

Abstract
Regenerative medicine aims to replace the lost or damaged cells in the human body through a new source of healthy transplanted cells or by endogenous repair. Although human embryonic stem cells were first thought to be the ideal source for cell therapy and tissue repair in humans, the discovery by Yamanaka and colleagues revolutionized the field. Almost any differentiated cell can be sent back in time to a pluripotency state by expressing the appropriate transcription factors. The process of somatic reprogramming using Yamanaka factors, many of which are oncogenes, offers a glimpse into how cancer stem cells may originate. In this review we discuss the similarities between tumor dedifferentiation and somatic cell reprogramming and how this may pose a risk to the application of this new technology in regenerative medicine.
Keywords: cancer stem cells, dedifferentiation, somatic reprogramming, tumor plasticity
Introduction
A new branch of medicine will develop that attempts to change the course of chronic disease and in many instances will regenerate tired and failing organ systems [1, 2].
Many human diseases are caused by deficits in the quantity or functionality of particular cells. These diseases include neurodegenerative disorders, certain types of blindness and deafness, diabetes, and some types of liver and heart disease. The idea that degenerative as well as genetic diseases can be treated by regenerating the diseased organ or replacing the “damaged” cells with “healthy” new cells is fast becoming a reality since the first isolation of human embryonic stem cells (ESCs) and generation of induced pluripotent stem cells (iPSCs) from terminally differentiated somatic cells [3, 4]. Remarkably, the process of dedifferentiation or reprogramming of the somatic cells by Yamanaka factors, many of which are oncogenes, offers a new insight into cancer stem cells (CSCs). They may be the product of dedifferentiation of somatic cells following oncogenic insult. Cancer cells are the ultimate survivors and will exploit and subvert the cellular machinery to achieve that goal, by proliferation, dedifferentiation, and even transdifferentiation. Here, we will highlight some human cancers that may be the product of somatic cell reprogramming and as such may even pose some risk to the application of iPSCs in regenerative medicine.
From ESC to iPSC
Embryonic stem cells have attracted special attention by virtue of their pluripotent nature, that is, the ability to self-renew indefinitely in culture while retaining the capacity to differentiate into nearly all cell types in the body. Since their isolation, ESCs have been regarded as gold standard for their unique properties and extraordinary potential in regenerative medicine. Despite these important properties and remarkable qualities, ESC-based therapy has many limitations in treating human diseases. ESCs are derived from the inner cell mass of pre-implantation embryos, and hence patient-specific, and cannot be used as a general cell source for transplantation to other patients in need due to the risk of immune rejection. Historically, the solution to overcome this obstacle comes from seminal frog studies when Briggs and King [5] demonstrated the reversal of cell differentiation by transplantation of a viable cell nucleus into an enucleated frog egg. They succeeded in producing normal swimming tadpoles of Rana pipiens by transplanting the nuclei of embryo (blastula) cells. Later, similar experiments were carried out with eggs of the South African frog Xenopus laevis using nuclei from fully differentiated cells [6]. Collectively, these results challenged the unidirectional developmental model: Cells now can go back in time, dedifferentiate by changes in nuclear gene expression while maintaining their genome intact. Somatic cell nuclear transfer (SCNT), or nuclear reprogramming, is the technology by which the nucleus of the donor somatic cell is removed and transferred into an enucleated oocyte, where undefined factors in the cytoplasm of this oocyte are able to reprogram the somatic donor nucleus to a pluripotent state.
In a remarkable experiment, Takahashi and Yamanaka [3] demonstrated that introduction of mere four genes (Oct-3/4, Sox2, c-Myc, and KLF4) into an adult mouse fibroblast population can generate colonies with the characteristics of ESCs. These colonies were capable of differentiation to endodermal, ectodermal, and mesodermal lineages upon transplantation in immunodeficient mice. The authors termed them as induced pluripotent cells, or iPSCs. In the subsequent years, a variety of approaches and a very wide variety of differentiated cells have now been used to generate iPSCs [7–9].
The accumulated knowledge in the mechanisms and pathways involved in cellular reprogramming and induced pluripotency clearly shows the important connections between protein and transcriptional networks and how these factors affect the chromatin landscape. Changes in chromatin structure clearly affect global gene expression directly contributing to cell fate transitions. The key pluripotency transcription factors described before are intertwined with chromatin-associated factors to form sophisticated networks with complex regulatory interactions responsible for the maintenance of stemness and differentiation states. Chromatin marks linked to gene activation, particularly HK4me3, are highly enriched in genes expressed in ESCs. Conversely, upon differentiation, cells acquire chromatin marks associated with transcriptional repression such as H3K27me3 [10]. Not surprisingly, chromatin markers of iPSCs resemble those found in ESCs [11]. Chromatin changes in both promoters and enhancers are early events in response to the core transcription factors during the reprogramming process [12]. We are still learning how the epigenetic landscape is reset during reprogramming of somatic cells to iPSC and how these modifications determine the cell fate.
Interestingly, the acquisition of stem cell properties has also been reported in the case of normal differentiated cells in certain organs. The non-stem cell compartment seems to be the source of a new pool of cells with stem-like characteristics very similar to the endogenous stem cell counterparts in the organ.
Examples of this interconversion between stem and non-stem cell compartments have been shown in a subpopulation of basal-like human mammary epithelial cells [13], a rare subpopulation of somatic cells from breast tissue [14]. Differentiated airway epithelial cells have also been reported to dedifferentiate to a stem-like state [15], and upon tissue damage, Dll1+ intestinal secretory progenitor cells can also acquire cell plasticity and regain stemness [16]. If terminally differentiated cells can regain stem cell traits to maintain a balanced equilibrium between non-stem and stem cell compartments or to be able to regenerate damage tissue, it is fair to assume that this process can be adopted in disease states like cancer.
Dedifferentiation in cancer cells
There are multiple levels of heterogeneity associated with cancer, and this heterogeneity is one of the hallmarks of cancers arising in several organs. Genetic heterogeneity in the majority of the cancers is reflected by genome instability, and in addition to these genetic alterations, the state of the cell may be changed epigenetically. Phenotypic heterogeneity refers to the diverse functional properties and expression of different lineage markers that tumor cells can adopt along cancer progression. Based on cell surface markers, we can identify distinct subpopulations of neoplastic cells within the same tumors, suggesting that irrespective of their genetic alterations, cancer cells may exist in different states of differentiation [17]. The latest has been defined as intratumor heterogeneity, to explain the variations found within individual tumors, and intertumoral heterogeneity refers to the molecular differences that occur between tumors initiated in the same organ, which allows the classification of these tumors in different subtypes and may even represent biologically distinct disease entities [18].
Cancer stem cell can be defined as the cells within a tumor that possess the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor. CSCs are thus a biologically unique subpopulation of cells that can perpetuate indefinitely as oppose to the bulk of cells that reside in the tumor, and are mostly insensitive to currently used cancer therapies. The CSC model assumes that this unique subpopulation of cells sustain malignant growth by means of their ability to self-renew and the possibility to give rise to progeny with self-limited proliferative capacity. This suggests a hierarchical organization where CSCs are responsible for the generation of the heterogeneity found within tumors. Although CSCs exhibit the stem cell properties of self-renewal and differentiation, they do not necessarily originate from the transformation of normal tissue stem cells [18].
Several recent studies now suggest that not all cancers strictly conform to the unidirectional hierarchical CSC model, and entertain the possibility of “tumor cell plasticity”, where non-CSC can dedifferentiate and acquire CSC-like properties under certain conditions as demonstrated by examples below:
Glioblastoma (GBM), the most common and aggressive subtype of the malignant gliomas, is characterized by intense proliferation, invasion, and intratumor heterogeneity. A decade ago, Ronald DePinho's group demonstrated that the combined loss of p16INK4a and p19ARF enables mature astrocyte dedifferentiation in response to EGFR activation [19]. Moreover, transduction of Ink4a/Arf(−/−) neural stem cells (NSCs) or astrocytes with constitutively active EGFR induces a common high-grade glioma phenotype. These findings identify neural stem cells and astrocytes as equally permissive compartments for gliomagenesis. The identification of TUJ1-positive neurons in the tumors originating from the transformed astrocytes suggested that dedifferentiation may be so complete as to generate a pluripotent cell with the potential to make neurons as well as glia. More recently, our group showed that GBM can originate from a variety of cells in the brain, including terminally differentiated cortical astrocytes and neurons [20]. Transduction by oncogenic Cre-inducible lentiviruses in the cortex of synapsinI-Cre or GFAP-Cre transgenic mice, which drive the expression of Cre specifically in neurons and glial cells, respectively, induced the formation of gliomas. Interestingly, these tumors mostly expressed markers of progenitor/neural stem cells, nestin and Sox2. In a study aimed to follow the kinetic expression of some of these markers during tumor development, we observed that at early stages, differentiation markers are progressively diminished, while nestin, a marker of NSC, undetectable a few days after transduction, increased significantly with tumor progression (Fig 1). We proposed that oncogenic-induced dedifferentiation of mature cells in the brain to a stem-/progenitor-like state leads to heterogeneous glioma tumors (Fig 2). The genetically acquired plasticity of these cells allows progression and maintenance of this aggressive tumor and even formation of its own blood vessels by transdifferentiation [21]. These data also supported the view originally proposed by Ronald DePinho and his group [19] that dysregulation of specific genetic pathways, rather than cell of origin, dictates the emergence and phenotype of high-grade gliomas.
Figure 1.
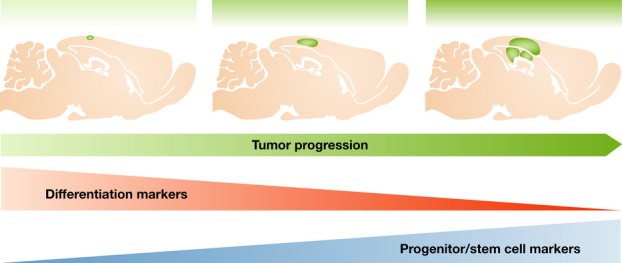
Glioblastoma tumors induced by oncogenic lentivirus either in neurons or in glia in the cortex initially express differentiation markers (e.g., Tuj1 and GFAP, respectively), but as tumor progresses, these markers decrease and stem/progenitor markers become predominantly expressed (like nestin and Sox2) [20].
Figure 2.
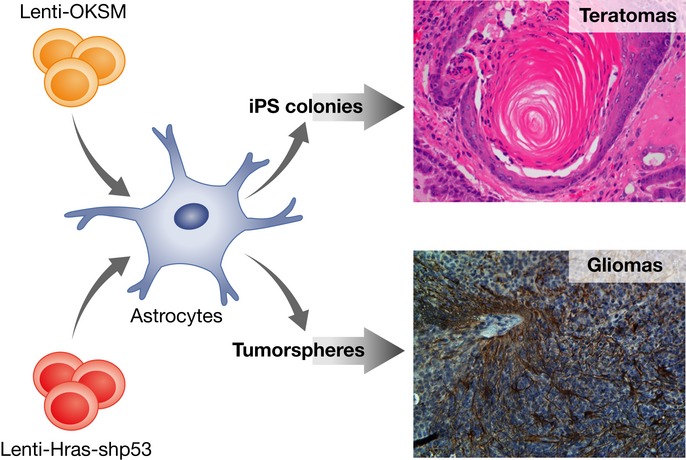
An astrocyte transduced with LV-Hras-shp53 dedifferentiates/reprograms to a progenitor/stem cell state, leading to tumorsphere formation. These tumorspheres when transplanted orthotopically in the brain of mice lead to glioma tumors. The same astrocytes transduced with a lentivector carrying the four transcription factors (Oct-4, Klf-4, Sox2, and myc) reprogram and form iPS colonies than when transplanted s.c. into mice develop teratomas.
The bidirectional conversion between CSCs and non-CSCs was also found in intestinal tumors, where an inflammatory stroma, through NF-κB activation, enhances Wnt signaling and leads to the reprogramming process. If any differentiated cell can be reprogrammed to a pluripotent state through the right combination of transcriptions factors [3], then, following the same line of reasoning and in theory, intestinal epithelial cells (IEC) can dedifferentiate to a progenitor/stem cell state given an appropriate transcription factor, for example Wnt signaling, is strongly activated. In the study of Florian Greten's group, the combination of an oncogenic hit like Kras and the activation of NF-κB induces the stabilization of β-catenin and thereof the activation of the β-catenin/tcf transcription complex, leading to the conversion of non-stem IEC into IEC with stem cell properties [22]. The implication of NF-κB activation in the context of inflammation and cancer has already been shown previously [23]. But in this study, both Kras and TNF-α-dependent NF-κB activation enhances β-catenin/Tcf-mediated transcriptional activity and induces dedifferentiation of non-stem IEC into tumor-initiating cells, supporting a model of bidirectional interconvertibility rather than the strict unidirectional model of the stem differentiation hierarchy (reviewed by [24]). It remains unclear whether this plasticity of tumor cells is specific to certain types of cancer, how frequently this process of interconversion occurs in vivo, and what is the mechanism that regulates the dynamic equilibrium that exists between non-CSC and CSC. Recently, Robert Weinberg's group addressed some of these questions and found that the switch between non-CSC to CSC state is frequently common in certain types of breast cancer, and proposed a mechanism responsible for this transition. Based on the notion that the epithelial–mesenchymal transition (EMT) generates cells with stem cell properties [25], they found ZEB1, a transcription factor known to be involved in the EMT program, to be required for the conversion from non-CSC to CSC and also for the maintenance of the CSC-like activity [26]. More specifically, the chromatin configuration in a bivalent/poised state associated with the ZEB1 promoter enables de novo generation of CSCs from non-CSC populations. Not surprisingly, the tumor microenvironment provides the stimuli, in this case by the secretion of TGF-β that enhances the rate of transition from non-CSC to CSC by inducing ZEB1 expression. The implications of tumor plasticity can go beyond the primary tumor and play an important role in tumor metastasis. It has been suggested that CSCs are endowed with multiple traits that are required for most of the steps related to the invasion–metastasis cascade [27] and therefore might be key players in metastatic tumors. Although it still needs to be proven, non-CSC too may leave a primary tumor and, upon arrival to the secondary site, dedifferentiate and create a new pool of CSCs, hence becoming founders of new metastatic colonies.
The study from Oleksi Petrenko's laboratory used a model of conditional expression of oncogenic KrasG12D in mice to show the phenotypic changes at the molecular and cellular level required in the process of transformation. Their results support the concept of tumor plasticity and the existence of a controlled balance between differentiated tumor cells and tumor cells in a stem-like state [28]. The acquisition of CSC characteristics induced by oncogenic Ras and one of its downstream targets, Myc, is essential to initiate the malignant transformation. According to their results, genome reprogramming and dedifferentiation are important early steps in pancreatic tumor initiation, progression, and even predispose early-stage pancreatic tumor cells to metastatic spread. Activating Kras mutation occurs at a frequency of 90% in pancreatic cancer and is the most frequent mutation among all cancers [29–32]. Kras signals via downstream effector pathways, and in their study, the transcription factor Myc is implicated in the regulation of self-renewal and development of metastatic pancreatic cancer cells. Collectively, their data support the studies described before in this section, wherein any cell in the tumor, regardless of its differentiation state, has the potential to become a CSC given that an appropriate oncogenic insult induces this plasticity. The big challenge in the field now will be to elucidate the mechanism responsible for this process.
Although we have focused so far on tumor reprogramming as a consequence mostly of cell-intrinsic changes, the tumor microenvironment plays a very important role in this process. To this extent, two independent studies showed that cancer stemness can be regulated by extrinsic factors generated in the tumor microenvironment. The study of Jean Paul Medema's group suggested that colorectal stroma cells surrounding the tumor, specifically myofibroblasts, by secreting hepatocyte growth factor can induce nuclear β-catenin localization, Wnt signaling activity, and the generation of stem cell features in more differentiated tumor cells [33]. In this scenario, myofibroblasts create a niche that support dedifferentiation of colon cancer cells to a CSC phenotype, and this dynamic conversion toward stemness is regulated by the intensity of Wnt activity. As we discussed previously, Wnt activity was an important cell-intrinsic feature responsible for the reprogramming of IEC into CSC. But the results of Medema's group suggest that stroma cells can not only sustain high Wnt activity in the surrounding CSC but can also induce reprogramming of differentiated tumor cells into CSC by secreting extrinsic factors and activating their Wnt signaling pathway. An inflammatory tumor microenvironment can also shape the response of the tumor cells to adoptive cell transfer therapy (ACT). TNF-α secreted by macrophages and T cells in the tumor microenvironment can induce an interconversion between differentiated and dedifferentiated melanoma tumor cells [34]. This induced inflammation plasticity allows the tumor cells to escape immune surveillance and in the context of ACT explains relapse of the tumors after the initial remission. Changes in the differentiation state of the tumor cells also change the lineage markers expressed on the surface of these tumor cells, generating a mechanism of resistance and escape from cytotoxic T-cell recognition.
Versatility of cancer stem cells: transdifferentiation
Stem cells have acquired the plasticity of not only self-sustenance and differentiation into expected lineages, but can also transdifferentiate into other lineages and cell types. Despite the fact that GBMs are highly vascularized, inhibitors of angiogenesis, like avastin, are not very effective [35, 36]. Several laboratories have discovered that in GBMs, the cancer cells can transdifferentiate to endothelial cells (TDECs), leading to the formation of blood vessels, which are functional [21, 37, 38]. In some human GBMs, over 70% of the endothelial cells forming the blood vessels were derived from cancer cells [37]. More recently, it has been shown that in human GBMs, the cancer cells can transdifferentiate to pericytes [39]. In some ways, it is not surprising, because in mice, NSCs have been shown to differentiate into endothelial cells which then presumably provide the niche for NSCs to differentiate to various cells of CNS lineage [40]. In the context of glioma tumors, the transdifferentiation process seems to be reversible, with tumor endothelial cells being able to reverse to a CSC state (Yasushi Soda and Amy Rommel, personal communication; Fig 3). Clearly, the emphasis will now be to understand the molecular mechanisms of transdifferentiation. Although there is not as yet evidence of transdifferentiation of CSCs to other cell types, it may be prudent to further analyze other cell types, especially the stromal cells surrounding the tumors, some of which may actually be of tumor origin.
Figure 3.

Normal mechanism of neuronal differentiation: Neural stem cell can self-renew, go through an intermediate progenitor cell, and differentiate into oligodendrocytes, astrocytes, neurons, and endothelial cells. In the formation of glioblastoma, the transformed neurons, astrocytes, and possibly oligodendrocytes can dedifferentiate/reprogram to become cancer stem cells (CSCs), which can then continue to self-proliferate and differentiate to more transformed neurons and astrocytes. The transformed neurons and astrocytes can also transdifferentiate into endothelial cells (TDECs), which can again dedifferentiate to CSCs.
Parallels between reprogramming of somatic cells and dedifferentiation in cancer
The concept that differentiated cells become “dedifferentiated” in cancer has been controversial, but the latest studies described above have provided sufficient data supporting the existence of this process and even the requirement of this transition for tumorigenesis. In this context, dedifferentiation should apply only to a situation where a more specialized cell type looses expression of lineage-specific genes of specialized tissue function, in favor of expression of the more primitive signature of the related tissue development. Indeed, in some types of cancer, the alterations in differentiated cells result in reversion to a stem cell phenotype. The process of iPS generation also implies a reversion from a differentiated state to a pluripotent state, and following this reprogramming, the differentiated somatic cells acquire unlimited proliferating properties and self-renewal capacity. As discussed below, this is only one of the many characteristics that iPSCs share with cancer development.
Oncogenic transformation frequently involves de novo acquisition of developmental programs, analogous to cellular reprogramming, and yields cells with unlimited self-renewal potential, a feature shared with iPSCs. This implies that similar pathways can be associated with both the induction of pluripotency and oncogenesis. The appropriation of specific ESC-associated regulators and gene expression pathways by poorly differentiated solid tumors has been described [30]. Indeed, molecular analysis of gene sets associated with ESC identity in various human tumor types highlights the fact that tumorigenesis can hijack embryonic pathways of tissue development. Another program that is being replayed in the evolution of primary tumors toward metastatic phenotypes and, as previously discussed, shared common transcription players in tumor dedifferentiation is the EMT. The EMT and the reverse process, termed the mesenchymal–epithelial transition (MET), play central roles in embryogenesis [25]. Some of the transcription factors orchestrating EMTs have been found to confer malignant traits. Furthermore, it has been shown previously that EMT can reprogram differentiated mammary epithelial cells into a less differentiated epithelial stem cell with mesenchymal traits, establishing a link between EMT and the acquisition of stem cell properties [25].
When focusing on dedifferentiation processes and comparing those with dedifferentiation leading to tumor cells, and the relevant role that CSCs play in tumor malignancy and growth, it is inescapable to appreciate the similarities between somatic cell reprogramming and tumorigenesis. Each of the iPSC reprogramming factors has established roles in oncogenesis. Oct-4 plays a driving role in initiating germ cell tumors and has been proposed to be a useful marker for germ cell tumors such as seminomas and embryonal carcinomas [41]. Although Oct-4 is highly expressed in seminomas, other non-germ-cell-originated tumors show detectable levels compared to their normal cell counterparts, like breast carcinomas and papillary carcinomas of the thyroid [42], as well as esophageal cell carcinoma [43] and prostate cancer [44]. The notion that Oct-4 induction affects epigenetic regulations and contributes to the maintenance of undifferentiated proliferating cells [45] may provide a possible link between transcription factor-mediated reprogramming and oncogenesis. Sox2 is amplified in lung and esophagus cancer and is an essential driver of CSCs subpopulations in GBM, breast cancer, and Ewing sarcoma [46, 47]. A large variety of human malignancies express high levels of MYC. Its expression may explain the observation that most of the mice generated with iPSC clones spontaneously developed tumors [48]. Myc is an important transcriptional regulator in ESC, and it significantly promotes the process of iPSC derivation. Its role as a global amplifier of gene expression not surprisingly also drives a wide range of malignant programs [49]. The list can go on including KLF4, Nanog, Lin28, and other pluripotency factors and transcription factors that mediate direct lineage conversion, emphasizing the link between reprogramming and oncogenesis [50, 51].
Changes in the epigenetic landscape have also been implicated in both reprogramming and oncogenic transformation. Epigenetics can be defined as the external modifications to DNA that regulate gene expression without changes in the underlying DNA sequence. Two major epigenetic regulations are DNA methylation and histone modifications. DNA methylation is a relatively stable epigenetic modification that mediates silencing of repetitive elements and certain gene promoters [52]. Changes in DNA methylation are required to achieve nuclear reprogramming, evidenced by the loss of promoter methylation in key pluripotency genes during iPSC generation [3]. Several studies have reported de novo DNA methylation during reprogramming of differentiated cells to iPSC. DNA methyltransferases (DNMTs) are involved in the establishment and maintenance of DNA methylation, and high expression of DNMTs has been reported during the induction of reprogramming as well as in ESCs [53]. Tet1 proteins facilitate the hydroxylation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), and this modification in DNA methylation plays an important role during the reprogramming process. The enrichment of 5hmC in the Oct-4 loci facilitates DNA demethylation and the transcriptional reactivation required for the induction of reprogramming by the core transcription factors (OKSM) [54]. Abnormal patterns of genomic methylation in cancer are characterized by global losses of genomic methylation and hypermethylation, predominantly in CpG islands, a well-recognized epigenetic event in cancer [52]. Both inactivation and higher expression of DNMTs have been reported in cancer, and previous studies have suggested that such altered expressions of DNMTs could partly explain the abnormal methylation patterns observed in cancer cells [55]. Chromatin regulators (CRs) have also been involved both in cellular reprogramming and in oncogenesis. Like the transcription factors described before, CRs have also been implicated in tumorigenesis, either acting as oncogenes or as tumor suppressor genes. CRs are associated with both repressive and active chromatin states. Epigenetic silencing is associated with the following histone modifications: H3K27me3, H3K9me2, and H3K9me3. As an example, inhibition of CRs that catalyze H3K9 methylation, including Suv39h1, Setdb1, and G9a, leads to a higher reprogramming efficiency [56, 57] and all three have established roles in different malignancies [58, 59]. There are also several examples of CRs involved in active chromatin states that play important roles both in reprogramming and in cancer (e.g., MLL and Dot1l [60]). In our GBM model system, another common feature between our dedifferentiated tumorspheres and iPSCs is their chromatin state. It is well accepted that ESCs as well as iPSC have an “open” chromatin, while differentiated cells have a “close” chromatin [10]. Using a qRT-PCR designed in our laboratory to detect highly repetitive DNA elements in the murine pericentric heterochromatin (e.g., minor and major satellites [61]), we showed that both dedifferentiated tumorspheres and NSCs have a relaxed chromatin that resulted in derepression of normally silenced genes in the heterochromatin regions [20].
There are many parallels between reprogramming and cancer. The similarities between the process of reprogramming cells to iPCS and differentiated tumor cells to CSCs suggest that some of these mechanisms, like epigenetic resetting, can render cells in a susceptible state where genetic alterations are only the next step toward transformation and tumor progression. Understanding the mechanisms governing cellular reprogramming and induced pluripotency may shed light into deciphering the processes involved in tumorigenesis.
Prospects of eliminating cancer stem cells
Treatments of tumors, which have resident stem cells, will not be very effective, leading to recurrence, unless the stem cells are also eliminated. Because stem cells have unique expression of genes required for self-perpetuation, like chromatin remodelers, perhaps they could be the targets of drug therapy. In preliminary experiments, we have shown that if GBM cells are transduced with an shRNA targeting Bmi1, an essential gene for self-replication, upon transplantation, these cells are unable to form tumors. In GBMs, additional molecules that prevent differentiation to various CNS lineages can also be targets of therapy, though blocking normal differentiation process in the brain may also have deleterious side effects. In case of GBMs, approaches to block transdifferentiation to endothelial cells may also be another avenue to explore as a therapeutic agent. In other CSCs, it will be very beneficial to find specific cell surface proteins/receptors which can serve as therapeutic targets. Identification of unique drugable targets (like kinases, transcription factors) in CSC will be very helpful in eliminating them from the tumors. In addition to interfering with the proliferation of CSCs, alteration of microenvironment should also be considered to prevent both formation and proliferation of CSC. Because many of these interfering strategies will also have an impact on normal stem cells, it will be important to ensure relative safety of the treated patients.
The risks and limitations of iPS-based cell therapy
The first therapeutic success using iPSCs was reported for the mouse model of sickle cell anemia, a blood disorder disease [62]. The defective β-globin gene was corrected by homologous recombination in a mutant iPSC line and the transplantation of these cells into the mutant mice cured the disease. This is a very good example and model of iPSC-mediated regenerative medicine: a genetic disorder disease caused by a single defective gene that can be corrected by replacement in autologous cells. The first limitation that comes to mind when thinking of autologous iPSC for individualized medicine is the associated high medical costs, the lack of large-scale culture technologies, and the timeframe needed to prepare the cells for transplantation (crucial, for example, for spinal cord injuries). Another important aspect when considering using iPSC in the clinics is the quality of these cells, mostly derived from somatic cells of aged individuals. The risk that comes with this source of cells is the incidence of spontaneously occurring tumors, which commonly increases exponentially with aging. Although it has been reported that epigenetic changes and telomerase activity in cells of aging individuals can be reversed during the reprogramming process [63], somatic mutations and chromosomal aberrations acquired by these cells are not corrected in the reprogramming process. These abnormalities may lead to iPSCs with reduced functionality and higher risk of developing cancer.
Other problems, mostly associated with the first generation of human iPSCs, were the integration site of retroviral vectors, the risk of insertional mutagenesis and hence the risk of tumorigenicity, and the use of undefined serum-containing media to support iPSC generation. In addition, the use of oncogenic transgenes, such as MYC, can also increase the risk of tumor development. As mentioned above, new and safer technologies for the generation of iPSCs have emerged in the past few years that diminished these risks.
Although it is well accepted today that iPSCs are pluripotent, the findings in the past few years have been controversial in regard to whether ESCs and iPSCs are distinct cell types. Some groups argued that these two populations are undistinguishable [64–66]; others have reported that they differ in their molecular signature [67–70], DNA methylation [51, 70–72], and their potential for differentiation [73]. Yamanaka's group recently reported a subset of iPSC lines that have aberrant gene expression and defective potential in neural differentiation [74]. They performed a large-scale analysis of human iPSCs and ESCs and found that although they have overlapping variations in gene expression and DNA methylation, some iPSC clones retained a significant number of undifferentiated cells, even after neural differentiation, and formed teratomas when transplanted in vivo. These differentiation-defective iPSC clones express high levels of LTRs of endogenous retroviruses and retain a substantial number of undifferentiated cells after in vitro directed neural differentiation. Clearly, prior to applications in regenerative medicine, these defective iPSC clones need to be identified and eliminated.
Sidebar A. In need of answers
Do all cancers have CSCs?
Do all CSCs originate by dedifferentiation or reprogramming?
Does the microenvironment influence the maintenance or frequency of CSCs?
What is the mechanism of transdifferentiation?
Do CSCs contribute toward tumor invasiveness?
What is the role of epigenetic changes in tumor plasticity?
Concluding remarks
It has long been known that many cancer cells show markers and properties of ESCs, and some of these have often been targets of therapy. The discovery of CSCs further points to this notion. In organismal development, events are deterministic and move forward in one direction, without ability to reverse the process. The famous Waddington's landscape for development visualizes the developmental history of a cell in an embryo, “by a ball rolling down the ‘landscape’ making several ‘choices’ as to which way to go—just as the developing embryo is influenced down certain ‘paths’ by various genetic and environmental factors—and by the time it reaches the bottom of the landscape, it will have made several such choices”. The ball eventually lands at the bottom, signaling that being pushed upward will be difficult, thereby hinting that the process is essentially one directional. The discovery of Yamanaka and colleagues, however, shows that terminally differentiated cells can be pushed upwards, going back to the original pluripotent cell, a situation created by oncogenic insult to terminally differentiated cells like glia or neuron in the case of GBM (Fig 3). The convergence and commonality of CSCs and iPSCs opens a new avenue to develop therapeutic approaches to combat recurring cancers.
Acknowledgments
We thank Jamie Simon for the graphic design. IMV is an American Cancer Society Professor of Molecular Biology and holds the Irwin and Joan Jacobs Chair in Exemplary Life Science. This work was supported in part by grants from the NIH (HL053670), Cancer Center Core Grant (P30 CA014195-38), Ipsen, the Leona M. and Harry B. Helmsley Charitable Trust, and the H.N. and Frances C. Berger Foundation. The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- Bmi1
B-lymphoma Mo-MLV insertion region 1 homolog
- CNS
central nervous system
- Cre
Cre recombinase
- CSC
cancer stem cell
- Dll1
Delta-like gene 1
- Dot1l
DOT1-like, histone H3 methyltransferase
- EGFR
epidermal growth factor receptor
- EMT
epithelial–mesenchymal transition
- ESC
embryonic stem cell
- GBM
glioblastoma
- H3K27me3
histone H3 trimethyl Lys27
- H3K9me2
histone H3 dimethyl Lys9
- H3K9me3
histone H3 trimethyl Lys9
- iPSC
induced pluripotent stem cell
- Klf4
Kruppel-like factor 4
- Kras
Ki-ras2 Kirsten rat sarcoma viral oncogene
- LTR
long terminal repeat
- MET
mesenchymal–epithelial transition
- Nanog
Nanog homeobox
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NSC
neural stem cell
- Oct-3/4
octamer-binding transcription factor 3/4
- Ras
rat sarcoma
- Setdb1
Set domain, bifurcated 1 methyltransferase
- Sox2
sex-determining region Y-related box2
- Suv39h1
suppressor of variegation 3–9 homolog 1
- TDEC
tumor-derived endothelial cell
- Tet1
Ten-eleven translocation methylcytosine dioxygenase 1
- TGF-β
transforming growth factor beta
- TNF-α
tumor necrosis factor alpha
- TUJ1
neuron-specific class III beta-tubulin
- Wnt
Wingless integrase-1
- ZEB1
zinc finger E-box-binding homeobox 1
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Kaiser LR. The future of multihospital systems. Top Health Care Financ. 1992;18:32–45. [PubMed] [Google Scholar]
- 2.Lysaght MJ, Crager J. Origins. Tissue Eng Part A. 2009;15:1449–1450. doi: 10.1089/ten.tea.2007.0412. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 5.Briggs R, King TJ. Transplantation of living nuclei from blastula cells into enucleated frogs' eggs. Proc Natl Acad Sci USA. 1952;38:455–463. doi: 10.1073/pnas.38.5.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurdon JB. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol. 1962;10:622–640. [PubMed] [Google Scholar]
- 7.Sommer CA, Mostoslavsky G. The evolving field of induced pluripotency: recent progress and future challenges. J Cell Physiol. 2013;228:267–275. doi: 10.1002/jcp.24155. [DOI] [PubMed] [Google Scholar]
- 8.Malik N, Rao MS. A review of the methods for human iPSC derivation. Methods Mol Biol. 2013;997:23–33. doi: 10.1007/978-1-62703-348-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez F, Boue S, Izpisua Belmonte JC. Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat Rev Genet. 2011;12:231–242. doi: 10.1038/nrg2937. [DOI] [PubMed] [Google Scholar]
- 10.Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, Bernstein BE, Meissner A. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy S, Gascard P, Dumont N, Zhao J, Pan D, Petrie S, Margeta M, Tlsty TD. Rare somatic cells from human breast tissue exhibit extensive lineage plasticity. Proc Natl Acad Sci USA. 2013;110:4598–4603. doi: 10.1073/pnas.1218682110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, Law BM, Vinarsky V, Cho JL, Breton S, Sahay A, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–223. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Es JH, Sato T, van de Wetering M, Lyubimova A, Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J, et al. Dll1 + secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–1104. doi: 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 19.Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 20.Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, Verma IM. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, Pastorino S, Yang M, Hoffman RM, Kesari S, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci USA. 2011;108:4274–4280. doi: 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 23.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 24.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 25.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D'Alessio AC, Young RA, Weinberg RA. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154:61–74. doi: 10.1016/j.cell.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. Direct reprogramming by oncogenic Ras and Myc. Proc Natl Acad Sci USA. 2013;110:3937–3942. doi: 10.1073/pnas.1219592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–389. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anokye-Danso F, Trivedi CM, Juhr D, Gupta M, Cui Z, Tian Y, Zhang Y, Yang W, Gruber PJ, Epstein JA, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 34.Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412–416. doi: 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- 35.Jain RK, Tomaso di E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 36.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Wagner M, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 37.Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 38.Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 39.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wurmser AE, Nakashima K, Summers RG, Toni N, D'Amour KA, Lie DC, Gage FH. Cell fusion-independent differentiation of neural stem cells to the endothelial lineage. Nature. 2004;430:350–356. doi: 10.1038/nature02604. [DOI] [PubMed] [Google Scholar]
- 41.Gidekel S, Pizov G, Bergman Y, Pikarsky E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell. 2003;4:361–370. doi: 10.1016/s1535-6108(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 42.Madjd Z, Hashemi F, Shayanfar N, Farahani E, Zarnani A, Shrifi A, Akbari M. OCT-4, embryonic Stem cell marker expressed in breast, brain and thyroid carcinomas compared to testicular carcinoma. Iran J Cancer Prev. 2009;2:167–173. [Google Scholar]
- 43.Zhou X, Huang GR, Hu P. Over-expression of Oct4 in human esophageal squamous cell carcinoma. Mol Cells. 2011;32:39–45. doi: 10.1007/s10059-011-2315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Resende MF, Chinen LT, Vieira S, Jampietro J, da Fonseca FP, Vassallo J, Campos LC, Guimaraes GC, Soares FA, Rocha RM. Prognostication of OCT4 isoform expression in prostate cancer. Tumour Biol. 2013;34:2665–2673. doi: 10.1007/s13277-013-0817-9. [DOI] [PubMed] [Google Scholar]
- 45.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 46.Riggi N, Suva ML, De Vito C, Provero P, Stehle JC, Baumer K, Cironi L, Janiszewska M, Petricevic T, Suva D, et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010;24:916–932. doi: 10.1101/gad.1899710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell. 2012;10:678–684. doi: 10.1016/j.stem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 49.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris SA, Daley GQ. A blueprint for engineering cell fate: current technologies to reprogram cell identity. Cell Res. 2013;23:33–48. doi: 10.1038/cr.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daley GQ. Common themes of dedifferentiation in somatic cell reprogramming and cancer. Cold Spring Harb Symp Quant Biol. 2008;73:171–174. doi: 10.1101/sqb.2008.73.041. [DOI] [PubMed] [Google Scholar]
- 52.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao Y, Chen J, Li K, Wu T, Huang B, Liu W, Kou X, Zhang Y, Huang H, Jiang Y, et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell. 2013;12:453–469. doi: 10.1016/j.stem.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Linhart HG, Lin H, Yamada Y, Moran E, Steine EJ, Gokhale S, Lo G, Cantu E, Ehrich M, He T, et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21:3110–3122. doi: 10.1101/gad.1594007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, Liang H, Chen Y, Wu Y, Guo L, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- 58.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene. 2010;29:576–588. doi: 10.1038/onc.2009.361. [DOI] [PubMed] [Google Scholar]
- 60.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477:179–184. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, Cassady JP, Beard C, Brambrink T, Wu LC, Townes TM, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 63.Marion RM, Strati K, Li H, Tejera A, Schoeftner S, Ortega S, Serrano M, Blasco MA. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009;4:141–154. doi: 10.1016/j.stem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 64.Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guenther MG, Frampton GM, Soldner F, Hockemeyer D, Mitalipova M, Jaenisch R, Young RA. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell. 2010;7:249–257. doi: 10.1016/j.stem.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Newman AM, Cooper JB. Lab-specific gene expression signatures in pluripotent stem cells. Cell Stem Cell. 2010;7:258–262. doi: 10.1016/j.stem.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 67.Chin MH, Mason MJ, Xie W, Volinia S, Singer M, Peterson C, Ambartsumyan G, Aimiuwu O, Richter L, Zhang J, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marchetto MC, Yeo GW, Kainohana O, Marsala M, Gage FH, Muotri AR. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PLoS ONE. 2009;4:e7076. doi: 10.1371/journal.pone.0007076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghosh Z, Wilson KD, Wu Y, Hu S, Quertermous T, Wu JC. Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PLoS ONE. 2010;5:e8975. doi: 10.1371/journal.pone.0008975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, Qi Z, Downey SL, Manos PD, Rossi DJ, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol. 2011;13:541–549. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park IH, Yu J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Asciutti S, Akiri G, Grumolato L, Vijayakumar S, Aaronson SA. Diverse mechanisms of Wnt activation and effects of pathway inhibition on proliferation of human gastric carcinoma cells. Oncogene. 2011;30:956–966. doi: 10.1038/onc.2010.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koyanagi-Aoi M, Ohnuki M, Takahashi K, Okita K, Noma H, Sawamura Y, Teramoto I, Narita M, Sato Y, Ichisaka T, et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proc Natl Acad Sci USA. 2013;110:20569–20574. doi: 10.1073/pnas.1319061110. [DOI] [PMC free article] [PubMed] [Google Scholar]