

Abstract
The objective of the present study was to determine the impact of simulated apnea with intermittent hypoxia (IH) on endothelial barrier function and assess the underlying mechanism(s). Experiments were performed on human lung microvascular endothelial cells exposed to IH-consisting alternating cycles of 1.5% O2 for 30s followed by 20% O2 for 5 min. IH decreased transendothelial electrical resistance (TEER) suggesting attenuated endothelial barrier function. The effect of IH on TEER was stimulus dependent and reversible after reoxygenation. IH-exposed cells exhibited stress fiber formation and redistribution of cortactin, vascular endothelial-cadherins, and zona occludens-1 junction proteins along with increased intercellular gaps at cell-cell boundaries. Extracellular signal-regulated kinase (ERK) and c-jun NH2-terminal kinase (JNK) were phosphorylated in IH-exposed cells. Inhibiting either ERK or JNK prevented the IH-induced decrease in TEER and the reorganization of the cytoskeleton and junction proteins. IH increased reactive oxygen species (ROS) levels, and manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride, a membrane-permeable antioxidant, prevented ERK and JNK phosphorylation as well as IH-induced changes in endothelial barrier function. These results demonstrate that IH via ROS-dependent activation of MAP kinases leads to reorganization of cytoskeleton and junction proteins resulting in endothelial barrier dysfunction.
Keywords: oxidative stress, microvascular endothelial cells, cytoskeleton proteins, junction proteins
lung microvascular endothelial cells line the blood vessels of the microcirculatory bed forming a semipermeable barrier, selectively permitting alveolar gas exchange, while preventing fluid and solute flow across the alveolar capillary wall. The barrier function of the endothelial cells is regulated by the structural integrity of the cytoskeleton as well as the tight and adherens junctions with adjacent endothelial cells (8, 23). Given that endothelial cells constitute the first layer of the vasculature that is in direct contact with the blood, any perturbations in blood flow and/or its composition such as hypoxia might affect the barrier function. Systemic hypoxia (i.e., decreases in blood oxygen level) occurs under many different circumstances profoundly impacting a variety of physiological systems (20). Continuous exposure to hypoxia lasting 24 h has been shown to disrupt endothelial barrier function by activating proinflammatory cytokines (1), heat shock protein (HSP)27 (13), and hypoxia-inducible factor (HIF)-1α (21). Intermittent hypoxia (IH) is a hallmark manifestation of recurrent apnea patients with obstructive sleep apnea (OSA). Recent studies showed that OSA patients exhibit pulmonary edema, which could be prevented by treating OSA with continuous positive airway pressure (Refs. 4–6). Mimicking obstructive sleep apnea in anesthetized dogs also produce pulmonary edema (10). The pulmonary edema seen in OSA patients might reflect endothelial barrier dysfunction by IH. However, the effect of IH on endothelial barrier function and the underlying mechanisms are not known.
Emerging evidence suggests that systemic and cellular responses to IH are mediated by oxidative stress resulting from increased generation of reactive oxygen species (ROS) (12, 19). Since oxidative stress results in endothelial barrier dysfunction (3), we hypothesized that IH disrupts endothelial barrier function via ROS-dependent activation of signaling pathway(s). We tested this possibility in human lung microvascular endothelial cells exposed to a previously established in vitro IH paradigm (18), which recapitulates many effects of chronic IH in intact experimental animals.1
MATERIALS AND METHODS
Cell Culture
Human lung microvascular endothelial cells (Lonza Group) were plated at a density of ∼1 × 105/cm2 and cultured in EBM-2 complete medium supplemented with 10% fetal bovine serum, antibiotics, ascorbic acid, and growth factors under 20% O2 and 10% CO2 at 37°C. Before experiments, the cells were placed in serum-free medium for 18 h.
Exposure of Cells to IH
Endothelial cell cultures were exposed to alternating cycles of 1.5% O2 for 30 s followed by 20% O2 for 5 min, with a total duration of 330 s per cycle, in a humidified Lucite chamber at 37°C as described previously (31). Briefly, the chamber was equilibrated with gases at a flow rate of 2.4 l/min. The durations of hypoxia and normoxia were maintained using timer-controlled solenoid valves. The O2 levels in the culture medium and the ambient O2 levels in the chamber were monitored with an O2 electrode (Lazar Research Laboratories) and by an O2 analyzer (Beckman LB2), respectively. Cells exposed to repetitive normoxia (i.e., alternating cycles of 20% O2 for 30 s followed by 20% O2 for 5 min) served as controls. In experiments testing the effects of various drugs, cells were incubated with the indicated concentration of drugs for 30 min before exposure to either repetitive normoxia or IH without changing the medium.
Determination of Transendothelial Electrical Resistance
Transendothelial electrical resistance (TEER) was measured in an electrical cell-substrate impedance sensing system (Applied BioPhysics, Troy, NY) as described previously (26). Briefly, endothelial cells were grown to ∼95% confluence in polycarbonate wells containing gold electrodes connected to a phase-sensitive lock-in amplifier. Electrodes containing cells were placed in an electrical cell-substrate impedance incubator for 1 h to stabilize basal electrical resistance. The total electrical resistance across the endothelial monolayer was determined by the combined resistance between the basal surface of the cell and the electrode, providing a measure of alterations in cell-cell or cell-matrix adhesion. In the experiments assessing the time course of the response, TEER is expressed as normalized resistance (i.e., ratio of resistance at a given time to resistance at “zero” time).
Measurement of ROS
Fluorescence microscopy.
Cells were loaded with 25 μM 6-carboxy- 2′,7′-dichloro-dihydrofluorescein diacetate (DCFDA) in EBM-2 medium for 30 min at 37°C. After loading with DCFDA, cells were exposed to IH. At the end of IH exposure, cells were washed twice with warm phosphate-buffered saline (PBS) and examined under a Zeiss Axiovert100TV inverted fluorescence microscope using excitation and emission wavelengths of 495 and 520 nm, respectively. The fields were randomly selected for imaging using a high-resolution, cooled CCD camera system with a ×20 objective lens. In experiments assessing the effect of antioxidant, cells were pretreated with manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP), a membrane-permeable ROS scavenger for 30 min before loading with DCFDA and exposure to IH. The concentration of MnTMPyP was 50 μM, which was based on preliminary experiments.
Malondialdehyde levels.
To further confirm ROS generation, malondialdehyde (MDA) levels were determined as a measure of lipid peroxidation in cell homogenates as previously described (22). The results were expressed as microgram of MDA formed per milligram of protein.
Immunofluorescence Microscopy
Cells were prepared for immunofluorescence analysis as described previously (28). Briefly, cells were sequentially treated with the following reagents and were rinsed with PBS before each step. The treatments include 1) 3.7% formaldehyde in PBS for 10 min (fixation); 2) 0.25% Triton X-100 in Tris-buffered saline containing 0.01% Tween 20 (TBST) for 2 min (permeabilization); 3) 10% donkey serum in PBS for 60 min (blocking nonspecific binding sites); 4) either rabbit anti-vascular endothelial cadherin (VE-cadherin; 1:200 dilution; Cayman Chemicals) or rabbit anti-zona occludens-1 (ZO-1; 1:200 dilution; Invitrogen) or rabbit anti-Cortactin (1:200 dilution; Santa Cruz Biotechnology) in PBS containing 1% serum overnight at 4°C (primary); and 5) either Alexa Fluor 488 or 568 conjugated goat anti-rabbit antibody (1:1,000 dilution; Molecular Probes) in TBST for 1 h at room temperature (secondary). Cells treated with secondary antibody were used as negative controls. Actin stress fibers were stained with Alex Fluor Phalloidin 568. The slides were prepared with mounting medium containing 4′6′-diamidino-2-phenylindole (DAPI; Vector Laboratories) to stain the nucleus. Cells were examined by a Nikon Eclipse TE 2000-S fluorescence microscope with a Hamamatsu digital camera (Japan) using a ×60 oil-immersion objective. All postacquisition image processing was performed with Fiji (ImageJ) v.1.47H software (http://fiji.sc/Fiji) with the standard plug-in bundle.
Analysis of Distribution of Cortactin, VE-Cadherins, and ZO-1 at the Cytoplasm Periphery
The image analyses were performed on endothelial cells that were immunostained individually for either cortactin or VE-cadherins or ZO-1 in a double-blinded manner by an individual who had no prior knowledge of sample history using Fiji image software (http://fiji.sc/Fiji). Before the analysis, all images were background subtracted and summed fluorescence intensities (raw integrated density) were independently calculated for the entire endothelial cell (FTotal) as well as for the cell periphery defined manually by corresponding region of interest. The ratio of the summed fluorescence intensity of cortactin or VE-cadherins or ZO-1 in cell periphery (F) to that in the entire cell (FTotal) was calculated. The ratio of F/FTotal for a given protein provides a measure of its relative distribution in a specific region of the cell. The IH-induced change in the distribution of a given protein was expressed as percentage of the control cells exposed to repetitive normoxia instead of IH.
Analysis of Colocalization of Cortactin and Actin
The degree of colocalization of cortactin and actin was determined using a “colocalization threshold” plug-in developed for Fiji image analysis software. This plug-in involves spatial correlation analysis along with an algorithm that permits automatic threshold identification (7, 14). Briefly, the areas for colocalization analysis were selected by drawing the corresponding region of interest in the endothelial monolayer that was doubly immunostained for actin and cortactin. For each cell, colocalization coefficient for cortactin (M1) and for actin (M2) was calculated using the following equation:
where T1 is automatic threshold for channel 1 below which the Pearson correlation coefficient is negative; T2 is automatic threshold for channel 2 below which the Pearson correlation coefficient is negative; I1 is pixel intensity for channel 1; I2 is pixel intensity for channel 2; All I1 is all pixel intensities for channel 1; and All I2 is all pixel intensities for channel 2. A 0 value for M indicates absence of colocalization whereas a value between 0 and 1 represents fractional colocalization.
Western Blot Analysis
Cells were extracted in lysis buffer containing 25 mM Tris·HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM sodium orthovanadate, PMSF, and protease inhibitor cocktail. Cell extracts containing equal amounts of proteins (1 mg/ml) were separated on a 10% SDS-PAGE gel and transferred to PVDF membrane. Membranes were probed with either anti-extracellular signal-regulated kinase 1/2 (ERK1/2; 1:1,000 dilution) or anti-phosphorylated ERK (1:1,000 dilution) or anti-total SAPK/c-jun NH2-terminal kinase (JNK; 1:1,000 dilution) or anti-phosphorylated JNK (1:1,000 dilution) polyclonal antibody obtained from Cell Signaling and then with horseradish peroxidase-conjugated secondary antibody. The immunocomplexes were visualized using an ECL detection system (Amersham). The blots were scanned and quantified using Scion Image Software (National Institutes of Health).
Experimental Protocols
Series 1.
The TEER was determined in cells exposed to repetitive normoxia (control) or 10 or 30 or 60 cycles IH (n = 8–16 channels in each group).
Series 2.
ROS levels in cells (∼5 × 106) exposed to either repetitive normoxia (control) or 30 cycles of IH were monitored by DCFDA fluorescence and MDA levels (n = 5 experiments in each group). A double-blinded approach was used to ensure unbiased evaluation of DCFDA fluorescence data.
Series 3.
The effect of antioxidant on TEER was determined in cells treated with either vehicle (control) or MnTMPyP (50 μM) before exposure to either repetitive normoxia or 30 cycles of IH (n = 10–15 channels in each group).
Series 4.
Cells were exposed to either repetitive normoxia (control) or 30 cycles of IH. The distribution of cortactin, VE-cadherins, and ZO-1 proteins as well as the colocalization of cortactin and actin was assessed by immunocytochemistry using anti-cortactin, anti-actin, anti-VE-cadherins, and anti-ZO-1 antibodies. A double-blinded approach was used for the evaluation of immunostaining patterns (n = 16–20 cells in each group).
Series 5.
In experiments assessing the role of ROS in IH-induced redistribution of cortactin, VE-cadherins, and ZO-1 proteins as well as the colocalization of cortactin and actin, cells were treated with either vehicle (control) or MnTMPyP (50 μM) before exposure to repetitive normoxia or 30 cycles of IH (n = 16–17 cells in each group). The other experimental details are same as described in series 4.
Series 6.
To assess the effect of IH on the levels of phosphorylated forms of ERK1/2 and JNK, cells (∼5 × 106) were exposed to repetitive normoxia (control) or 30 cycles of IH and the cell extracts were subjected to immunoblot analysis using anti-ERK1/2, anti-JNK, anti-phospho-ERK, and anti-phospho-JNK antibodies (n = 5 experiments in each group).
Series 7.
To assess the role of MAP kinases in IH-induced endothelial barrier dysfunction, PD98059, an ERK1/2 inhibitor (30 μM); SP600125, a JNK inhibitor (30 μM); or vehicle (control) was added to cell culture medium and the cells were subsequently exposed to either repetitive normoxia or 30 cycles of IH (n = 10–12 channels in each group).
Series 8.
To assess the role of MAP kinases in IH-induced redistribution of cortactin, VE-cadherins, and ZO-1 as well as on the colocalization of cortactin and actin, cells were first treated with PD98059 (30 μM) or SP600125 (30 μM) or vehicle (control) and then exposed to either repetitive normoxia or 30 cycles of IH (n = 15–18 cells in each group). The other experimental details are same as described in series 4.
Data Analysis
In all experiments, the samples were analyzed in triplicate. The data were expressed as means ± SE. Statistical significance was evaluated by one-way ANOVA followed by Tukey's test. P values of < 0.05 were considered significant.
RESULTS
Effect of IH on Endothelial Barrier Function
Endothelial barrier function was determined by monitoring TEER in lung microvascular endothelial cells before and immediately after termination of IH exposure. Thirty cycles of IH decreased TEER compared with pre-IH, which gradually returned to control levels during 1 h of reoxygenation (Fig. 1, A and B). The effect of IH on TEER was stimulus dependent and the response reached a plateau with 30 cycles of IH (Fig. 1C). Based on these observations, the following studies were performed on cells exposed to 30 cycles of IH.
Fig. 1.

Effect of intermittent hypoxia (IH) on endothelial barrier function. A: representative examples of transendothelial electrical resistance (TEER) expressed as normalized resistance from control and IH-exposed human lung microvascular endothelial cells (HLMVEC). Cells were exposed to repetitive normoxia (control) or 30 cycles of IH. B: average data of TEER, expressed as normalized resistance of cells before IH exposure (pre-IH), immediately after IH exposure (post-IH, 0 h), and after 1 h of reoxygenation (post-IH, 1 h) are shown; n = 16 channels. C: effect of increasing cycles of IH from 0–60 cycles on TEER. Average data of normalized resistance from control cells (n = 10 channels) and cells exposed to IH 10 (n = 8 channels), 30 (n = 12 channels), and 60 cycles (n = 9 channels) are presented. B and C: data are presented as means ± SE from 3 independent experiments. *P < 0.01; **P < 0.001.
ROS Mediate IH-Induced Endothelial Barrier Dysfunction
We then determined whether IH increases ROS levels in lung microvascular endothelial cells. ROS levels were determined by two approaches: one by monitoring DCFDA fluorescence and the other by measuring MDA levels. Cells exposed to IH exhibited increased DCFDA fluorescence and elevated MDA levels, and these effects were prevented by pretreatment with MnTMPyP (50 μM), a membrane-permeable ROS scavenger (Fig. 2, A–C). Pretreatment with MnTMPyP markedly attenuated IH-induced reduction in TEER, whereas it had no effect on barrier function in control cells (Fig. 3, A and B).
Fig. 2.
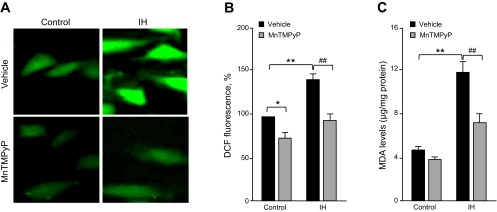
IH increases reactive oxygen species (ROS) generation in human lung microvascular endothelial cells. A: representative fluorescence images of cells exposed to repetitive normoxia (control; left) and 30 cycles of IH (right). Cells were loaded with DCFDA, a ROS sensitive fluorescent dye before IH exposure. Cells were treated with either vehicle or 50 μM of manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP), a ROS scavenger. B: effect of IH (30 cycles) on DCFDA fluorescence levels. Data are presented as mean %control ± SE from 5 independent experiments. *P < 0.05 compared within the controls; **P < 0.001, compared with control; ##P < 0.001, compared with vehicle-treated cells exposed to 30 cycles of IH. C: effect of IH (30 cycles) on malondialdehyde (MDA) levels, a marker of oxidative stress. Data are presented as means ± SE from 5 independent experiments. **P < 0.001, compared with control; ##P < 0.001, compared with vehicle-treated cells exposed to 30 cycles of IH.
Fig. 3.

Effect of ROS scavenger on IH-induced endothelial barrier dysfunction. A: Representative examples of normalized resistance (TEER). Control: cells exposed to repetitive normoxia; IH + vehicle: cells exposed to IH 30 cycles pretreated with vehicle; IH + MnTMPyP: cells pretreated with 50 μM MnTMPyP and then exposed to IH 30 cycles. Note that MnTMPyP markedly attenuated IH-induced decrease in TEER. B: average data of normalized TEER from control (n = 15 channels), cells exposed to IH 30 cycles treated with vehicle (n = 10 channels) or 50 μM MnTMPyP (n = 12 channels). Data are presented as means ± SE from 4 independent experiments. **P < 0.001; ##P < 0.001; n.s., not significant, P > 0.05.
Reorganization of the Cytoskeleton and Junction Proteins by IH
Endothelial barrier function depends on the integrity of cytoskeleton and junction proteins (8, 23). We, therefore, determined the effects of IH on actin and cortactin, the major cytoskeletal proteins. Immunofluorescence analysis revealed reorganization of cortactin and actin as well as stress fiber formation in cell exposed to IH (Fig. 4A). Quantitative analysis of the fluorescence showed that cortactin redistributed to cell periphery (Fig. 4B). Colocalization of actin and cortactin was markedly increased in IH-exposed cells compared with controls (Fig. 4, A and C). Reoxygenation for 1 h restored the effects of IH on actin and cortactin (Fig. 4, A and C). Pretreatment with MnTMPyP prevented IH-induced reorganization of actin and cortactin (Fig. 4, A and C).
Fig. 4.
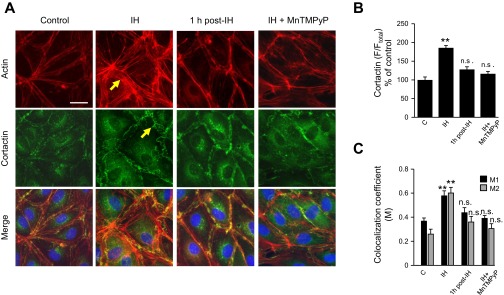
Effect of IH on cytoskeleton proteins in human lung microvascular endothelial cells. A: representative photomicrographs of HLMVEC monolayer immunostained with anti-actin (top) and anti-cortactin antibodies (middle). Merged images to identify actin-cortactin interaction are shown at bottom. Control: cells exposed to repetitive normoxia; IH: cells exposed to IH 30 cycles; 1 h post-IH: cells exposed to IH 30 cycles and reoxygenated for 1 h; IH + MnTMPyP: cells pretreated with 50 μM MnTMPyP and then exposed to IH 30 cycles. Yellow arrows indicate actin stress-fibers and cortical redistribution of cortactin. Bar in the control at top = 20 μm. B: quantitative analysis of cortactin distribution in lung microvascular endothelial cell monolayers. Control (C; n = 16 cells), IH (n = 19 cells), 1 h post-IH (n = 18), and IH + MnTMPyP (n = 20 cells). Data are expressed as percentage of control. C: quantitative analysis of the colocalization of actin and cortactin in lung microvascular endothelial cell monolayers monolayers. Control (n = 16 cells), IH (n = 18 cells), 1 h post-IH (n = 16), and IH + MnTMPyP (n = 20 cells). Colocalization coefficient for cortactin (M1) and actin (M2) are shown. B and C: data are presented as means ± SE from 4 independent experiments. **P < 0.001, compared with vehicle-treated control.
VE-cadherins and ZO-1 representing tight and adherens junction proteins, respectively, regulate endothelial cell permeability by modulating membrane adhesion of neighboring cells (8, 27, 28). In control endothelial cells, ZO-1 and VE-cadherins are primarily localized at the cell boundaries exhibiting a continuous distribution, which was markedly disrupted in IH-exposed cells (Fig. 5A). Both ZO-1 and VE-cadherins were redistributed partially to the cytoplasm in IH-exposed cells (Fig. 5, B and C). As a consequence, intercellular gaps and regions of membrane ruffling appeared on the cell-cell interface in IH-exposed cells (Fig. 5A), and these effects disappeared following reoxygenation for one h (Fig. 5, A–C). MnTMPyP prevented reorganization of junction proteins (Fig. 5, A–C).
Fig. 5.

Effect of IH on junction proteins in human lung microvascular endothelial cells A: immunofluorescence analysis of the distribution of zona occludens-1 (ZO-1) and vascular endothelial (VE)-cadherins in control (left), IH (left middle), 1 h post-IH (right middle), and IH + MnTMPyP-treated cells (right). Nuclei were stained with DAPI. The yellow arrows indicate sites of membrane ruffling as well as cytoplasmic redistribution of VE-cadherins and ZO-1. Bar in the control at top = 20 μm. B and C: quantitative analyses of the distribution of ZO-1 (B) and VE-cadherins (C) are shown. Control (n = 17 cells), IH (n = 16 cells), 1 h post-IH (n = 15), and IH + MnTMPyP (n = 16 cells). Data (means ± SE) are expressed as percentage of control. **P < 0.001, compared with vehicle-treated control.
MAP kinase Activation by IH
ERK1/2 and JNK contribute to endothelial cell barrier function via phosphorylation of junction proteins (27). Therefore, the roles of ERK1/2 and JNK in IH-induced alteration in endothelial barrier function were examined. Cells exposed to IH showed increased levels of phosphorlylated forms of ERK1/2 and JNK. Either reoxygenation or treatment with MnTMPyP decreased IH-induced ERK1 and 2 phosphorylation, and completely prevented JNK phosphorylation (Fig. 6, A and B). Treatment of cells with either 30 μM of PD98059, an ERK1/2 inhibitor, or 30 μM of SP600125, a JNK inhibitor, prevented IH-induced decrease in TEER (Fig. 7, A and B) and the reorganization of cytoskeleton and junction proteins (Fig. 8, A–D).
Fig. 6.

IH increases ERK and JNK phosphorylation. Representative immunoblots (top) and average data of densitometric analysis (bottom) of ERK1/2 (A) and JNK (B) phosphorylation in control and in cells exposed to 30 cycles of IH with and without MnTMPyP (50 μM) treatment, and in the cells subjected to reoxygenation after IH exposure (1 h post-IH) are presented. Average data are presented as a ratio of phosphorylated and unposphorylated ERK1/2 and JNK protein. Data are presented as means ± SE from 5 independent experiments. *#P < 0.01 and **##P < 0.001.
Fig. 7.

Mean arterial pressure (MAP) kinase inhibitors prevent IH-induced endothelial barrier dysfunction Representative examples (A) and average data (B) of normalized resistance (TEER) from control (n = 12 channels); IH + vehicle (n = 12 channels); IH + 30 μM of PD98059, a JNK inhibitor (n = 10 channels); and IH+ 30 μM of SP600125, a ERK inhibitor (n = 11 channels) cells. Data represent means ± SE from 3 independent experiments. **P < 0.001; ##P < 0.001.
Fig. 8.

MAP kinase inhibitors prevent IH-induced reorganization of cytoskeleton and cell junction proteins in human lung microvascular endothelial cells. A: immunofluorescence analysis of the distribution of actin, cortactin, ZO-1, and VE-cadherins in cells exposed to IH 30 cycles treated with either vehicle (left); 30 μM SP600125, an ERK inhibitor (middle); or 30 μM PD98059, a JNK inhibitor (right). Nuclei were stained with DAPI. Bar at top left = 20 μm. B–D: quantitative analyses of cortactin (B), ZO-1 (C), and VE-cadherins (D) distribution in IH-exposed cells are shown. V: vehicle treated (n = 18 cells each); SP: SP600125 treated (n = 17 cells each); PD: PD98059 treated (n = 15 cells each) cells. Data (means ± SE) are expressed as percentage of control. **P < 0.001.
DISCUSSION
In this study, we determined the impact of IH on lung microvascular endothelial barrier function and assessed the underlying mechanisms. Our results demonstrate that exposure to IH disrupts the endothelial barrier dysfunction via ROS-dependent activation of ERK1/2 and JNK-mediated reorganization of cytoskeleton and junction proteins. The effects of IH were reversed following reoxygenation. IH-induced reorganization of cytoskeleton and junction proteins is unlikely due to shear stress caused by gas flows, because alternating cycles of normoxia, which were used as controls, virtually had no effect on endothelial barrier function. It would have been ideal to monitor TEER during exposure to IH, but technical difficulties precluded performing these experiments. However, we believe that TEER measurements performed immediately after the last episode of hypoxia represents the impact of IH on endothelial permeability. The cumulative, total duration of hypoxia during 30 cycles of IH is 15 min (30 s/cycle × 30 cycles). Despite this short duration, IH exposure led to marked changes in barrier function and reorganization of cytoskeleton and junction proteins. In sharp contrast, 24 h of continuous hypoxic exposure is required to produce similar changes in barrier function and cytoskeletal reorganization (2, 21). These observations demonstrate that IH is a more potent stimulus than continuous hypoxia in disrupting endothelial barrier function.
What makes IH a more potent stimulus than continuous hypoxia? Using two independent approaches we demonstrated that IH increases ROS generation in lung microvascular endothelial cells. MnTMPyP, a membrane-permeable ROS scavenger, prevented ROS generation and blocked IH-induced endothelial barrier dysfunction. In a previous study, we reported that a comparable cumulative duration of continuous hypoxia had no effect on ROS generation in cell cultures (29). The central role for ROS in mediating endothelial barrier dysfunction by IH is further supported by a previous study showing that H2O2, an oxidant, also produces similar barrier dysfunction (11). Therefore, it is likely that ROS generation by IH makes it a more potent stimulus than a given duration of continuous hypoxia.
How might ROS contribute to IH-induced endothelial barrier dysfunction? MAP kinase-mediated phosphorylation of scaffold and junction proteins of the endothelial cells has been implicated in endothelial barrier dysfunction (25, 26). IH-activated ERK1/2 and JNK and MnTMPyP prevented this effect suggesting that MAP kinase activation is mediated by ROS. Quantitative immunofluorescence analysis demonstrated increased interaction between actin and cortactin, and actin polymerization resulting in stress fiber formation. The contribution of cortactin redistribution to endothelial barrier function is complex. Previous studies reported redistribution of cortactin correlates with increased (9) and in certain conditions to decreased (24) endothelial barrier function. Our results with IH showed that cortactin redistribution was associated endothelial barrier dysfunction. However, besides cortactin, endothelial barrier function is also regulated by a complex interplay of other molecules including junction proteins. Indeed, we observed translocation of VE-cadherins and ZO-1 to the cytosol in IH-exposed cells, resulting in formation of intercellular gaps and membrane ruffling, which are characteristic of endothelial barrier dysfunction. Remarkably, the reorganization of cytoskeletal and junction proteins was associated with phosphorylation of ERK1/2 and JNK and these effects were prevented by pretreatment with MnTMPyP, a membrane-permeable ROS scavenger. Furthermore, inhibitors of ERK1/2 or JNK blocked reorganization of cytoskeleton and junction proteins with a concomitant restoration of endothelial barrier function in IH-exposed cells. These results taken together suggest that IH-induced barrier dysfunction is mediated by ROS-dependent activation of MAP kinases, which initiates reorganization of cytoskeletal and junction proteins.
The Microvascular endothelial barrier is critical for gas and fluid exchange in the lung, and disruption of barrier function not only affects pulmonary gas exchange but also leads to pulmonary edema. Previous studies reported the occurrence of pulmonary edema in OSA patients (4–6) and also in experimental models of OSA (10). Although several other pathologies are associated with OSA, the current study suggests that IH, which is a hallmark manifestation of OSA, is a potent stimulus for disrupting endothelial barrier function, which might account for the development of pulmonary edema. Recent studies showed that increased transcription and activation of the prooxidant enzyme Nox2 and decreased transcription and activity of the antioxidant enzyme (e.g., Sod2) contribute to increased ROS levels by IH (15, 16). Analysis of the molecular mechanisms revealed that activation of HIF-1 mediates the increased Nox2 transcription (17, 30). On the other hand, IH decreases HIF-2 activity, which in turn results in insufficient transcription of the antioxidant enzyme Sod2 (15). Thus the imbalance between HIF-1 and HIF-2 and resulting changes in pro- and antioxidant enzyme expressions contribute to IH-induced oxidative stress. However, further studies are needed to demonstrate that IH-induced imbalance in HIF-1 and HIF-2 contributes to endothelial barrier dysfunction via ROS.
GRANTS
This work was supported by grants from National Institutes of Health, National Heart, Lung and Blood Institute RO1-HL-76537 and PO1-HL-90554 (to N. R. Prabhakar) and P01-HL-58064 (to V. Natarajan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.R.P., conception and design of research; V.V.M., P.V.U., G.Y., M.M.L., J.N., performed experiments; V.V.M., M.M.L., J.N., V.N. analyzed data; V.V.M., N.R.P. interpreted results of experiments; V.V.M. prepared figures; V.V.M., G.K.K., drafted manuscript; G.K.K., N.R.P. edited and revised manuscript; N.R.P. approved final version of manuscript.
Footnotes
This article is the topic of an Editorial Focus by Kimberly A. Smith and Jason X.-J. Yuan (22a).
REFERENCES
- 1. Ali MH, Schlidt SA, Chandel NS, Hynes KL, Schumacker PT, Gewertz BL. Endothelial permeability and IL-6 production during hypoxia: role of ROS in signal transduction. Am J Physiol Lung Cell Mol Physiol 277: L1057–L1065, 1999 [DOI] [PubMed] [Google Scholar]
- 2. Ali MH, Schlidt SA, Hynes KL, Marcus BC, Gewertz BL. Prolonged hypoxia alters endothelial barrier function. Surgery 124: 491–497, 1998 [PubMed] [Google Scholar]
- 3. Boueiz A, Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc Res 77: 26–34, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Chaudhary BA, Chaudhary TK, Speir W. Progressive dyspnea and pulmonary edema in an obese somnolent man. J Med Assoc Ga 76: 133–135, 1987 [PubMed] [Google Scholar]
- 5. Chaudhary BA, Ferguson DS, Speir WA. Pulmonary edema as a presenting feature of sleep apnea syndrome. Chest 82: 122–124, 1982 [DOI] [PubMed] [Google Scholar]
- 6. Chaudhary BA, Nadimi M, Chaudhary TK, Speir WA. Pulmonary edema due to obstructive sleep apnea. South Med J 77: 499–501, 1984 [DOI] [PubMed] [Google Scholar]
- 7. Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J 86: 3993–4003, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001 [DOI] [PubMed] [Google Scholar]
- 9. Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem 279: 24692–24700, 2004 [DOI] [PubMed] [Google Scholar]
- 10. Fletcher EC, Proctor M, Yu J, Zhang J, Guardiola JJ, Hornung C, Bao G. Pulmonary edema develops after recurrent obstructive apneas. Am J Respir Crit Care Med 160: 1688–1696, 1999 [DOI] [PubMed] [Google Scholar]
- 11. Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H2O2-mediated permeability: role of MAPK and occludin. Am J Physiol Cell Physiol 279: C21–C30, 2000 [DOI] [PubMed] [Google Scholar]
- 12. Lavie L, Lavie P. Molecular mechanisms of cardiovascular disease in OSAHS: the oxidative stress link. Eur Respir J 33: 1467–1484, 2009 [DOI] [PubMed] [Google Scholar]
- 13. Liu T, Guevara OE, Warburton RR, Hill NS, Gaestel M, Kayyali US. Modulation of HSP27 alters hypoxia-induced endothelial permeability and related signaling pathways. J Cell Physiol 220: 600–610, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manders EM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169: 8, 1993 [DOI] [PubMed] [Google Scholar]
- 15. Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA 106: 1199–1204, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci 29: 4903–4910, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol 577: 705–716, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prabhakar NR, Kumar GK, Nanduri J. Intermittent hypoxia-mediated plasticity of acute O2 sensing requires altered red-ox regulation by HIF-1 and HIF-2. Ann NY Acad Sci 1177: 162–168, 2009 [DOI] [PubMed] [Google Scholar]
- 19. Prabhakar NR, Kumar GK, Nanduri J, Semenza GL. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxid Redox Signal 9: 1397–1403, 2007 [DOI] [PubMed] [Google Scholar]
- 20. Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92: 967–1003, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qi H, Wang P, Liu C, Li M, Wang S, Huang Y, Wang F. Involvement of HIF-1α in MLCK-dependent endothelial barrier dysfunction in hypoxia. Cell Physiol Biochem 27: 251–262, 2011 [DOI] [PubMed] [Google Scholar]
- 22. Ramanathan L, Gozal D, Siegel JM. Antioxidant responses to chronic hypoxia in the rat cerebellum and pons. J Neurochem 93: 47–52, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22a. Smith KA, Yuan JX. Breaking barriers in obstructive sleep apnea. Focus on “Intermittent hypoxia-induced endothelial barrier dysfunction requires ROS-dependent MAP kinase activation.” Am J Physiol Cell Physiol (March 13, 2014). 10.1152/ajpcell.00072.2014 [DOI] [PubMed] [Google Scholar]
- 23. Stevens T, Garcia JG, Shasby DM, Bhattacharya J, Malik AB. Mechanisms regulating endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol 279: L419–L422, 2000 [DOI] [PubMed] [Google Scholar]
- 24. Usatyuk PV, Fomin VP, Shi S, Garcia JG, Schaphorst K, Natarajan V. Role of Ca2+ in diperoxovanadate-induced cytoskeletal remodeling and endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol 285: L1006–L1017, 2003 [DOI] [PubMed] [Google Scholar]
- 25. Usatyuk PV, Natarajan V. Regulation of reactive oxygen species-induced endothelial cell-cell and cell-matrix contacts by focal adhesion kinase and adherens junction proteins. Am J Physiol Lung Cell Mol Physiol 289: L999–L1010, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Usatyuk PV, Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J Biol Chem 279: 11789–11797, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 281: 35554–35566, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Usatyuk PV, Singleton PA, Pendyala S, Kalari SK, He D, Gorshkova IA, Camp SM, Moitra J, Dudek SM, Garcia JG, Natarajan V. Novel role for non-muscle myosin light chain kinase (MLCK) in hyperoxia-induced recruitment of cytoskeletal proteins, NADPH oxidase activation, and reactive oxygen species generation in lung endothelium. J Biol Chem 287: 9360–9375, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol 557: 773–783, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol 226: 2925–2933, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol 217: 674–685, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]