holding potential of 0 mV at 4 s intervals.
Ca2+ measurements
For measurement of Ca2+ influx in HEK cells transfected with GFP-TPC2-L11L12/AA or mCherry-TRPML1(ΔNΔC), the cells were incubated with 1 μM PI(3,5)P2, 0.3 μM carrier or PI(3,5)P2 and carrier for 1 h at 37°C in Ca2+-free DMEM solution containing 0.2 mM EGTA. During the last 20 min the cell were loaded with Fura2 and Ca2+ was measured as the 340/380 ratio. External Na+ was reduced by replacing NaCl with NMDG-Cl. Ca2+ measurement in SKBR3 cells was as before (Brailoiu et al, 2009). Fura2 loaded cells on coverslips were mounted in an open chamber in a Nikon Eclipse TiE microscope equipped with a Perfect Focus System and a Photometrics CoolSnap HQ2 CCD camera. TPC2-transfected cells were identified by the YFP fluorescence and when TPC2 was not expressed, the P38 and JNK kinases and their mutants were co-expressed with YFP to identify the transfected cells. Fura2 fluorescence was acquired at a frequency of 0.25 Hz and images were analyzed using NIS-Elements AR software. Injections were as before (Brailoiu et al, 2009). Pipettes were back-filled with an intracellular solution composed of (mM) 110 KCl, 10 NaCl, and 20 HEPES (pH 7.2) and with or without NAADP and Mg2+. The cells to be injected were Z scanned and the cellular volume was calculated using Nikon NIS-Elements AR software prior to injection. The injection time was 0.4 s at 60 h Pascal with a compensation pressure of 20 h Pa to maintain the microinjected volume to < 1% of cell volume.
Statistics
All experiments were repeated at least three times and the results are given as means ± s.e.m. Differences between the groups were analyzed for statistical significance by un-paired Student's t-test. P < 0.05 or better was considered statistically significant.
Acknowledgments
This work was supported in part by National Institutes of Health Grants HL090804 to E.B. and by NIH/NIDCR intramural grant DE000735-03 to S.M.
Author contributions
AJ, MA and EB performed and analyzed the experiments, SP provided essential material, EB and SM designed and directed the studies, SM drafted the manuscript with input from all authors.
Conflict of interest
All authors declare that they have no conflict of interests.
Supplementary information
for this article is available online: http://emboj.embopress.org
References
- Aarhus R, Dickey DM, Graeff RM, Gee KR, Walseth TF, Lee HC. Activation and inactivation of Ca2+ release by NAADP+ J Biol Chem. 1996;271:8513–8516. doi: 10.1074/jbc.271.15.8513. [DOI] [PubMed] [Google Scholar]
- Bertl A, Blumwald E, Coronado R, Eisenberg R, Findlay G, Gradmann D, Hille B, Kohler K, Kolb HA, MacRobbie E, Meinsser G, Miller C, Neher E, Palade P, Pantoja O, Sanders D, Schroeder J, Slayman C, Spanswick R, Walker A, et al. Electrical measurements on endomembranes. Science. 1992;258:873–874. doi: 10.1126/science.1439795. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–209. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu E, Rahman T, Churamani D, Prole DL, Brailoiu GC, Hooper R, Taylor CW, Patel S. An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J Biol Chem. 2010;285:38511–38516. doi: 10.1074/jbc.M110.162073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang C, Zhou Y, Navarro B, Seo YJ, Aranda K, Shi L, Battaglia-Hsu S, Nissim I, Clapham DE, Ren D. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell. 2013;152:778–790. doi: 10.1016/j.cell.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churamani D, Hooper R, Rahman T, Brailoiu E, Patel S. The N-terminal region of two-pore channel 1 regulates trafficking and activation by NAADP. Biochem J. 2013;453:147–151. doi: 10.1042/BJ20130474. [DOI] [PubMed] [Google Scholar]
- Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111:703–708. doi: 10.1016/s0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Dionisio N, Albarran L, Lopez JJ, Berna-Erro A, Salido GM, Bobe R, Rosado JA. Acidic NAADP-releasable Ca(2+) compartments in the megakaryoblastic cell line MEG01. Biochim Biophys Acta. 2011;1813:1483–1494. doi: 10.1016/j.bbamcr.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galione A, Ruas M. NAADP receptors. Cell Calcium. 2005;38:273–280. doi: 10.1016/j.ceca.2005.06.031. [DOI] [PubMed] [Google Scholar]
- Genazzani AA, Empson RM, Galione A. Unique inactivation properties of NAADP-sensitive Ca2+ release. J Biol Chem. 1996;271:11599–11602. doi: 10.1074/jbc.271.20.11599. [DOI] [PubMed] [Google Scholar]
- Gunther T. Concentration, compartmentation and metabolic function of intracellular free Mg2+ Magnes Res. 2006;19:225–236. [PubMed] [Google Scholar]
- Guse AH, Lee HC. NAADP: a universal Ca2+ trigger. Sci Signal. 2008;1:re10. doi: 10.1126/scisignal.144re10. [DOI] [PubMed] [Google Scholar]
- Ho CY, Alghamdi TA, Botelho RJ. Phosphatidylinositol-3,5-bisphosphate: no longer the poor PI. Traffic. 2012;2:1–3. doi: 10.1111/j.1600-0854.2011.01246.x. [DOI] [PubMed] [Google Scholar]
- Jaeschke A, Karasarides M, Ventura JJ, Ehrhardt A, Zhang C, Flavell RA, Shokat KM, Davis RJ. JNK2 is a positive regulator of the cJun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133–139. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci. 2013;126:60–66. doi: 10.1242/jcs.118836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnear NP, Boittin FX, Thomas JM, Galione A, Evans AM. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem. 2004;279:54319–54326. doi: 10.1074/jbc.M406132200. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, Soyombo A. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280:43218–43223. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Wang X, Shin DM, Zang W, Muallem S. Calcium signaling complexes in microdomains of polarized secretory cells. Cell Calcium. 2006;40:451–459. doi: 10.1016/j.ceca.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Kiselyov KK, Ahuja M, Rybalchenko V, Patel S, Muallem S. The intracellular Ca(2)(+) channels of membrane traffic. Channels. 2012;6:344–351. doi: 10.4161/chan.21723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Aarhus R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem. 1995;270:2152–2157. doi: 10.1074/jbc.270.5.2152. [DOI] [PubMed] [Google Scholar]
- Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S, Marchant JS. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem. 2011;287:2296–2307. doi: 10.1074/jbc.M111.305813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YY, Hao BX, Graeff R, Wong CW, Wu WT, Yue J. TPC2 signaling inhibits autophagosomal-lysosomal fusion by alkalizing lysosomal pH. J Biol Chem. 2013;288:24247–24263. doi: 10.1074/jbc.M113.484253. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Davis LC, Wagner SK, Lewis AM, Parrington J, Churchill GC, Galione A. Bidirectional Ca(2)(+) signaling occurs between the endoplasmic reticulum and acidic organelles. J Cell Biol. 2013;200:789–805. doi: 10.1083/jcb.201204078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J. 2011;439:349–374. doi: 10.1042/BJ20110949. [DOI] [PubMed] [Google Scholar]
- Patel S, Marchant JS, Brailoiu E. Two-pore channels: regulation by NAADP and customized roles in triggering calcium signals. Cell Calcium. 2010;47:480–490. doi: 10.1016/j.ceca.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Muallem S. Acidic Ca(2+) stores come to the fore. Cell Calcium. 2011;50:109–112. doi: 10.1016/j.ceca.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Pereira GJ, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, Stilhano RS, Ureshino RP, Bloor-Young D, Churchill G, Piacentini M, Patel S, Smaili SS. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J Biol Chem. 2011;286:27875–27881. doi: 10.1074/jbc.C110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt SJ, Funnell TM, Sitsapesan M, Venturi E, Rietdorf K, Ruas M, Ganesan A, Gosain R, Churchill GC, Zhu MX, Parrington J, Galione A, Sitsapesan R. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+ J Biol Chem. 2010;285:35039–35046. doi: 10.1074/jbc.M110.156927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor PR, Reimann F, Gribble FM, Luzio JP. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic. 2006;7:1388–1398. doi: 10.1111/j.1600-0854.2006.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalchenko V, Ahuja M, Coblentz J, Churamani D, Patel S, Kiselyov K, Muallem S. Membrane potential regulates nicotinic acid adenine dinucleotide phosphate (NAADP) dependence of the pH-and Ca2+-sensitive organellar two-pore channel TPC1. J Biol Chem. 2012;287:20407–20416. doi: 10.1074/jbc.M112.359612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- Schieder M, Rotzer K, Bruggemann A, Biel M, Wahl-Schott CA. Characterization of two-pore channel 2 (TPCN2)-mediated Ca2+ currents in isolated lysosomes. J Biol Chem. 2010;285:21219–21222. doi: 10.1074/jbc.C110.143123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugba Durlu-Kandilci N, Ruas M, Chuang KT, Brading A, Parrington J, Galione A. TPC2 proteins mediate nicotinic acid adenine dinucleotide phosphate (NAADP)-and agonist-evoked contractions of smooth muscle. J Biol Chem. 2010;285:24925–24932. doi: 10.1074/jbc.M110.129833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Kirchhoff F. NMDA receptors in glia. Neuroscientist. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Walseth TF, Lin-Moshier Y, Jain P, Ruas M, Parrington J, Galione A, Marchant JS, Slama JT. Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide 2′-phosphate (NAADP) proteins in sea urchin egg. J Biol Chem. 2011;287:2308–2315. doi: 10.1074/jbc.M111.306563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, Xu H. TPC proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell. 2012;151:372–383. doi: 10.1016/j.cell.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Jha A, Li Q, Soyombo AA, Dickinson GD, Churamani D, Brailoiu E, Patel S, Muallem S. Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J Biol Chem. 2011;286:22934–22942. doi: 10.1074/jbc.M110.210930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Xu M, Han WQ, Li PL. Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(−/−) cells. Am J Physiol Cell Physiol. 2011;301:C421–C430. doi: 10.1152/ajpcell.00393.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rotzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C. The two-pore channel TPCN2 mediates NAADP-dependent Ca(2+)-release from lysosomal stores. Pflugers Arch. 2009;458:891–899. doi: 10.1007/s00424-009-0690-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.