

Abstract
The dynamic network of mitochondria fragments under stress allowing the segregation of damaged mitochondria and, in case of persistent damage, their selective removal by mitophagy. Mitochondrial fragmentation upon depolarisation of mitochondria is brought about by the degradation of central components of the mitochondrial fusion machinery. The OMA1 peptidase mediates the degradation of long isoforms of the dynamin-like GTPase OPA1 in the inner membrane. Here, we demonstrate that OMA1-mediated degradation of OPA1 is a general cellular stress response. OMA1 is constitutively active but displays strongly enhanced activity in response to various stress insults. We identify an amino terminal stress-sensor domain of OMA1, which is only present in homologues of higher eukaryotes and which modulates OMA1 proteolysis and activation. OMA1 activation is associated with its autocatalyic degradation, which initiates from both termini of OMA1 and results in complete OMA1 turnover. Autocatalytic proteolysis of OMA1 ensures the reversibility of the response and allows OPA1-mediated mitochondrial fusion to resume upon alleviation of stress. This differentiated stress response maintains the functional integrity of mitochondria and contributes to cell survival.
Keywords: mitochondria, mitochondrial fusion, OMA1, OPA1, stress
Introduction
Mitochondria continuously divide and fuse. This dynamic behaviour is required for the inheritance of mitochondria, ensures mitochondrial trafficking and the distribution of mitochondria-derived metabolites throughout the cell and contributes to mitochondrial quality control (Chen & Chan, 2010; Westermann, 2010; Nunnari & Suomalainen, 2012; Youle & van der Bliek, 2012; Anand et al, 2013). Fusion and fission of mitochondrial membranes are mediated by dynamin-like GTPases that act on both the mitochondrial inner (IM) and outer membrane (OM; Hoppins et al, 2007; Ishihara et al, 2013). Mutations in these GTPases are associated with various neurodegenerative disorders highlighting the importance of mitochondrial dynamics for neuronal survival (Knott & Bossy-Wetzel, 2008; Chan, 2012).
Both mitochondrial fusion and fission are highly regulated in response to various physiological cues. Starvation and stress conditions inhibit mitochondrial fission and induce the fusion of mitochondrial membranes to form a highly interconnected network (Tondera et al, 2009; Gomes et al, 2011; Rambold et al, 2011). On the other hand, severe mitochondrial damage and depolarisation impair fusion (Duvezin-Caubet et al, 2006; Ishihara et al, 2006; Cereghetti et al, 2008). Unopposed fission leads to fragmentation of the mitochondrial network under these conditions, which allows the selective autophagic removal of damaged mitochondria and which is associated with necrotic and apoptotic cell death (Twig & Shirihai, 2011; Youle & Narendra, 2011; Youle & van der Bliek, 2012).
The dynamin-like GTPase OPA1 mediates the fusion of the mitochondrial IM and maintains cristae morphology (Olichon et al, 2003; Cipolat et al, 2004; Griparic et al, 2004; Frezza et al, 2006; Merkwirth et al, 2008; Song et al, 2009). Mounting evidence revealed that its activity is regulated in response to mitochondrial stress (Duvezin-Caubet et al, 2006; Ishihara et al, 2006). Under normal conditions, proteolytic cleavage of long isoforms of OPA1 (L-OPA1) results in the balanced accumulation of long and short OPA1 (S-OPA1) forms (Ishihara et al, 2006; Griparic et al, 2007; Song et al, 2007). Constitutive processing of OPA1 is required to maintain a normal morphology of mitochondria. However, mitochondrial depolarisation induces the complete conversion of L-OPA1 into S-OPA1 (Duvezin-Caubet et al, 2006; Ishihara et al, 2006; Song et al, 2007; Guillery et al, 2008). Stress-induced OPA1 processing inhibits fusion and causes mitochondrial fragmentation and the segregation of damaged mitochondria (Twig et al, 2008). A tubular mitochondrial network is restored upon removal of stress stimuli (Ishihara et al, 2006; Griparic et al, 2007; Guillery et al, 2008; Head et al, 2009), whereas irreversibly damaged mitochondria are selectively degraded by mitophagy if stress persists (Narendra et al, 2008; Twig et al, 2008). The plasticity of the mitochondrial network thus allows a differentiated response to mitochondrial dysfunction and is central to maintain the functional integrity of mitochondria.
A number of proteases have been linked to constitutive and stress-induced OPA1 processing. These include the i-AAA protease YME1L that constitutively cleaves L-OPA1 isoforms at the proteolytic site S2 (Griparic et al, 2007; Song et al, 2007; Stiburek et al, 2012), while the metallopeptidase OMA1 processes OPA1 at site S1 (Ehses et al, 2009; Head et al, 2009; Quiros et al, 2012). OMA1 was first identified in yeast as a peptidase with overlapping activities with the m-AAA protease and was shown to degrade misfolded membrane proteins (Käser et al, 2003; Khalimonchuk et al, 2012). Members of the OMA1 family of membrane-embedded peptidases contain a conserved M48 metallopeptidase domain [according to the MEROPS database, (Rawlings et al, 2012)] and are found in eukaryotes as well as eubacteria. The analysis of OMA1-deficient mice revealed an unexpected metabolic phenotype characterised by hepatic steatosis, decreased energy expenditure and impaired thermogenic regulation, pointing to important functions of stress-induced OPA1 processing for metabolic homeostasis (Quiros et al, 2012).
It is presently not understood how the activity of OMA1 is regulated to allow an immediate and reversible response to mitochondrial stress. Various stress stimuli were found to trigger mitochondrial fragmentation affecting the membrane potential across the inner membrane (Δψ) and intramitochondrial ATP levels (Baricault et al, 2007), but the role of OMA1 under these conditions remained speculative. Here, we demonstrate a general role of OMA1 for stress-induced OPA1 processing and identify a critical sensory region outside the metallopeptidase domain of OMA1, which is required for OMA1 activation. Activation of OMA1 is accompanied by its autocatalytic turnover ensuring the reversibility of the stress response.
Results
OMA1 mediates constitutive and stress-induced OPA1 processing
At least five different forms of OPA1 accumulate in mouse embryonic fibroblasts (MEFs; Griparic et al, 2007; Guillery et al, 2008; Ishihara et al, 2006; Fig 1A): long (L) forms a and b derived from OPA1 splice variants 1 and 7, respectively, and short (S) forms c, d and e that are generated upon proteolytic processing of L-OPA1 forms at sites S1 and S2. Cleavage of L-OPA1 forms a and b at S1 results in the formation of c and e, respectively, while processing at S2 generates d. To unambiguously establish the role that OMA1 plays in the processing of OPA1, we monitored the accumulation of OPA1 forms in MEFs isolated from Oma1−/− mice by immunoblotting. Both L-and S-OPA1 forms accumulated in Oma1−/− cells; however, the proteolytic forms c and e were absent (Fig 1B). Thus, OMA1 mediates constitutive OPA1 processing of both splice variants 1 and 7 at S1. The morphology of the mitochondrial network appears largely normal in MEFs lacking OMA1 (Ehses et al, 2009; Head et al, 2009; Quiros et al, 2012; see also Fig 1G), suggesting that OPA1 forms c and e are dispensable for mitochondrial fusion.
Figure 1.

- Linear depiction of the OPA1 splice variants 1 and 7. L-OPA1 splice variant 1 (b) and splice variant 7 (a) are shown with cleavage sites for OMA1 (S1) or YME1L (S2). Processing generates S-OPA1 form e (OMA1) or S-OPA1 forms c (OMA1) and d (YME1L). MTS, mitochondrial targeting sequence; TM, transmembrane domain.
- OMA1 is required for inducible and constitutive OPA1 processing. Oma1+/+ and Oma1−/− MEFs expressing OMA1-myc or OMA1E324Q-myc as indicated were incubated in the absence or presence of CCCP for 2 h.
- Stress-induced OPA1 processing by OMA1. Oma1+/+ and Oma1−/− MEFs were incubated with CCCP, valinomycin, oligomycin (Oligo) and/or antimycin A (Anti) for 1 h.
- Determination of ΔΨ in Oma1+/+ MEFs under various stress conditions by JC-1 staining. Untreated MEFs were used as a control.
- Heat stress-induced OPA1 processing depends on OMA1. Oma1+/+ and Oma1−/− MEFs were cultured at 42°C for the times indicated.
- H2O2 induces OMA1-mediated OPA1 processing. Oma1+/+ and Oma1−/− MEFs were incubated in the presence of H2O2 (0.5 mM) for the times indicated. In all panels, cellular proteins were extracted and analysed by SDS–PAGE and immunoblotting. a-e, OPA1 forms.
G,H The mitochondrial network in Oma1−/− MEFs is protected against H2O2-induced fragmentation. Oma1+/+ and Oma1−/− MEFs were incubated for 4 h in the presence or absence of H2O2 as indicated. Mitochondrial morphology was visualised by immunofluorescence microscopy using cytochrome c-specific antibodies. Scale bar, 15 μm (inset 5 μm). A quantification of mitochondrial morphology in 100 cells is shown in (H).
Source data are available online for this figure.
Depolarisation of the mitochondrial membrane potential (ΔΨ) using the ionophore carbonyl cyanide m-chlorophenylhydrazone (CCCP) results in loss of L-OPA1 in an OMA1-dependent manner (Fig 1B; Ehses et al, 2009; Head et al, 2009; Quiros et al, 2012), suggesting that OMA1 regulates L-OPA1 stability in response to the bioenergetic status of mitochondria in the cell. To further define conditions for OMA1 activation, we first examined the role of the electrical component (ΔV) of ΔΨ. Treatment of cells with valinomycin, a potassium ionophore dissipating ΔΨ similar to CCCP, triggered proteolysis of L-OPA1 in an OMA1-dependent fashion (Fig 1C). Addition of antimycin A, which blocks transfer of electrons from cytochrome b to cytochrome c, did not efficiently stimulate OPA1 processing by OMA1, while treatment of the cells with oligomycin, which inhibits the ATP synthase, stimulated OMA1-mediated OPA1 processing at least to some extent under the conditions used (Fig 1C). However, addition of antimycin A and oligomycin in combination destabilised L-OPA1 and resulted in a more efficient degradation of L-OPA1 by OMA1 (Fig 1C). These results indicate that the loss of the proton motive force and the collapse of ΔΨ activate OMA1 and lead to the degradation of L-OPA1. However, OMA1 activation does not strictly correlate with dissipation of ΔΨ (Fig 1D). In agreement with previous findings (Baricault et al, 2007), oligomycin treatment stimulated proteolysis of L-OPA1, but does not significantly affect ΔΨ. Conversely, ΔΨ was lost, but L-OPA1 remained stable upon short incubation with antimycin A (Fig 1D). Similar effects on ΔΨ were observed in Oma1−/− MEFs (Supplementary Fig S1A).
To ascertain whether OMA1 is activated following other types of stress, we analysed L-OPA1 stability under heat stress. In agreement with recent observations (Sanjuan Szklarz & Scorrano, 2012), incubation of MEFs at 42°C accelerated processing of L-OPA1 (Fig 1E). However, OPA1 processing was completely abolished in Oma1−/− MEFs (Fig 1E), demonstrating that heat stress activates OMA1. Heat stress did not dissipate ΔΨ as revealed by TMRM staining of mitochondria (Supplementary Fig S1B). We also addressed whether reactive oxygen species (ROS) affect OMA1 activity. Following the addition of H2O2 to induce ROS, we observed loss of ΔΨ (Fig 1D), OMA1-dependent proteolysis of L-OPA1 (Fig 1F) and mitochondrial fragmentation (Fig 1G and H). OMA1 activation under oxidative stress was further substantiated incubating wild-type and OMA1-deficient MEFs with paraquat or rotenone (Supplementary Fig S2).
We conclude from these experiments that OMA1 mediates both constitutive and stress-induced OPA1 processing. Multiple stress conditions that impinge on mitochondria activate OMA1, which degrades L-OPA1 and causes fragmentation of the mitochondrial network. These findings identify stress sensing by OMA1 as a critical cellular response to dysfunction of mitochondria.
OPA1 processing by OMA1 in the intermembrane space of mitochondria
To define the localisation of OMA1 within mitochondria, we exploited a lentiviral system to express OMA1 or C-terminally myc-tagged variants of OMA1 in Oma1−/− MEFs (OMA1-myc). In parallel, we expressed OMA1E324Q-myc harbouring a point mutation in its proteolytic centre. The OPA1 forms c and e were absent in cells lacking OMA1 but accumulated upon expression of OMA1-myc in these cells (Fig 1B). In contrast, OMA1E324Q-myc did not promote formation of c and e, demonstrating that OMA1-myc is active and that OPA1 processing at S1 depends on the proteolytic activity of OMA1 (Fig 1B).
Mitochondria were isolated from Oma1−/− MEFs expressing OMA1-myc and subfractionated by osmotic disruption of the OM or solubilisation of mitochondrial membranes (Fig 2A). OMA1 remained largely resistant to externally added protease in intact mitochondria (Fig 2A). However, it became protease-accessible following osmotic disruption of the OM, while matrix-localised Hsp60 remained protected against proteolytic attack (Fig 2A). In contrast, solubilisation of mitochondrial membranes allowed degradation of OMA1-myc and Hsp60 by externally added protease (Fig 2A).
Figure 2.

- Submitochondrial localisation of OMA1. Mitochondria and mitoplasts, which were generated by hypotonic disruption of the OM (swelling), were incubated with or without proteinase K (PK; 50 μg/ml) and analysed by SDS–PAGE and immunoblotting. The IMS protein Smac and the matrix protein Hsp60 served as controls. Mitochondrial membranes were solubilised with Triton X-100 where indicated. The slightly reduced level of Smac upon PK treatment of mitochondria indicates partial disruption of the OM upon purification of mitochondria.
- OMA1 is an integral membrane protein. Alkaline extracts of mitochondria in Na2CO3 (pH 11.5) were separated into soluble (S) and membrane (P) fractions by centrifugation and analysed by SDS–PAGE and immunoblotting. The integral inner membrane protein PHB2 and the soluble intermembrane space protein Smac served as controls. T, total.
- Topological model of mature OMA1 in the IM. The C-terminal M48 metallopeptidase domain is exposed to the IMS, whereas the N-terminal domain is present at the matrix side of the IM.
- FLAG-tagged OPA1 Sp1 or OPA1 Sp1-TCS, which contains a TEV-cleavage site (TCS) instead of S1, were transfected into cell lines allowing tetracycline-inducible expression of HA-tagged Su9-TEV (targeted to the matrix) or Smac-TEV (targeted to the IMS). Cells were treated with tetracycline when indicated and analysed by SDS–PAGE and immunoblotting.
Source data are available online for this figure.
Hydrophobicity plots reveal the presence of several hydrophobic regions within the amino acid sequence of OMA1 that may form transmembrane helices. We therefore examined the solubility of OMA1 and other mitochondrial marker proteins upon alkaline extraction of mitochondrial membranes isolated from OMA1-myc-expressing MEFs (Fig 2B). While Smac/DIABLO, a soluble protein of the intermembrane space (IMS), accumulated in the supernatant fraction, OMA1 was recovered from the pellet fraction, similar to the integral IM protein PHB2 (Fig 2B). Collectively, these results reveal that OMA1 is integral part of the IM, exposing its C-terminal domain to the IMS (Fig 2C).
Previous experiments revealed that the S1 cleavage site of OPA1 is localised in the matrix (Ishihara et al, 2006), that is, in a different subcompartment of mitochondria than the catalytic domain of OMA1. To address these apparently conflicting data, we revisited the previous experiments and replaced S1 in OPA1 splice variant 1 (Sp1; Fig 1A) with the cleavage site for the tobacco etch virus (TEV) protease (Sp1-TCS). Sp1 or Sp1-TCS was co-expressed in HEK293 cells with TEV protease, which was expressed under the control of a tetracycline-inducible promoter and directed either to the mitochondrial matrix (Su9-TEV) or to the IMS (Smac-TEV; Fig 2D). Expression of Sp1 containing the S1 site allowed the formation of S-OPA1, independent of the presence of TEV (Fig 2D). In contrast, Sp1-TCS lacking the S1 site accumulated in the long form only (Fig 2D). While tetracycline-induced expression of matrix-localised TEV in these cells did not allow OPA1 processing, we observed cleavage of Sp1-TCS in the presence of TEV in the IMS (Fig 2D). These findings strongly suggest that the S1 processing site of OPA1 resides in the IMS and therefore is accessible for proteolytic attack by OMA1.
OMA1 is unstable in depolarised mitochondria
In agreement with previous findings (Head et al, 2009), we observed that OMA1-myc accumulated at a molecular mass of approximately 45 kDa within mitochondria. However, bioinformatic analysis of the amino acid sequence of OMA1 revealed potential targeting sequences of 28 or 86 amino acids, which would result in the formation of a significant larger mature form of OMA1 (55 or 49 kDa, respectively). To unambiguously identify the proteolytically active form of OMA1, we first assessed the import of nuclear-encoded OMA1 into mitochondria. Synthesis of OMA1 in a cell-free system in the presence of 35S-methionine generated a product of approximately 60 kDa, consistent with the predicted molecular mass of OMA1 (Fig 3A). Incubation of 35S-labelled OMA1 with mitochondria isolated from MEFs was accompanied by a time-and ΔΨ-dependent accumulation of shorter forms of OMA1, indicating proteolytic processing upon import into mitochondria (Fig 3A). The molecular mass of these proteolytic products was approximately 45 kDa and thus of a similar size as OMA1-myc expressed in vivo, suggesting that they represent the mature form of OMA1. Therefore, we sought to determine the N-terminal sequence of mature OMA1 accumulating in vivo. We employed a HEK293 cell line to overexpress proteolytically inactive OMA1E324Q-myc in a tetracycline-inducible manner and purified OMA1E324Q-myc by immunoprecipitation using myc-specific antibodies (Fig 3B). A single protein of the expected molecular weight was detected in the eluate and subjected to N-terminal sequencing (Fig 3B). In contrast to in silico predictions for processing sites in OMA1, this analysis identified alanine at position 140 as the N-terminal amino acid of mature OMA1 (Fig 3C), which thus has a molecular mass of approximately 43 kDa.
Figure 3.
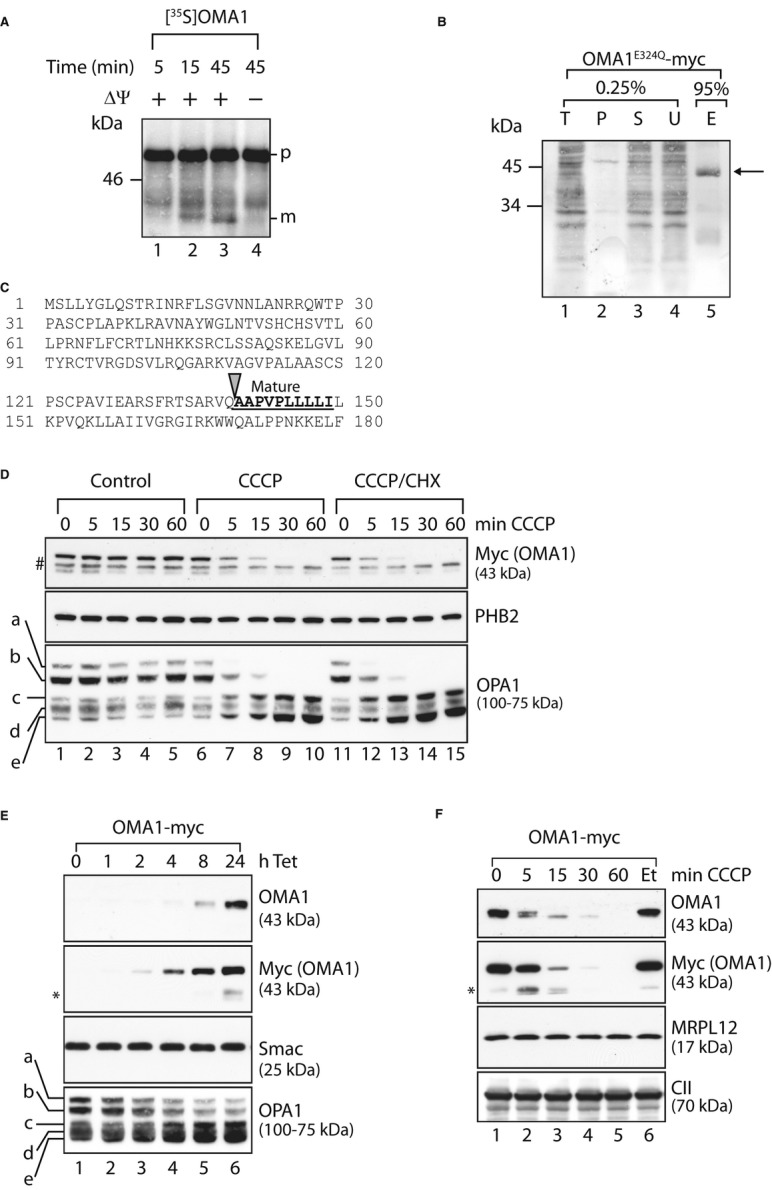
- In vitro import of OMA1 into mitochondria. Mitochondria were isolated from MEFs and incubated with [35S]-labelled OMA1 that was synthesised in a cell-free system for the time points indicated in the presence or absence of membrane potential (ΔΨ). Mitochondrial proteins were analysed by SDS–PAGE and autoradiography. p, precursor form; m, mature form.
- N-terminal sequencing of mature OMA1. Mitochondria isolated from HEK293 cells expressing OMA1E324Q-myc were solubilised in 1% (v/v) NP-40 and subjected to immunoprecipitation using myc-specific antibodies. Total (T), pellet (P), supernatant (S) and unbound (U) fractions (0.25%) and the eluate (E; 95%) were analysed by SDS–PAGE and Coomassie staining. The arrow indicates OMA1E324Q-myc in the eluate.
- N-terminal amino acid sequence of OMA1. Mature OMA1 is generated upon proteolytic cleavage at the site indicated with an arrow. The bold, underlined sequence represents the N-terminal sequence of mature OMA1 determined by Edman degradation.
- Degradation of pre-existing OMA1 following mitochondrial stress. MEFs stably expressing OMA1-myc were treated with CCCP, with cyclohexamide (CHX) or with ethanol and DMSO (control) as indicated. Samples were isolated and analysed via SDS–PAGE and immunoblotting. #, non-specific cross reaction; a-e, OPA1 forms.
- Overexpression of OMA1-myc in HEK293 cells. OMA1-myc expression was induced by addition of tetracycline (Tet) for the time points shown. Cell extracts were analysed by SDS–PAGE and immunoblotting. *, proteolytic fragments of OMA1.
- Overexpressed OMA1 is degraded following mitochondrial dysfunction. After tetracycline-induced expression of OMA1-myc, HEK293 cells were treated with CCCP or, for control, ethanol (Et) as indicated. Samples were assessed by SDS–PAGE and immunoblotting. *, C-terminal, proteolytic fragments of OMA1.
Source data are available online for this figure.
The mechanism of OMA1 activation is presently not understood. However, it was previously reported that dissipation of ΔΨ caused the accumulation of an approximately 60 kDa form of OMA1 at the mitochondrial surface, while the approximately 45 kDa form disappeared. This has led to the hypothesis that the approximately 60 kDa species is the proteolytically active form of OMA1, which is localised to the OM upon depolarisation of mitochondria and degrades L-OPA1 (Head et al, 2009). Therefore, we used the Oma1−/− cell line stably expressing OMA1-myc to investigate the requirement of newly synthesised OMA1 for OPA1 processing. Mitochondrial depolarisation induced rapid degradation of L-OPA1 and led to the accumulation of OPA1 forms c and e, while the OPA1 pattern was not altered in control cells (Fig 3D). Importantly, inhibition of cytosolic protein synthesis by cyclohexamide did not inhibit CCCP-induced OPA1 processing (Fig 3D). Thus, OMA1 pre-existing within mitochondria is activated under mitochondrial stress, demonstrating that the approximately 43 kDa form of OMA1 is proteolytically active.
In the course of these experiments, we noted that mitochondrial depolarisation was accompanied by the loss of OMA1-myc (Fig 3D). This suggested that a dysfunction of mitochondria induces proteolysis of OMA1-myc, which is either completely degraded or C-terminally cleaved under these conditions. To further explore this proteolytic process and exclude indirect effects of the C-terminal protein tag, we generated antibodies directed against the proteolytic domain of mouse OMA1. However, we were unable to detect endogenous OMA1 in MEFs or HEK293 cells. We therefore turned to HEK293 cells, which allow tetracycline-inducible expression of OMA1-myc. Following addition of tetracycline, OMA1-myc accumulated in these cells as monitored using myc-specific antibodies. Increased expression of OMA1-myc resulted in processing of L-OPA1 (Fig 3E), consistent with proteolytic activity of OMA1 in polarised mitochondria.
The overexpressed protein was detected using OMA1-specific antibodies in cells grown in the presence of tetracycline (Fig 3E), allowing us to monitor proteolysis of OMA1 upon dissipation of ΔΨ. We induced OMA1-myc expression and then activated OMA1 by adding CCCP (Fig 3F). Two smaller OMA1-derived proteolytic fragments were observed with myc-specific antibodies pointing to degradation of OMA1 from the N-terminus (Fig 3F). Immunoblotting using OMA1-specific antibodies revealed rapid degradation of OMA1 (Fig 3F), indicating that OMA1 is completely degraded upon mitochondrial depolarisation, without the accumulation of stable intermediates. Using OMA1-specific antibodies, we detected a second form of OMA1 that was slightly smaller than mature OMA1 but degraded similarly to mature OMA1 (Fig 3F). This likely reflects clipping of C-terminal amino acids of OMA1-myc, as only the larger band was recognised by myc-specific antibodies. We conclude from these experiments that mitochondrial depolarisation induces the degradation of OMA1 from both termini offering a mechanism to attenuate the stress response.
Autocatalytic proteolysis of OMA1
To identify the protease(s) mediating OMA1 turnover, we first tested various protease inhibitors for their ability to block the degradation of OMA1 in depolarised mitochondria. Pepstatin A (aspartic protease inhibitor), E64D (cysteine protease inhibitor) and pefabloc (serine protease inhibitor) did not stabilise OMA1 under these conditions (Fig 4A). In contrast, OMA1 accumulated in depolarised mitochondria in the presence of o-phenanthroline, indicating that a metallopeptidase is responsible for OMA1 turnover (Fig 4A).
Figure 4.
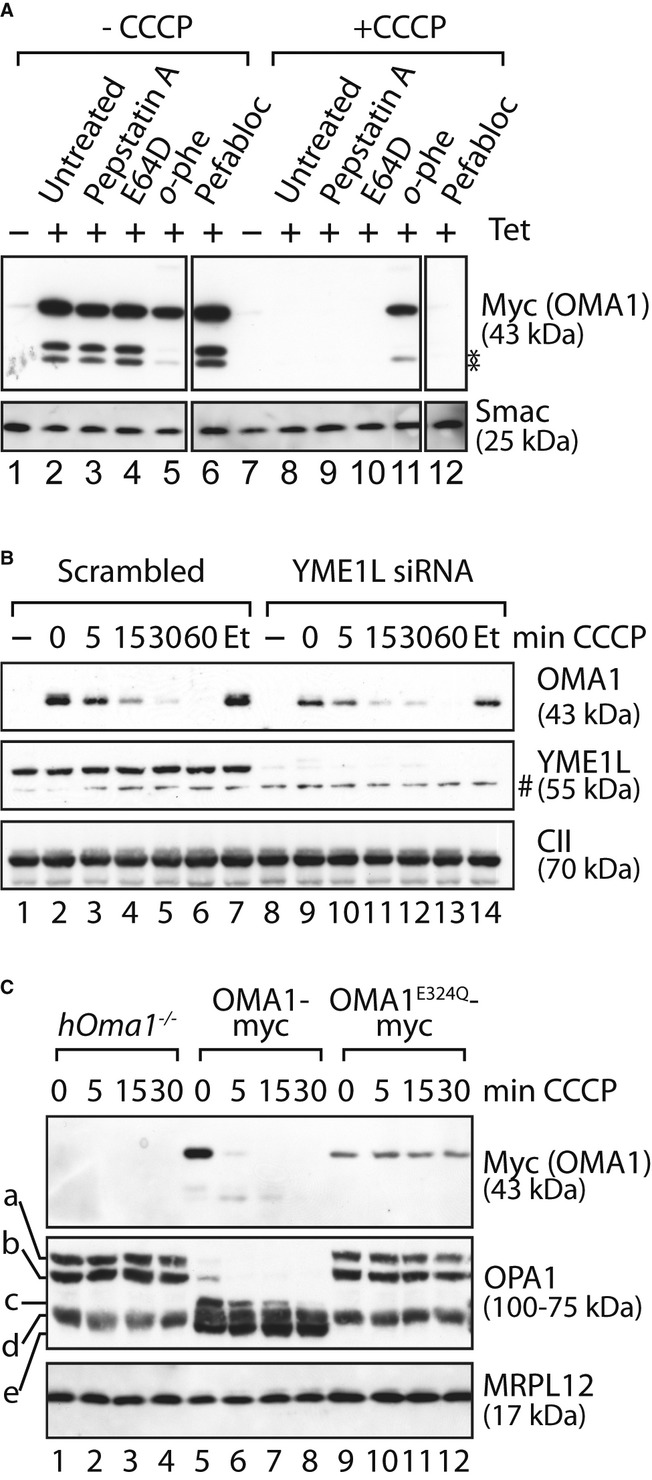
- Stability of OMA1-myc in the presence of various protease inhibitors. OMA1-myc expression was induced in HEK293 cells expressing OMA1-myc. Cells were incubated for 5 h with pepstatin A, E64D, o-phenanthroline (o-phe) or pefabloc before addition of CCCP or ethanol (-CCCP) for 1 h. *, proteolytic fragments of OMA1.
- Stress-induced OMA1 turnover does not depend on YME1L. HEK293 cells expressing OMA1-myc were transfected with YME1L-specific or scrambled siRNA control for 72 h and subsequently incubated in the presence of CCCP or, for control, ethanol (Et) for the time points indicated. #, non-specific cross reaction.
- OMA1E324Q-myc is stable following stress. hOma1−/− cells expressing OMA1-myc or OMAE324Q-myc were incubated in the presence of CCCP as indicated. In all panels, samples were analysed by SDS–PAGE and immunoblotting using the indicated antibodies.
Source data are available online for this figure.
We therefore examined the involvement of mitochondrial metallopeptidases in OMA1 turnover using RNAi. Following tetracycline-induced OMA1-myc expression, we depleted cells of the i-AAA protease, YME1L, which, like OMA1, faces its active site to the IMS. However, this did not affect the proteolytic breakdown of OMA1 in depolarised mitochondria (Fig 4B). Similarly, OMA1 was degraded under these conditions in m-AAA protease-deficient mitochondria (Supplementary Fig S3). As OMA1 is a member of the M48 metallopeptidase family and itself might be inhibited by o-phenanthroline, we also examined the stability of a proteolytically inactive OMA1E324Q-myc. To exclude proteolysis by endogenous OMA1, we deleted Oma1 in HEK293 cells using zinc finger nucleases and expressed in the obtained OMA1-deficient human cell line (hOma1−/−) OMA1-myc or OMA1E324Q-myc under the control of a tetracycline-inducible promoter (Fig 4C). Strikingly, OMA1E324Q-myc accumulated stably in these cells upon dissipation of ΔΨ, while OMA1-myc was rapidly turned over (Fig 4C). Thus, OMA1 is degraded in an autocatalytic fashion following mitochondrial depolarisation.
Yeast and mammalian OMA1 respond differently to mitochondrial stress
Sequence alignments between OMA1 homologues in different organisms revealed conservation within the M48 metallopeptidase domain, but significant variations in the N-and C-terminal regions. We therefore compared the stability of yeast and mouse OMA1 in dysfunctional mitochondria by expressing myc-tagged variants of both proteins in Δoma1 yeast cells. As deletion of OMA1 has no strong effect on yeast cell growth (Käser et al, 2003), we used Δoma1Δyme1 yeast cells, whose growth on glucose-containing media is severely impaired (Supplementary Fig S4). Mouse OMA1 was targeted to yeast mitochondria. However, growth defects of Δoma1Δyme1 yeast cells were only restored upon expression of yeast Oma1, but not of mouse OMA1 pointing to functional differences between both homologues (Supplementary Fig S4). We compared the stability of both proteins following depolarisation in cellular protein extracts. Mouse OMA1-myc but not OMA1E324Q-myc was rapidly degraded under these conditions (Fig 5A). These experiments confirm the proteolytic activity of mouse OMA1 expressed in yeast and demonstrate its autocatalytic turnover upon depolarisation of yeast mitochondria. In contrast, addition of CCCP did not destabilise yeast Oma1 (Fig 5B), highlighting functional differences between yeast and mouse OMA1 in response to mitochondrial stress.
Figure 5.

OMA1 activation is regulated by accessory domains outside the metallopeptidase domain.
A, B Mouse OMA1, but not yeast Oma1, is autocatalytically degraded upon mitochondrial depolarisation in yeast. Δoma1 yeast cells expressing mouse OMA1-myc or mouse OMA1E324Q-myc (A), or yeast Oma1-myc or yeast yOma1E205Q-myc (B) were incubated with CCCP or, for control, ethanol (Et) as indicated. Samples were analysed by SDS–PAGE and immunoblotting.
C Domain structure of OMA1 homologues from Mus musculus, Homo sapiens,Rattus norvegicus, Saccharomyces cerevisiae, Schizosaccharomyces pombe and Yarrowia lipolytica. The consensus metal-binding motif is shown with a light grey bar and the conserved hydrophobic regions with dark grey bars. An N-terminal hydrophobic domain (white striped grey bar) followed by a positively charged domain (+++) is only present in OMA1 homologues of higher eukaryotes.
D, E N-and C-terminal regions outside the metallopeptidase domain affect OMA1 activity. hOma1−/− cells expressing OMA1-myc, OMA1E324Q-myc and either OMA1ΔC-myc (lacking amino acids 493–521) (D) or OMA1Δ144–163-myc (E) were incubated with CCCP for the times indicated. Cell extracts were analysed by SDS–PAGE and immunoblotting using the indicated antibodies. The mature OMA1Δ144–163-myc exhibited a slightly larger molecular mass, indicating an altered processing site of the mutant variant.
Source data are available online for this figure.
Susceptibility to mitochondrial stress depends on the N-terminal domain of OMA1
Given the different domain architecture between mouse and yeast OMA1, we hypothesised that divergent domains between both homologues may impart different functions and responsiveness to mitochondrial depolarisation. In addition to an extended C-terminal domain, OMA1 from higher eukaryotic species contain an additional N-terminal hydrophobic stretch of amino acid residues, which is followed by a positively charged region (Fig 5C). While conserved among OMA1 homologues in higher eukaryotes, both the hydrophobic region and the adjacent cluster of positively charged amino acids are absent in yeast homologues of OMA1 (Fig 5C).
To investigate the importance of these domains of OMA1, we first deleted C-terminal amino acids 493–521 of OMA1 (OMA1ΔC) and expressed myc-tagged variants of OMA1, proteolytically inactive OMA1E324Q and OMA1ΔC in hOma1−/− cells (Fig 5D). Expression of OMA1 but not of OMA1E324Q allowed constitutive and stress-induced processing leading to the accumulation of the proteolytic OPA1 forms c and e in these cells (Fig 5D). Similarly, OMA1ΔC was proteolytically active and constitutively cleaved L-OPA1 to forms c and e (Fig 5D). Depolarisation of mitochondria induces OPA1 processing and autocatalytic turnover of OMA1ΔC (Fig 5D). Proteolysis of OMA1ΔC occurred with slower kinetics when compared to OMA1 and was accompanied by a more rapid processing of OPA1, pointing to a critical role of the C-terminal region for the regulation of OMA1 activity.
In further experiments, we deleted amino acid residues 144–163 of OMA1 to disrupt the N-terminal hydrophobic region and expressed myc-tagged OMA1Δ144–163 in hOma1−/− cells (Fig 5E). OMA1Δ144–163 accumulated in mitochondria but was functionally inactive, despite being present at higher levels than wild-type OMA1. In addition to L-OPA1 forms, we only detected the proteolytic product d of OPA1, while c and e generated by OMA1 did not accumulate in these cells (Fig 5E). Moreover, dissipation of ΔΨ did not promote OPA1 processing nor autocatalytic OMA1 turnover, demonstrating that the proteolytic activity of OMA1 depends on amino acid residue 144–163 outside of its M48 metallopeptidase domain.
To assess the function of positively charged amino acids following this hydrophobic stretch, we replaced lysine and arginine residues in this region by glutamate in a stepwise manner (OMA1−1, OMA1−3, OMA1−4, OMA1−6). The resulting OMA1 variants were fused with a myc-tag and stably expressed in hOma1−/− cells (Fig 6A). All variants were proteolytically active and constitutively cleaved L-OPA1 to forms c and e (Fig 6A). However, we observed a strikingly different response of the OMA1 mutant variants upon depolarisation of mitochondria. Decreasing the net positive charge of this region was accompanied with decreased kinetics of stress-induced OPA1 processing, which was almost completely inhibited in the presence of OMA1−6 (Fig 6A). Similarly, autocatalytic turnover was increasingly impaired upon removal of positively charged amino acids in the N-terminal region of OMA1. Two N-terminal OMA1 fragments of approximately 35 kDa accumulated at different levels in depolarised mitochondria harbouring OMA1−1, OMA1−3 or OMA1−4 (Fig 6A). OMA1−6 was the most stable variant, which was degraded only very slowly and was not converted into smaller forms (Fig 6A). Thus, the positively charged amino acids in the N-terminal region of OMA1 are crucial for OMA1 activation upon dissipation of ΔΨ.
Figure 6.

- Myc-tagged OMA1 variants harbouring point mutations within the N-terminal cluster of positively charged amino acids (OMA1−1, OMA1−3, OMA1−4, OMA1−6) were stably expressed in hOma1−/− cells by addition of tetracycline. Cells were treated with CCCP for the times indicated, and cell extracts were analysed by SDS–PAGE and immunoblotting using the indicated antibodies.
- The mitochondrial morphology of Oma1−/− MEFs expressing Oma1E324Q-myc or Oma1−6-myc was visualised by immunofluorescence microscopy upon co-expression of a GFP-variant targeted to the mitochondrial matrix. Cells were either treated with ethanol (not shown) or with 20 μM CCCP for 2 h as indicated.
- Quantification of mitochondrial morphology in Oma1−/− MEFs expressing Oma1−6. Approximately 150 cells were scored in three independent experiments.
Source data are available online for this figure.
To substantiate these findings, we monitored the morphology of the mitochondrial network upon dissipation of ΔΨ in Oma1−/− MEFs expressing OMA1, OMA1E324Q or OMA1−6 (Fig 6B and C). As expected, mitochondrial depolarisation resulted in mitochondrial fragmentation in Oma1+/+ MEFs, whereas tubular mitochondria were maintained in Oma1−/− MEFs (Fig 6B and C). Expression of OMA1 but not of OMA1E324Q in Oma1−/− MEFs allowed fragmentation of the mitochondrial network upon addition of CCCP (Fig 6B and C). Strikingly, tubular mitochondria were maintained upon mitochondrial depolarisation in Oma1−/− MEFs expressing OMA1−6 (Fig 6B and C).
We conclude from these experiments that positively charged amino acids in the N-terminal region of OMA1 are dispensable for the proteolytic activity of OMA1, but are required for the responsiveness of OMA1 to mitochondrial depolarisation triggering proteolysis of L-OPA1 and mitochondrial fragmentation.
Cooperation of OMA1 subunits upon oligomerisation
Yeast Oma1 oligomerises into a complex of 200–300 kDa (Käser et al, 2003). This complex is lost in respiratory-deficient yeast cells, leading to the hypothesis that Oma1 activation upon mitochondrial depolarisation may relate to alterations in its assembly (Khalimonchuk et al, 2012). We therefore assessed a potential role of OMA1 oligomerisation for its activation in depolarised mitochondria. Mitochondria isolated from OMA1-myc-expressing cells were solubilised and extracts fractionated by density gradient centrifugation (Fig 7A). OMA1 was recovered in fractions corresponding to a molecular weight of approximately 200–300 kDa; thus, it forms a similar-sized complex as yeast Oma1 (Fig 7A). However, in contrast to yeast Oma1 (Khalimonchuk et al, 2012), dissipation of ΔΨ did not interfere with the formation of the OMA1 complex in human mitochondria (Fig 7A), suggesting that OMA1 is active in an oligomeric state.
Figure 7.
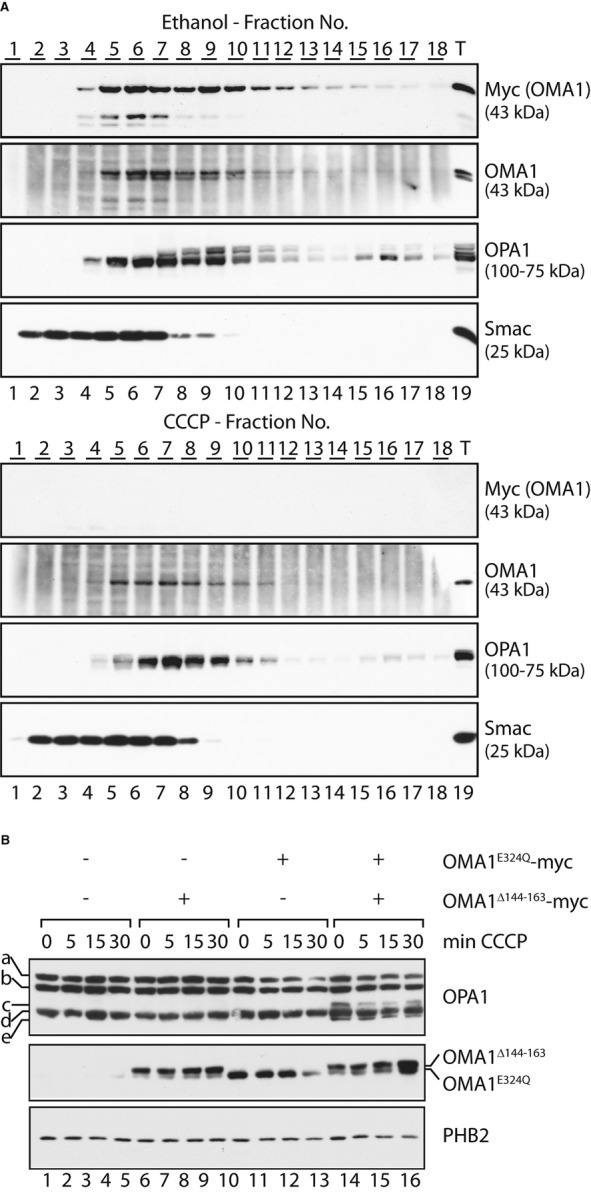
- ΔΨ-independent oligomerisation of OMA1. HEK293 cells expressing OMA1-myc were incubated for 15 min in the presence of CCCP or ethanol (control) for 15 min. Isolated mitochondria were solubilised and subjected to density gradient centrifugation. Fractions were collected, TCA-precipitated and subjected to SDS–PAGE and immunoblotting.
- Reconstitution of OMA1 activity upon oligomerisation of proteolytically inactive OMA1 variants. hOma1−/− cells allowing the tetracycline-inducible expression of OMA1E324Q-myc were transfected with YME1L-specific siRNA and with a plasmid encoding OMA1Δ144–163-myc. OMA1E324Q-myc expression was induced by addition of tetracycline where indicated. OPA1 processing was induced upon mitochondrial depolarisation using CCCP for the time points indicated. Cell extracts were analysed by SDS–PAGE and immunoblotting. a-e, OPA1 forms.
Source data are available online for this figure.
C-terminal proteolytic fragments of OMA1-myc comigrated with assembled OMA1, indicating that the N-terminal region of OMA1 is dispensable for OMA1 oligomerisation (Fig 7A). Consistently, the loss of proteolytic activity of OMA1Δ144–163 or the impaired activation of OMA1−6 cannot be explained by deficiencies in the assembly of these OMA1 variants. OMA1 variants lacking the hydrophobic region (OMA1Δ144–163) or positively charged amino acids in the N-terminal region (OMA1−6) formed similar-sized complexes as OMA1 or proteolytically inactive OMA1E324Q (Supplementary Fig S5).
To demonstrate proteolytic activity of oligomeric OMA1, we examined the possibility of an intersubunit communication within the OMA1 complex. We transiently expressed OMA1Δ144−163 in hOma1−/− cells which allowed tetracycline-inducible genomic expression of OMA1E324Q to examine the possibility that proteolytic activity of OMA1 is restored upon assembly into an oligomeric complex (Fig 7B). As the proteolytic OPA1 forms c and e accumulate generally at very low levels in these cells, it is difficult to conclusively assess OPA1 processing in the presence of OMA1 variants. We therefore exploited the observation that the proteolytic OPA1 form d decreases while c and e accumulate upon knock-down of YME1L. S-OPA1 forms c and e did not accumulate in the presence of either OMA1Δ144–163 or OMA1E324Q in these cells, demonstrating the dependence of OPA1 processing at S1 on the proteolytic activity of OMA1 (Fig 7B). Strikingly, co-expression of OMA1Δ144–163 and OMA1E324Q revealed formation of S-OPA1 forms c and e (Fig 7B), demonstrating that OMA1 activity is restored at least to some extent upon assembly of these proteolytically inactive OMA1 variants. Notably, we did not observe an increased accumulation of S-OPA1 forms upon mitochondrial depolarisation (Fig 7B). These findings indicate that activation of the mutant OMA1 complex is impaired, consistent with an important function of the N-terminal domain of OMA1 for activation rather than proteolytic activity per se.
To examine complex formation directly, we co-expressed myc-tagged OMA1E324Q and flag-tagged OMA1Δ144–163 in hOma1−/− cells and performed coimmunoprecipitation experiments using either myc-or flag-specific antibodies (Supplementary Fig S5). Although with low efficiency, both OMA1 variants were precipitated from these cells, but not from control cells, regardless of the antibodies used for immunoprecipitation (Supplementary Fig S5). Thus, both OMA1 variants can assemble into a functionally active complex, suggesting intersubunit communication and positive cooperation between different OMA1 subunits.
Discussion
Our results identify OMA1-mediated processing of OPA1 as a general cellular response under stress. Different stress stimuli were found to activate OMA1. Activation of OMA1 critically depends on positively charged amino acids within the N-terminal domain of OMA1 and is accompanied by its autocatalytic degradation, which is initiated from both the N-and C-terminal ends and results in the complete turnover of OMA1. This unusual regulatory mechanism ensures the reversibility of the response and allows restoration of the mitochondrial network upon alleviation of stress stimuli or repair of mitochondrial damage.
We observed striking differences in the response to mitochondrial stress in yeast and mammalian mitochondria, which enabled us to identify critical stress-responsive regions in mammalian OMA1. The maintenance of mitochondrial morphology depends on homologous dynamin-like GTPases (OPA1 in mammals and Mgm1 in yeast) and their balanced proteolytic conversion into short forms (Herlan et al, 2003; McQuibban et al, 2003; Song et al, 2007). However, mitochondrial depolarisation interferes only with the stability of OPA1 and not with that of Mgm1. Notably, Mgm1 is cleaved by the rhomboid protease Pcp1 in the IM, but not by yeast Oma1 (Herlan et al, 2003; McQuibban et al, 2003). Expression of mammalian OMA1 in yeast cells lacking endogenous Oma1 revealed another striking difference: while yeast Oma1 remained stable in depolarised mitochondria, we observed autocatalytic degradation of mammalian OMA1 under these conditions, suggesting that intrinsic properties of the protein itself cause its destabilisation upon mitochondrial depolarisation. Both yeast and mammalian peptidases contain a conserved M48 metallopeptidase domain, but yeast Oma1 lacks N-and C-terminal regions conserved in mammalian OMA1. Our mutational analysis revealed important functions of these regions for OMA1 activity and its regulation under stress.
OMA1 is active under normal conditions and constitutively cleaves L-OPA1 at S1 (Quiros et al, 2012). However, the specific activity of OMA1 appears to be low as we observed only inefficient L-OPA1 degradation even if OMA1 is highly overexpressed. In contrast, various stress stimuli activate OMA1 and result in the rapid and complete conversion of L-OPA1 into S-OPA1. Several models have been proposed for the activation of OMA1 under stress. While changes in the oligomeric state of yeast Oma1 were discussed to occur under stress, we did not observe an alteration of the native molecular mass of OMA1 in human cells. Mitochondrial depolarisation was proposed to lead to a re-localisation of OMA1 to the OM where it accumulates in an approximately 60 kDa form (Head et al, 2009). However, our results identify this form as the precursor protein of OMA1 before maturation occurs upon import into mitochondria. Cleavage after amino acid 139 results in the formation of proteolytically active OMA1 in the IM. These findings are consistent with the recent observation that complete OPA1 degradation can be induced upon depolarisation of isolated mitochondria (Hoppins et al, 2011). Metallopeptidases are often synthesised as zymogens and activated upon proteolytic removal of an inhibitory pro-domain (Lopez-Pelegrin et al, 2013). Although we observed the transient accumulation of proteolytic OMA1 fragments upon activation, OMA1 appears to be completely degraded, suggesting an alternative mechanism of activation. Our findings demonstrate that OMA1 activity under stress critically depends on an N-terminal domain outside the conserved M48 metallopeptidase domain, which is composed of a hydrophobic region and a cluster of positively charged amino acids. While the proteolysis by OMA1 depends on hydrophobic amino acids within this domain, the following cluster of positively charged amino acids was found to be critical for OMA1 activation in depolarised mitochondria. We propose that this domain functions as sensor domain, perhaps stabilised by ionic interactions with negatively charged phospholipid head groups, which induces conformational changes in OMA1 upon membrane depolarisation leading to its activation and autocatalytic turnover. Clearly, structural information on OMA1 is required to substantiate this hypothesis. It is noteworthy, however, that the structural characterisation of distantly related metallopeptidases from thermophilic prokaryotes suggests novel intra-and intermolecular latency mechanisms that are consistent with our findings (Lopez-Pelegrin et al, 2013). These include intermolecular interactions of C-terminal helices within an oligomer that impair proteolytic activity. Such a scenario may explain the accelerated processing of OPA1 upon deletion of C-terminal amino acid residues in OMA1.
Signal(s) leading to OMA1 activation remains to be defined and may even vary under different stress conditions. It is conceivable that the loss of the proton motive force interferes with the N-terminal sensor domain and activates OMA1. However, we did not observe a strict correlation between mitochondrial depolarisation and OMA1 activation in vitro, suggesting that other mechanisms do exist. In agreement with our findings, previous studies excluded a role of ROS and suggested lowered ATP levels in mitochondria to promote L-OPA1 degradation (Baricault et al, 2007). Notably, Oma1−/− mice exposed to a high-fat diet show deficiencies in the brown adipose system that cause decreased energy expenditure and defective thermoregulation (Quiros et al, 2012), suggesting that metabolic stress activates OMA1 in vivo.
It is noteworthy that similar conditions triggering OMA1 activation in vitro were found to impair mitochondrial import of the serine threonine kinase PINK1 leading to its accumulation at the mitochondrial surface and the recruitment of the E3 ubiquitin ligase parkin (Lazarou et al, 2012). Parkin-mediated ubiquitylation triggers proteasomal degradation of mitofusins, dynamin-like GTPases mediating OM fusion (Tanaka et al, 2010; Vives-Bauza et al, 2010). While PINK1 import from the mitochondrial surface is resumed in repolarised mitochondria (Lazarou et al, 2012), autoproteolysis of OMA1 prevents continued L-OPA1 proteolysis under these conditions. Thus, mitochondrial stress conditions induce the proteolytic breakdown of dynamin-like GTPases in both membranes and inhibit fusion in a reversible manner. Only if stress is persisting, damaged mitochondria are primed for turnover through mitophagy. It will be intriguing to further examine the functional interdependence of stress response pathways in both mitochondrial membranes and their impact on mitochondrial integrity and in human disease.
Materials and Methods
Cell culture, transfection and cell treatments
Wild-type and Oma1−/− primary mouse embryonic fibroblasts (MEFs) were kindly provided by Carlos Lopéz-Otín (Oviedo) and immortalised by SV40 transformation. MEFs were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/l glucose and 10% (v/v) foetal calf serum (FCS), supplemented with sodium pyruvate (100 μg/ml), penicillin (100 μg/ml) and streptomycin (100 μg/ml). HEK293 cells were cultured in DMEM with 10% (v/v) tetracycline-reduced FCS. All cell lines were grown at 37°C, 5% CO2 and 95% humidity. For knock-down and ectopic expression, Lipofectamine RNAiMax and Lipofectamine 2000 were used, respectively, according to the manufacturer's guidelines (Invitrogen). Efficiency of transfection and knock-down was assessed by Western blotting. CCCP (20 μM), valinomycin (1 μM), oligomycin (1 μM), antimycin A (1 μM), H2O2 (0.5 mM), paraquat (5 mM), cyclohexamide (100 μg/ml), pefabloc SC (0.5 mM), pepstatin A (50 μM), E64D (50 μM) and o-phenanthroline (0.5 mM) were added to the cells as indicated.
Generation of OMA1 knock out and OMA1-expressing cell lines
HEK293 Flp-In T-REx cells lacking Oma1 were generated according the manufacturer's guidelines (Sigma) using a CompoZr Zinc finger Nuclease kit, which consists of zinc finger nucleases specifically targeted to the Oma1 locus and a DNA-cleavage domain containing the nuclease domain of FokI (zinc finger nuclease binding/cutting site: GTTACCATATA-gtaaat-AAGTATCAGGGACTG). The plasmid encoding the zinc finger nucleases was electroporated into HEK293 cells. Following 24-h expression, the cells were trypsinised and single cells were sorted into 96-well dishes. Surviving clones were picked, expanded and selected based on their inability to process OPA1 upon mitochondrial depolarisation.
Oma1−/− MEFs were transduced with an OMA1-myc-expressing lentivirus using the lentiviral expression system (Invitrogen). Following selection with Blasticidin S (10 μg/ml), stable clones were picked and expanded. Expression was verified using myc-specific antiserum. Generation of stable tetracycline-inducible cell lines was performed as described previously (Ehses et al, 2009). The open reading frame encoding Mus musculus OMA1 was cloned into pCDNA5/FRT/TO. Point mutations and deletions of the Oma1 ORF were introduced by inverse PCR using Phusion polymerase. The following amino acid substitutions were introduced in the N-terminal region of OMA1: K181E (OMA1−1), K176E K177E K181E (OMA1−3), K181E R185E K186E K188E (OMA1−4), K176E K177E K181E R185E K186E K188E (OMA1−6). Expression of OMA1-myc and variants was induced by addition of tetracycline to 1 μg/ml for 16 h.
The open reading frame encoding splice variant 7 of human OPA1 was amplified by PCR from human cDNA and cloned into p3xFLAG. For the expression of splice variant 1 of OPA1, the DNA segment corresponding to exon 5b was deleted by excision PCR. The segment encoding the S1 cleavage site (amino acids 190–200 in splice variant 1) was replaced with a TEV-cleavage site using PCR-mediated site-directed mutagenesis. For expression of matrix-or IMS-targeted TEV, the open reading frame encoding TEV was first cloned into pCDNA5/FRT/TO omitting a start codon. DNA fragments encoding the matrix targeting sequence of subunit 9 of ATP synthase of Neurospora crassa (amino acids 1–69) or the IMS targeting sequence of human Smac/DIABLO (amino acids 1–70) were cloned in front of the sequence encoding TEV. Using the pCDNA5/FRT/TO constructs, stable tetracycline-inducible HEK293 cell lines expressing the TEV variants were generated.
Subfractionation of mammalian mitochondria and in vitro import assays
Isolation of mitochondria was performed as described previously (Lazarou et al, 2007). Following isolation, mitochondrial pellets (50 μg) were resuspended in 10 mM HEPES-KOH pH 7.4, 250 mM sucrose, swelling buffer (10 mM HEPES-KOH pH 7.4) or in solubilisation buffer [10 mM HEPES-KOH pH 7.4, 250 mM sucrose, 0.5% (v/v) Triton X-100]. Samples were divided into two groups, one treated with proteinase K (50 μg/ml) for 10 min on ice and the other not treated. Samples were TCA-precipitated and analysed by SDS–PAGE and immunoblotting. For sodium carbonate extraction, mitochondrial pellets (50 μg) were resuspended in freshly prepared Na2CO3 (100 mM, pH 11.5) to 0.125 mg/ml. Samples were incubated on ice for 30 min and centrifuged at 100,000 g for 30 min. The supernatant was subjected to TCA precipitation, and the pellet fraction resuspended in SDS–PAGE loading dye. For in vitro import into isolated mitochondria, the pGEM vector carrying the OMA1 ORF was used for in vitro transcription using the mMessage SP6 transcription kit (Ambion). Radiolabelled precursor proteins were synthesised in vitro in rabbit reticulocyte lysate in the presence of [35S]-methionine/cysteine. The in vitro import reaction was performed as described previously (Lazarou et al, 2007) and analysed via SDS–PAGE.
Density gradient centrifugation of mitochondrial extracts
To determine the native molecular mass of OMA1 complexes, sucrose gradient centrifugation was employed. OMA1 expression was induced in the HEK293 OMA1-myc cell line by addition of tetracycline (1 μg/ml) for 16 h. Mitochondria (1 mg) were isolated and solubilised in 1% (w/v) digitonin in 20 mM Tris–HCl pH 7.4 and 50 mM NaCl. Samples were overlayed on top of a 5–25% sucrose gradient and centrifuged at 330,000 g for 16 h at 4°C. Following centrifugation, fractions were subjected to TCA precipitation and analysed via SDS–PAGE and immunoblotting. Bovine serum albumin (67 and 134 kDa), ferritin (440 kDa) and thyroglobulin (669 kDa) were used for calibration.
N-terminal sequencing of mature OMA1
Following induction of OMA1E324Q-myc with tetracycline (1 μg/ml) in HEK293 Flp-In T-REx OMA1E324Q-myc cells, mitochondria (10 mg) were isolated and resuspended in lysis buffer [1% (v/v) NP-40, 20 mM Tris–HCl pH 8.0, 150 mM magnesium acetate, 1 mM PMSF, 1× complete protease inhibitor (Roche)] and incubated end over end for 1 h at 4°C. Following solubilisation, mitochondria were spun at 48,000 g for 30 min to remove insoluble material and incubated with pre-equilibrated EZ-myc beads (Sigma) for 3 h at 4°C. Unbound material was removed, the beads washed three times with wash buffer [0.1% (v/v) NP-40, 20 mM Tris–HCl pH 8.0, 150 mM magnesium acetate], and OMA1E324Q-myc was eluted with freshly prepared glycine (100 mM pH 2.5; 1 ml). The elution fraction was TCA-precipitated and separated by SDS–PAGE. Following transfer to PVDF, the membrane was stained with Coomassie Blue R-250 and subjected to N-terminal sequencing via Edman degradation.
Fluorescence microscopy of the mitochondrial network
Mitochondrial morphology of Oma1+/+ and Oma1−/− MEFs was examined by immunofluorescence microscopy using cytochrome c-specific antibodies or by fluorescence microscopy after expression of a GFP-variant targeted to the mitochondrial matrix (Delta Vision; Applied Precision). Fluorescence images were taken using a camera (CoolSNAP HQ/ICX258, Sony) and a Plan-Aprochromat N 60× NA 1.42 oil objective (Olympus). Image stacks were deconvoluted with SoftWorx Imagaing Suite (Applied Precision, Merkwirth et al, 2008).
For determination of ΔΨ, cells were grown in DMEM/GlutaMax media in the presence of different drugs as indicated (20 μM CCCP for 30 min, 0.5 mM H2O2 for 8 h, 5 mM paraquat for 8 h, 1 μM valinomycin for 1 h, 1 μM oligomycin for 1 h, 1 μM antimycin A for 1 h). Cells were harvested, washed once with PBS and incubated in PBS containing 2 μM JC-1 (Invitrogen) for 25 min at 37°C. After the incubation period, cells were washed twice with PBS, and the ratio between absorbance at 585 nm (red) and 530 nm (green) was determined. To monitor alterations in ΔΨ, after heat stress, we performed live cell imaging using TMRM. 5 × 104 cells expressing a GFP-variant targeted to the mitochondrial matrix were grown in DMEM containing 10 nM TMRM (Invitrogen) either at 37°C (5% CO2) or at 42°C (5% CO2) for 4 h. Cells grown at 37°C (5% CO2) with DMEM treated with 20 μM CCCP for 30 min were used as control.
Yeast strains and growth media
Oma1 was disrupted using the KanMX4 deletion cassette in the Saccharomyces cerevisiae strain BY4741. Yeast Oma1 and mammalian OMA1 were expressed in these cells under the control of the constitutive ADH1 promoter using the plasmid YCplac111 (CEN4, LEU2). To monitor the stability of yeast Oma1 and mouse OMA1 in depolarised mitochondria, cells were grown to logarithmic phase in YPD and CCCP (20 μM) or ethanol was added to the cells for the time points indicated. Cellular proteins were extracted and analysed via SDS–PAGE and immunoblotting.
Generation of OMA1-specific antibodies
A domain of mouse OMA1 consisting of amino acid residues 211–521 and containing an N-terminal hexahistidine peptide was expressed in E. coli and purified by metal chelating chromatography under denaturing conditions. The eluted protein was dialysed against phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4). Insoluble protein was collected by centrifugation, resuspended in phosphate-buffered saline and employed for immunisation in rabbits. The serum was affinity-purified using the immobilised antigenic protein.
Acknowledgments
This work was supported by a fellowship of the Alexander-von-Humboldt-Foundation to M.B., an EMBO short-term fellowship to D.S. and grants of the Deutsche Forschungsgemeinschaft and the European Research Council (AdG No. 233078) to T.L..
Author contributions
MB, PL and TT performed conception and design of experiments, data acquisition, analysis and interpretation of data, drafting and revising the manuscript; DS, AK and RA contributed to data acquisition, analysis and interpretation of data and revised the manuscript; TL involved in conception and design of experiments, drafting and revising the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
for this article is available online: http://emboj.embopress.org
References
- Anand R, Langer T, Baker MJ. Proteolytic control of mitochondrial function and morphogenesis. Biochim Biophys Acta. 2013;1833:195–204. doi: 10.1016/j.bbamcr.2012.06.025. [DOI] [PubMed] [Google Scholar]
- Baricault L, Segui B, Guegand L, Olichon A, Valette A, Larminat F, Lenaers G. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp Cell Res. 2007;313:3800–3808. doi: 10.1016/j.yexcr.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010;1201:21–25. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, Strooper De B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Benedetto Di G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, Bliek van der AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Wel van der NN, Orozco IJ, Peters PJ, Bliek van der AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localised constrictions along the lengths of mitochondria. J Biol Chem. 2004;279:18792–18798. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- Guillery O, Malka F, Landes T, Guillou E, Blackstone C, Lombes A, Belenguer P, Arnoult D, Rojo M. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell. 2008;100:315–325. doi: 10.1042/BC20070110. [DOI] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, Bliek van der AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlan M, Vogel F, Bornhövd C, Neupert W, Reichert AS. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem. 2003;278:27781–27788. doi: 10.1074/jbc.M211311200. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, Nunnari J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41:150–160. doi: 10.1016/j.molcel.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Otera H, Oka T, Mihara K. Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid Redox Signal. 2013;19:389–399. doi: 10.1089/ars.2012.4830. [DOI] [PubMed] [Google Scholar]
- Käser M, Kambacheld M, Kisters-Woike B, Langer T. Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J Biol Chem. 2003;278:46414–46423. doi: 10.1074/jbc.M305584200. [DOI] [PubMed] [Google Scholar]
- Khalimonchuk O, Jeong MY, Watts T, Ferris E, Winge DR. Selective Oma1 protease-mediated proteolysis of Cox1 subunit of cytochrome oxidase in assembly mutants. J Biol Chem. 2012;287:7289–7300. doi: 10.1074/jbc.M111.313148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. 2008;1147:283–292. doi: 10.1196/annals.1427.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, McKenzie M, Ohtake A, Thorburn DR, Ryan MT. Analysis of the assembly profiles for mitochondrial-and nuclear-DNA-encoded subunits into complex I. Mol Cell Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pelegrin M, Cerda-Costa N, Martinez-Jimenez F, Cintas-Pedrola A, Canals A, Peinado JR, Marti-Renom MA, Lopez-Otin C, Arolas JL, Gomis-Ruth FX. A novel family of soluble minimal scaffolds provides structural insight into the catalytic domains of integral-membrane metallopeptidases. J Biol Chem. 2013;288:21279–21294. doi: 10.1074/jbc.M113.476580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuibban GA, Saurya S, Freeman M. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature. 2003;423:537–541. doi: 10.1038/nature01633. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, Kleist-Retzow von JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- Quiros PM, Ramsay AJ, Sala D, Fernandez-Vizarra E, Rodriguez F, Peinado JR, Fernandez-Garcia MS, Vega JA, Enriquez JA, Zorzano A, Lopez-Otin C. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40:D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan Szklarz LK, Scorrano L. The antiapoptotic OPA1/Parl couple participates in mitochondrial adaptation to heat shock. Biochim Biophys Acta. 2012;1817:1886–1893. doi: 10.1016/j.bbabio.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–3532. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, Houstek J, Zeman J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol Biol Cell. 2012;23:1010–1023. doi: 10.1091/mbc.E11-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Cruz Da S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14:1939–1951. doi: 10.1089/ars.2010.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, Vries de RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Bliek van der AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.