

Abstract
Haspin is an atypical mitotic kinase that phosphorylates histone H3 on threonine 3 (H3T3), which is required to target Aurora B to centromeres. However, how Haspin is activated upon mitotic entry remained unknown. Two independent studies, published in Molecular Cell and in this issue of EMBO reports by Ghenoiu et al [1] and Zhou et al [2], respectively, now show that Plk1 is responsible for Haspin activation as a H3T3 kinase. These results shed light on the spatiotemporal regulation of Aurora B to ensure mitotic fidelity.
The structural unit of chromatin is the nucleosome—a short segment of DNA wrapped around an octamer of histones. Histones are subjected to several post-translational modifications; in mitosis, these include phosphorylation, acetylation, methylation, and ubiquitination. Phosphorylation of histones has long been recognized as a hallmark of mitosis, but the function and regulation of this so called mitotic histone code remain poorly understood. Haspin is an atypical kinase responsible for the phosphorylation of histone H3 on threonine 3 (H3T3). H3T3 phosphorylation is required to target Aurora B to centromeres through another chromosomal passenger complex (CPC) subunit—survivin (see for example 3, 4). Aurora B is a key player in the correction mechanism that is responsible for repairing erroneous kinetochore–microtubule attachments during spindle assembly 5. Thus, H3T3 phosphorylation is likely to be required for faithful cell division through the proper recruitment of Aurora B to centromeres and subsequent regulation of kinetochore–microtubule attachments.
“…elucidate how Haspin is activated, and thereby understand how its kinase activity is restricted to mitosis”
Aurora B centromeric localization is not only required for its activation as a kinase, but also establishes a positive feedback loop in which Aurora B further increases the kinase activity of Haspin 6. Structural studies on the Haspin kinase domain have shown that it is an atypical kinase that does not require phosphorylation on the activation loop for activity, suggesting that it might be constitutively active 7. Paradoxically, H3T3 phosphorylation is only detected in mitosis. Key to solving this conundrum was to elucidate how Haspin is activated, and thereby understand how its kinase activity is restricted to mitosis.
Using different approaches, the Funabiki and Higgins groups first observed that impairing the activity of Xenopus and human Polo-like kinase 1 (Plx1 and Plk1, respectively) leads to a reduction in H3T3 phosphorylation. In addition, both groups were able to map a conserved threonine residue (T206 in xHaspin and T128 in hHaspin) that is specifically phosphorylated by Cdk1/cyclin B and recognized by the polo-box domain (PBD) of Plk1. Priming phosphorylation of specific residues by Cdk1/cyclin B is a well-established mechanism through which Plk1 recognizes and binds to its substrates. Both groups further demonstrate that not only this is the case for xHaspin and hHaspin, but also that Haspin phosphorylation by Plx1/Plk1 results in increased H3T3 kinase activity. Subsequently, Ghenoiu and collaborators 1 used liquid chromatography tandem mass spectrometry to identify more than 30 phosphorylation sites on the Haspin N-terminal domain (see also 6 for similar findings on human Haspin). Mutational analysis of several of these residues had an impact on the levels of H3T3 phosphorylation. Importantly, although some of these residues lie within consensus sequences for Cdk1 and Plk1 phosphorylation, others may be the result of autophosphorylation events. Altogether, these data reveal that heavy phosphorylation of Haspin N-terminus by Plk1 (after priming Cdk1/cyclin B phosphorylation), and perhaps by autophosphorylation, is required for its full mitotic activation, thereby explaining how Haspin kinase activity is restricted to mitosis (Fig 1).
Figure 1.
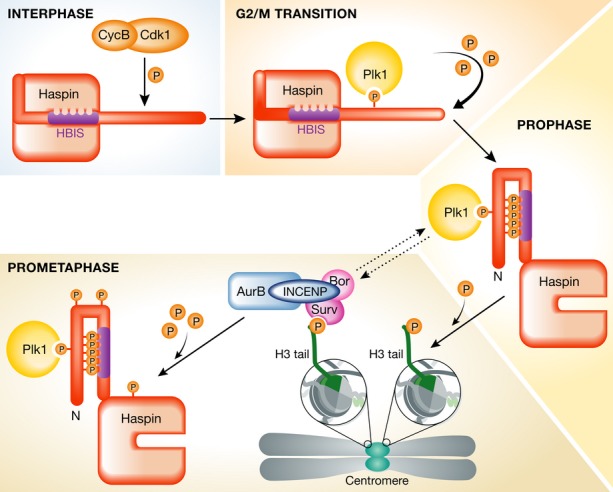
Cell cycle regulation of Haspin, the H3T3 kinase.
Haspin is an atypical kinase that is kept inactive during interphase through the interaction of a specific region in its N-terminus—enriched in basic amino acids and known as Haspin basic inhibitory segment (HBIS)—with its kinase domain. Upon mitotic entry, the Cdk1/cyclin B complex phosphorylates a specific threonine residue (T128) on human Haspin (T206 in Xenopus), generating a polo-box domain recognition site on Haspin. During prophase, Plk1 heavily phosphorylates the N-terminus of Haspin, leading to its activation and resulting in the phosphorylation of the threonine 3 on histone H3 tail on threonine 3 (H3T3). This specific histone post-translation modification is necessary for the recruitment of the chromosomal passenger complex (CPC) to centromeres. During prometaphase, Haspin is also phosphorylated by the catalytic subunit of CPC—the Aurora B kinase—which ensures that H3T3 phosphorylation at centromeres is maintained.
“…the activity of both [Plk1 and Aurora B] is necessary for robust H3T3 phosphorylation”
Both groups also investigated the role of Plk1 and Aurora B in H3T3 phosphorylation and concluded that the activity of both kinases—possibly through partially independent pathways—is necessary for robust H3T3 phosphorylation. Importantly, they show that although Haspin is a substrate for both Plk1 and Aurora B, each kinase phosphorylates specific residues on the protein [1, 2].
“…Haspin is regulated by allosteric auto-inhibition and activated through a Plx1-dependent mechanism”
Ghenoiu et al 1 also dissected the molecular mechanism that keeps Haspin silent during interphase. They identified an evolutionarily conserved, basic amino acid-rich stretch of residues (which they called HBIS, for Haspin basic inhibitory segment) that directly inhibits Haspin through binding to its C-terminally located kinase domain. Importantly, this group was able to show that HBIS interaction with the catalytic domain of Haspin is regulated by Plx1-dependent phosphorylation: HBIS has affinity for the kinase domain of Haspin, but heavy N-terminal Plk1-dependent phosphorylation relieves this inhibitory interaction (Fig 1).
How Plk1 and Aurora B cooperate in the activation of Haspin is analyzed in detail by Zhou et al 2. They propose two major, partially independent pathways that temporally regulate the activity of Haspin. Whereas Plk1 has a prominent role during prophase as a Haspin-activating kinase, Aurora B takes over in prometaphase and metaphase (Fig 1). Importantly, Higgins and collaborators put forward a model by which a single phosphorylation event catalyzed by Cdk1/cyclin B leads to the sequential action of Plk1 and Aurora B, with the consequent full activation of Haspin. Such a mechanism has implications for the regulation of Aurora B activity by Plk1, which has previously been reported by others (reviewed in 5). How H3T3 phosphorylation and Aurora B are excluded from the chromosome arms to concentrate on centromeres as cells progress from prophase to prometaphase remains to be determined.
Collectively, both studies shed light on the mitotic regulation of Haspin and its role in targeting Aurora B to centromeres through H3T3 phosphorylation. Surprisingly, inhibition of Haspin activity has only a relatively mild effect on chromosome congression and affects spindle assembly checkpoint (SAC) response to variable extents 8, 9, whereas the inhibition of Aurora B causes an apparently more penetrant phenotype (reviewed in 5). This reinforces the recent view that tension-based regulation of kinetochore–microtubule attachment stability is independent of Aurora B localization on centromeres 10. In agreement with this view, artificially targeting Aurora B to the centromeres does not restore a fully functional SAC upon Haspin inhibition 8, 9. It will be essential to investigate the effect of preventing Haspin activation by Plk1 on kinetochore–microtubule attachment stability. This could be used as a tool to dissect whether Aurora B-mediated error-correction mechanisms and SAC response depend on Aurora B centromeric targeting.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Ghenoiu C, Wheelock MS, Funabiki H. Mol Cell. 2013;52:734–745. doi: 10.1016/j.molcel.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Tian X, Zhu C, et al. EMBO Rep. 2014;15:273–281. doi: 10.1002/embr.201338080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AE, Ghenoiu C, Xue JZ, et al. Science. 2010;330:235–239. doi: 10.1126/science.1189505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Dai J, Daum JR, et al. Science. 2010;330:231–235. doi: 10.1126/science.1189435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena M, Wheelock M, Funabiki H, et al. Nat Rev Mol Cell Biol. 2012;13:789–803. doi: 10.1038/nrm3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Ulyanova NP, van der Waal MS, et al. Curr Biol. 2011;21:1061–1069. doi: 10.1016/j.cub.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswaran J, Patnaik D, Filippakopoulos P, et al. Proc Natl Acad Sci USA. 2009;106:20198–20203. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Ulyanova NP, Daum JR, et al. J Cell Biol. 2012;199:251–268. doi: 10.1083/jcb.201205106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Antoni A, Maffini S, Knapp S, et al. J Cell Biol. 2012;199:269–284. doi: 10.1083/jcb.201205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CS, Desai A. Nature. 2013;497:118–121. doi: 10.1038/nature12057. [DOI] [PMC free article] [PubMed] [Google Scholar]