

Abstract
The faithful segregation of chromosomes into daughter cells is essential for cellular and organismal viability. Errors in this process cause aneuploidy, a hallmark of cancer and several congenital diseases. For proper separation, chromosomes attach to microtubules of the mitotic spindle via their kinetochores, large protein structures assembled on centromeric chromatin. Kinetochores are also crucial for a cell cycle feedback mechanism known as the spindle assembly checkpoint (SAC) [1]. The SAC forces cells to remain in mitosis until all chromosomes are properly attached to microtubules. At the beginning of mitosis, the SAC proteins—Mad1, Mad2, Bub1, Bub3, BubR1, Mps1, and Cdc20—are recruited to kinetochores in a hierarchical and interdependent fashion (Fig 1A). There they monitor, in ways that are not fully clarified, the formation of kinetochore–microtubule attachments [1]. Two studies recently published in EMBO reports by the groups of Silke Hauf [2] and Jakob Nilsson [3], and a recent study by London and Biggins in Genes & Development [4], shed new light on the conserved SAC protein Mad1.
All three studies were inspired by the crucial question of how checkpoint components are recruited to unattached kinetochores and how they interact there to promote rapid formation of the mitotic checkpoint complex (MCC), which is the SAC effector 1. The MCC consists of BubR1, Bub3, Mad2, and Cdc20 and is a potent inhibitor of the anaphase-promoting complex/cyclosome (APC/C), a large E3 ubiquitin ligase required for anaphase onset 1. Cells whose APC/C is inhibited by MCC remain in mitosis, and as the abundance of MCC is a reflection of incomplete or defective kinetochore–microtubule attachment, these cells will stay mitotic until the process of attachment has come to a positive end (Fig 1A).
Figure 1.
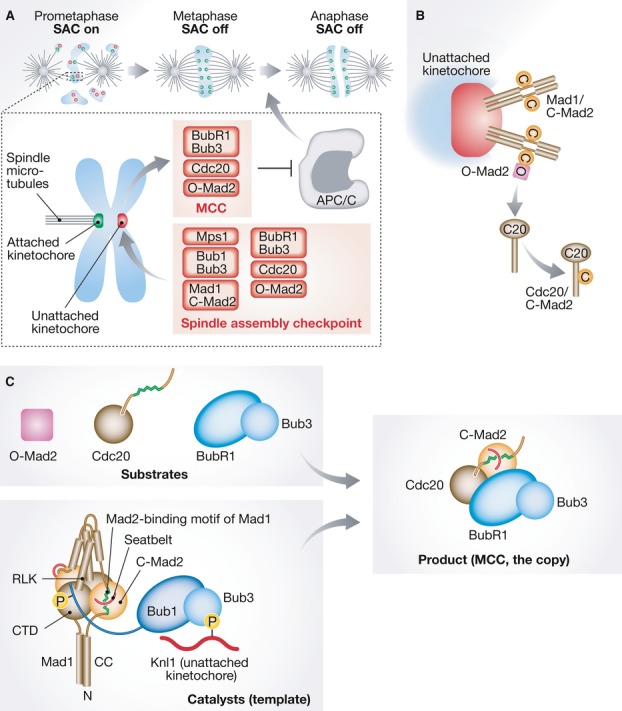
Control of mitotic progression by the SAC.
(A) The SAC is active in prometaphase and turns off when chromosomes are bi-oriented (metaphase). Kinetochores are shown as green or red dots, depending on whether or not they are bound to microtubules. SAC proteins are recruited to “red” kinetochores, where they assemble the MCC, which inhibits the APC/C. (B) Mad1/C-Mad2 forms a 2:2 tetramer. Mad1/C-Mad2 at kinetochores acts as a receptor for O-Mad2, facilitating the transformation of O-Mad2 into C-Mad2 bound to Cdc20. (C) Mad2 binds a Mad2-binding motif in Mad1 (green) with the help of its “seatbelt” (red). The CTD of Mad1 is near the Mad2-binding region, and the RLK motif neighbors the CTD. Please see the text for details of our proposed model of how Mad1/C-Mad2 and Bub1/Bub3 catalyze rapid MCC formation. The coloring scheme emphasizes the copy to template relationship of the MCC product with the catalytic platform.
The early observation that even a single unattached kinetochore is sufficient to halt mitotic progression spurred the idea that kinetochores act as catalytic platforms to assemble the SAC effector 1. Biochemical reconstitution and structural analyses identified the conformational plasticity of the Mad2 protein as a focal element of the mechanism of MCC assembly. Mad2 exists in an active closed conformation (C-Mad2) and an inactive open conformation (O-Mad2). During checkpoint activation, O-Mad2 is transformed into C-Mad2 and binds Cdc20, thus establishing a subcomplex of the MCC 1. Remarkably, the conversion of O-Mad2 to C-Mad2 is templated by a molecule of C-Mad2 that is constitutively bound to Mad1 5 (Fig 1B). Thus, the Mad1/C-Mad2 complex, which is recruited to unattached kinetochores, binds cytosolic O-Mad2 and acts as a template for its conversion into Cdc20-bound C-Mad2 5. In vitro, conformational dimerization with C-Mad2 accelerates the binding of O-Mad2 to Cdc20 by a factor of ∼10 without changing the equilibrium of this reaction 6.
It was also recently proposed that the predominant role of Mad2 might be as a catalyst for the assembly of a complex of BubR1/Bub3 with Cdc20. The latter, rather than the MCC, was identified as the crucial APC/C inhibitor 7. A direct catalytic role of Mad2 on the assembly of the BubR1/Bub3/Cdc20 complex, however, was not directly demonstrated 7. Furthermore, Mad2 is a bona fide APC/C inhibitor, a tight binding partner of Cdc20, and one of the most abundant checkpoint proteins (contrarily to Mad1/C-Mad2, which is significantly less abundant than free Mad2). These three facts are all at odds with a role of Mad2 as a catalyst of the interaction of BubR1/Bub3 with Cdc20.
A more plausible candidate to flank the Mad1/C-Mad2 complex as a catalyst for MCC assembly is Bub1. This large protein kinase, which forms a constitutive complex with Bub3, has been implicated in kinetochore recruitment of the Mad1/C-Mad2 and BubR1/Bub3 complexes, as well as Cdc20 1. Surprisingly, none of these functions of Bub1 depends on its kinase domain, which is dispensable for SAC function 1. As expected for a catalytic promoter of MCC assembly, Bub1—as Mad1/C-Mad2—binds unattached kinetochores more stably than the MCC subunits 8. Together with Mad1/C-Mad2, Bub1 might create a scaffold that orients the MCC subunits to facilitate their combination into the MCC.
Previous studies in budding yeast identified a conserved Arg-Leu-Lys (RLK) motif in Mad1 as required for interaction with Bub1 9. RLK mutations abolished binding of Mad1 to Bub1, kinetochore recruitment of Mad1, and checkpoint function 4, 9. In human cells, the Mad1 RLK motif was also shown to be required for kinetochore targeting of Mad1, and depletion of Bub1 led to a 50% reduction in the kinetochore levels of Mad1 10. Now, Heinrich et al confirm in fission yeast that mutations in the RLK motif impair kinetochore localization of Mad1 and Mad2 and abolish checkpoint signaling 2. They further show that mutations in a conserved region of Bub1 result in the same phenotype. Interestingly, London and Biggins 4 discovered that the interaction of Bub1 with Mad1 is controlled by Mps1 through Bub1 phosphorylation (Fig 1C).
Kruse et al 3 showed that, in human cells, forced kinetochore localization of a constitutively closed Mad2 mutant elicits a prolonged mitotic arrest that depends on the other SAC components, including Mad1. Complementary experiments using kinetochore-tethered Mad1 demonstrated that SAC is dependent, not only on the integrity of the RLK motif, but also of the Mad1 C-terminal domain (CTD) (Fig 1C). Indeed, mutations in a conserved patch in the C-terminal domain of Mad1 severely compromise checkpoint function in both human cells and fission yeast without affecting Mad1 localization or its interaction with Mad2 2, 3. Thus, the C-terminal region of Mad1 now joins the RLK motif as a region required for checkpoint signaling by Mad1, in addition to the well-established role of Mad1 as a “holder” of C-Mad2.
The CTD could contribute to Bub1 binding, in addition to the RLK motif (Fig 1C). However, neither the Hauf nor the Nilsson group were able to detect an interaction between Mad1 and Bub1 in fission yeast or in human cells 2, 3, suggesting that such interaction—if it exists—is short-lived in these organisms and difficult to observe by conventional affinity precipitation approaches. Thus, the molecular mechanism underlying the role of the Mad1 CTD in the SAC remains unknown and an interesting subject for future analyses. An important concept emerging from these recent studies is that the role of the RLK motif and the CTD of Mad1 is not limited to recruiting the Mad1/C-Mad2 complex to kinetochores: Artificial tethering to kinetochores of mutants in these regions of Mad1 did not restore checkpoint function, indicating that the mutations create a second deficit 2, 3. The mutations might disrupt an interaction, likely with Bub1, that places Mad1/C-Mad2 and Bub1 in the correct reciprocal position to catalyze MCC formation 2, 3.
The kinase activity of Mps1 is emerging as the generator of the “glue” that makes the putative catalytic apparatus of the SAC—Mad1/C-Mad2 and Bub1—stick to kinetochores and to each other 1 (Fig 1C). With the exception of Aurora B, which is required for Mps1 kinetochore localization, Mps1 acts upstream of all other known checkpoint proteins 1. Mps1 is recruited to unattached kinetochores, most likely through a regulated interaction with the Ndc80 complex—the main microtubule receptor at the kinetochore 1. Mps1 phosphorylates the kinetochore protein Knl1, leading to the recruitment of Bub1/Bub3. Subsequent phosphorylation of Bub1 by Mps1 might complete the process of assembly of the catalytic platform by promoting the recruitment of Mad1/C-Mad2. This, in turn, leads to the recruitment and rapid turnover of all other checkpoint proteins and catalytic assembly of the MCC. The provocative model emerging from these observations is that the template hypothesis, now also involving the related protein complexes Bub1/Bub3 and BubR1/Bub3 in addition to Mad1/C-Mad2 and Cdc20/C-Mad2, might have even more extensive implications than originally proposed 5 (Fig 1C).
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Lara-Gonzalez P, Westhorpe FG, Taylor SS. Curr Biol. 2012;22:R966–R980. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Heinrich S, Sewart K, Windecker H, et al. EMBO Rep. 2014;15:291–298. doi: 10.1002/embr.201338114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kruse T, Larsen MSY, Sedgwick GG, et al. EMBO Rep. 2014;15:282–290. doi: 10.1002/embr.201338101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.London N, Biggins S. Genes Dev. 2014;28:140–152. doi: 10.1101/gad.233700.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Antoni A, Pearson CG, Cimini D, et al. Curr Biol. 2005;15:214–225. doi: 10.1016/j.cub.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 6.Simonetta M, Manzoni R, Mosca R, et al. PLoS Biol. 2009;7:e10. doi: 10.1371/journal.pbio.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han JS, Holland AJ, Fachinetti D, et al. Mol Cell. 2013;51:92–104. doi: 10.1016/j.molcel.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howell BJ, Moree B, Farrar EM, et al. Curr Biol. 2004;14:953–964. doi: 10.1016/j.cub.2004.05.053. [DOI] [PubMed] [Google Scholar]
- 9.Brady DM, Hardwick KG. Curr Biol. 2000;10:675–678. doi: 10.1016/s0960-9822(00)00515-7. [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Sun H, Tomchick DR, et al. Proc Natl Acad Sci USA. 2012;109:6549–6554. doi: 10.1073/pnas.1118210109. [DOI] [PMC free article] [PubMed] [Google Scholar]