

Abstract
A number of growth factors and signaling pathways regulate matrix deposition and fibroblast proliferation in the lung. The epidermal growth factor receptor (EGFR) family of receptors and the transforming growth factor-β (TGF-β) family are active in diverse biological processes and are central mediators in the initiation and maintenance of fibrosis in many diseases. Transforming growth factor-α (TGF-α) is a ligand for the EGFR, and doxycycline (Dox)-inducible transgenic mice conditionally expressing TGF-α specifically in the lung epithelium develop progressive fibrosis accompanied with cachexia, changes in lung mechanics, and marked pleural thickening. Although recent studies demonstrate that EGFR activation modulates the fibroproliferative effects involved in the pathogenesis of TGF-β induced pulmonary fibrosis, in converse, the direct role of EGFR induction of the TGF-β pathway in the lung is unknown. The αvβ6 integrin is an important in vivo activator of TGF-β activation in the lung. Immunohistochemical analysis of αvβ6 protein expression and bronchoalveolar analysis of TGF-β pathway signaling indicates activation of the αvβ6/TGF-β pathway only at later time points after lung fibrosis was already established in the TGF-α model. To determine the contribution of the αvβ6/TGF-β pathway on the progression of established fibrotic disease, TGF-α transgenic mice were administered Dox for 4 wk, which leads to extensive fibrosis; these mice were then treated with a function-blocking anti-αvβ6 antibody with continued administration of Dox for an additional 4 wk. Compared with TGF-α transgenic mice treated with control antibody, αvβ6 inhibition significantly attenuated pleural thickening and altered the decline in lung mechanics. To test the effects of genetic loss of the β6 integrin, TGF-α transgenic mice were mated with β6-null mice and the degree of fibrosis was compared in adult mice following 8 wk of Dox administration. Genetic ablation of the β6 integrin attenuated histological and physiological changes in the lungs of TGF-α transgenic mice although a significant degree of fibrosis still developed. In summary, inhibition of the β6 integrin led to a modest, albeit significant, effect on pleural thickening and lung function decline observed with TGF-α-induced pulmonary fibrosis. These data support activation of the αvβ6/TGF-β pathway as a secondary effect contributing to TGF-α-induced pleural fibrosis and suggest a complex contribution of multiple mediators to the maintenance of progressive fibrosis in the lung.
Keywords: epidermal growth factor receptor, integrin, pleural fibrosis, pulmonary fibrosis, TGF-β
pulmonary fibrotic lesions are characterized pathologically by mesenchymal cell proliferation, differentiation, and increased deposition of extracellular matrix. A number of growth factors expressed in the lung activate the mesenchymal cell and experimental studies support targeting these fibrogenic growth factors and their receptors as potential therapeutic strategies to inhibit progression of pulmonary fibrosis. Although these profibrotic growth factors have unique receptors, there is a complex interface among their downstream signaling pathways. Many cell signaling pathways ultimately converge onto common pathways that regulate the cellular processes associated with pulmonary fibrosis including proliferation, differentiation, migration, and apoptosis. A more detailed understanding whether and where these fibrotic pathways overlap and interact will be important in designing future therapeutic strategies.
TGF-α, along with epidermal growth factor and amphiregulin, are ligands for the epidermal growth factor receptor (EGFR). Experimental studies in rodents support a role for EGFR activation in fibroproliferative processes following exposure to bleomycin, naphthalene, asbestos, silica, and hyperoxia (1, 38, 41, 53, 54). In addition, lung epithelial-specific overexpression of TGF-α leads to progressive and pronounced pulmonary fibrotic lesions in transgenic mice (21, 31). TGF-α knockout mice are protected from bleomycin-induced fibrosis, and the selective EGFR tyrosine kinase inhibitor gefitinib prevents the development of bleomycin-induced fibrosis and reverses transgenic TGF-α-induced fibrosis (20, 25, 39). EGFR receptor and ligands are elevated in idiopathic pulmonary fibrosis (IPF), cystic fibrosis, bronchopulmonary dysplasia, and the remodeled airways in asthma (8, 19, 40, 45, 46, 50, 51). Together these findings strongly support that EGFR signaling plays a central role in the pathogenesis of pulmonary fibrosis.
The transforming growth factor-β (TGF-β) family includes multifunctional cytokines that are active in diverse biological processes and are central mediators in the initiation and maintenance of fibrosis in many diseases. TGF-β1 is a potent activator of fibroblast recruitment, extracellular matrix deposition, and myofibroblast differentiation (7). TGF-β1 is a critical mediator of bleomycin-induced fibrosis and transgenic lung epithelial-specific overexpression of TGF-β1 leads to significant fibrotic disease (12, 33). Expression of TGF-β receptors are increased in fibroblasts from patients with scleroderma and increased active TGF-β1 is detected in the lungs of patients with IPF, further supporting antagonism of the TGF-β pathway as a logical strategy for the treatment of fibrosis (28–30).
TGF-β is ubiquitously expressed by all cell types where it is secreted and stored in latent form that is bound mostly to the extracellular matrix through noncovalent associations with its propeptide, the latency associated peptide (2). Activation of latent TGF-β is increasingly recognized as a critical step for its cellular effects (5). Integrins are transmembrane heterodimers of α and β subunits that bind a wide variety of physically constrained extracellular ligands and regulate cell growth, migration, and survival. A host of studies demonstrates that αvβ6 integrin is a primary mediator of TGF-β activation in vivo in the lung (2, 42, 59). Mice with deleted β6 integrin are completely protected from lung fibrosis induced through intratracheal bleomycin or radiation, and blocking β6 with a specific antibody protects mice from lung injury and fibrosis in multiple experimental models (16, 23, 26, 42, 47). Collectively these data show that inhibition of TGF-β signaling restricted to sites of β6-integrin-mediated TGF-β activation is an effective, selective strategy to treat lung fibrosis and avoid potential off-target effects of total TGF-β inhibition. Further evidence supporting this approach comes from the observation that αvβ6 is expressed at low levels in healthy adult tissues but is strongly upregulated on epithelial cells at fibrotic foci in lungs of patients with IPF or pulmonary fibrosis from scleroderma (10, 23, 42). Together these findings have led to a current phase 2 trial of humanized anti-αvβ6 antibody in IPF patients (16).
Although both the EGFR and TGF-β pathways are central in the pathogenesis of pulmonary fibrosis, the relationship or integration between these pathways remains under investigation. There is strong evidence that EGFR ligands and receptors are critical mediators in fibrogenic responses to TGF-β. TGF-β induces EGFR ligand upregulation and receptor expression through the canonical SMAD pathway specifically in fibroblasts (3). Furthermore, EGFR activation by TGF-β is essential for the induction of fibroblast morphological transformation and anchorage-independent growth (4). Recent studies by Zhou et al. (63) demonstrate that TGF-β1 significantly induced the expression of amphiregulin in lung fibroblasts in vitro and antagonism of either amphiregulin or EGFR significantly reduced TGF-β1-induced fibroblast proliferation and expression of smooth muscle actin and extracellular matrix genes. Consistent with these in vitro findings, amphiregulin expression was markedly increased in the lung of TGF-β1 transgenic mice and either siRNA of amphiregulin expression or pharmacological inhibition of EGFR significantly reduced TGF-β1-stimulated collagen accumulation in the lung (63).
Although EGFR activation modulates the fibroproliferative effects involved in the pathogenesis of TGF-β induced pulmonary fibrosis, potential contributions of TGF-β downstream of EGFR activation have not been reported. To examine the fibroproliferative role of TGF-β activity following EGFR activation in vivo, we measured changes in histology, biochemistry, and physiology in TGF-α transgenic mice where αvβ6-mediated TGF-β activation was prevented through both genetic and pharmacological inhibition of the αvβ6 integrin.
MATERIALS AND METHODS
Transgenic mice.
All mice were derived from the FVB/NJ inbred strain. TGF-α transgenic mice were generated and maintained as described previously (21, 52). Mice homozygous with the rtTA transgene driven by the Clara Cell Specific Protein-rtTA+/− promoter (abbreviated as CCSP) were mated with mice heterozygous with the human TGF-α transgene driven by a tetracycline-responsive promoter [(TetO)7-cmv TGF-α+/−], to produce bitransgenic (abbreviated as CCSP/TGF-α) and single transgenic pups (abbreviated as CCSP/-). CCSP/- litter-matched controls were used as controls in antibody studies.
Mice functionally deficient in the β6 integrin (abbreviated as β6−/−) have been previously characterized (24). To investigate the role of the β6 integrin subunit in TGF-α-induced fibrosis, β6−/− mice were mated with transgenic CCSP mice and transgenic (TetO)7-cmv TGF-α+/− mice. Single-transgenic offspring from each cross were then mated to generate CCSP/TGF-α/β6+/+ and CCSP/TGF-α/β6−/− mice. All mice were in a FVB/N background. Mice were genotyped by PCR analysis of tail DNA using primers specific for transgene constructs using conditions as previously described (21, 24).
All mice were housed under specific pathogen-free conditions and protocols were approved by the Institutional Animal Use and Care Committee of the Cincinnati Children's Hospital Research Foundation. To induce TGF-α expression, doxycycline (Dox) (Sigma, St. Louis, MO) was administered in food (62.5 mg/kg) and water (0.5 mg/kg).
Administration of neutralizing antibodies.
The inhibitory anti-αvβ6 mAb, 6.3G9, and isotype control antibody, 1E6, have previously been described (57). Antibodies were injected weekly at doses of 3 mg/kg with injection volumes of 200 μl by intraperitoneal injection. This dosing was based on previous dose-response studies administering these antibodies in mouse lung fibrosis models (23, 47). Dosing throughout the study was based on original baseline weights and not adjusted for weight changes.
Immunohistochemistry, TGF-β assay.
Immunohistochemical detection of β6 protein was performed as previously described (18). Briefly, paraffin-embedded lung tissue were deparaffinized and hydrated to distilled water. Proteolytic digestion was carried out by applying pepsin (Zymed 00-3009) to tissues for 10 min at 37°C. Slides were incubated in MeOH/H2O2 to inhibit peroxidase activity. Tissue was blocked in 4% normal goat serum/TBS-Tween/0.1% BSA for 20 min at room temperature. Slides were incubated for 1 h at room temperature in the anti-β6 chimeric monoclonal antibody, ch.2A1 (Biogen Idec, Cambridge, MA) at a concentration of 0.25 μg/ml and 0.5 μg/ml. Slides were then incubated in biotinylated anti-human IgG (Vector BA-3000) for 30 min at room temperature. Tissue was counterstained with Mayer's hematoxylin for 1 min then placed in lithium carbonate solution.
Bronchiolar lavage fluid collection and monitoring TGF-β activation.
Bronchoalveolar lavage (BAL) collection was performed on mice as previously described (22) and active TGF-β was determined by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer's recommendations.
RNA preparation and real-time PCR.
Total RNA was extracted from lung tissue and cells by use of the RNeasy Mini Kit from Qiagen (Qiagen Sciences, Valencia, CA) as described previously (36). Real-time PCR analysis was performed on a Step One Plus instrument (Applied Biosystems, Foster City, CA). Primer sequences for TGFbi, IGF1, and MMP10 are provided in Table 1.
Table 1.
Primer sequences for RT-PCR
| Forward | Reverse | |
|---|---|---|
| mTgfbi | GAGCTGCTTATCCCAGATTCA | GGCAGTGGAGACGTCAGATT |
| mIGF1 | TCGGCCTCATAGTACCCACT | ACGACATGATGTGTATCTTTATTGC |
| mMMP10 | GAGTCTGGCTCATGCCTACC | TGCAACCAGGAATAAGTTGGT |
Measurements of lung histology, fibrosis score, and pleural thickness.
Lung tissue sections were prepared and stained with Masson's trichrome as described previously (20). Lung tissue sections stained with Masson's trichrome from mice in both pharmacological and genetic studies were scored for the degree of lung fibrosis by using a novel scoring system developed specifically for the TGF-α transgenic mouse. Each tissue section was assigned a single integer score between zero and five. The number corresponds to the degree of lung fibrosis in the pleural surface, perivascular, and terminal peribronchial adventitial regions of the lung (Table 2). The intervention or genetic designation for each slide was masked to the readers, and the score assigned to each mouse was based on reviewing three separate lung sections.
Table 2.
Fibrosis score for TGF-α transgenic mice
| Score | Pleural Surface | Adventitia | |
|---|---|---|---|
| 0 | Normal pleura | AND | Normal adventitia |
| 1 | Pleural thickening scattered covering <50% of lung circumference | OR | Fibrosis covers <50% of vascular and terminal peribronchial adventitial circumference |
| 2 | Pleural thickening scattered covering <50% of lung circumference | AND | Fibrosis covers <50% of vascular and terminal peribronchial adventitial circumference |
| 3 | Pleural thickening covering >50% of lung circumference but thickness is inconsistent | AND | Fibrosis covers <50% of vascular and terminal peribronchial adventitial circumference |
| 4 | Pleural thickening covering entire lung circumference with near uniform thickness | AND | Fibrosis covers >50% of perivascular adventitial circumference but <50% of terminal peribronchial adventitial circumference |
| 5 | Pleural thickening covering entire lung circumference with near uniform thickness | AND | Fibrosis covers >50% of vascular and terminal peribronchial adventitial circumference |
The pleural thickness was measured by histomorphometric measurement of Masson's trichrome-stained lung sections by using the measured distance function of MetaMorph as previously described (37). Five random measures per lung section were obtained for each animal by using a Leica DM2700 M bright-field microscope (Leica Microsystems, Buffalo Grove, IL). High-magnification images (×40) were captured with a 3CCD color video camera and analyzed with MetaMorph imaging software (v6.2; Molecular Devices, Sunnyvale, CA).
Total lung collagen assay and pulmonary mechanics.
The total lung collagen was quantified by measuring hydroxyproline levels and total soluble collagen (measured by Sircol Collagen Assay, Biocolor) as previously described (35, 37). Lung mechanics were assessed on mice by using a computerized Flexi Vent system (SCIREQ, Montreal, Canada) as previously described (35).
Statistics.
All data were analyzed with Prism (Version 5; GraphPad, La Jolla, CA). One-way ANOVA with Tukey's multiple-comparison posttest was used to compare different experimental groups, and data were considered statistically significant for P values less than 0.05.
RESULTS
Expression of αvβ6 integrin during TGF-α-induced pulmonary fibrosis.
The αvβ6 integrin has been shown to contribute to the fibrotic remodeling in mouse models of chemical- and radiation-induced fibrosis. To assess αvβ6 integrin expression during TGF-α-induced pulmonary fibrosis, we immunostained the lung sections of control and TGF-α transgenic mice administered Dox for 4 and 8 wk using a monoclonal antibody (clone ch.2A1) that recognizes the β6 subunit. Minimal αvβ6 integrin staining is detected in control mice but there is obvious staining throughout the alveolar epithelium in the lungs of TGF-α transgenic mice following 4 wk of Dox administration with a marked increase in epithelial staining intensity in the lungs of TGF-α transgenic mice following 8 wk of Dox (Fig. 1). No expression is detected in the adventitial or pleural fibrotic regions.
Fig. 1.
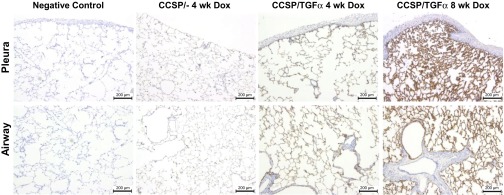
αvβ6 expression in the lung following TGF-α overexpression. The lungs of TGF-α mice were administered doxycycline (Dox) to induce TGF-α overexpression in the lung epithelium and immunostained with an antibody (ch.2A1) specific for the β6 subunit. αvβ6 expression is increased from controls after 4 wk of Dox and is highly expressed in the epithelium after 8 wk of Dox. No αvβ6 expression is detected in the pleural or adventitial fibrotic regions.
Increased expression of αvβ6 integrin has been shown to contribute to fibrotic remodeling via activation of TGF-β. In TGF-α transgenic mice we previously reported no evidence of active TGF-β in the lung homogenates following 1 and 4 days of the administration of Dox (21). To assess whether TGF-β activation evolves later during TGF-α-induced fibrosis, we measured active TGF-β in the BAL fluid (BALF) of TGF-α transgenic mice after fibrosis was already established. We observed a significant increase in active TGF-β levels in the BALF of TGF-α mice administered Dox for 6 wk compared with controls or TGF-α transgenic mice following 2 wk of Dox administration (Fig. 2A). We also assessed αvβ6/TGF-β pathway specific gene transcripts in lung homogenates of TGF-α transgenic mice administered Dox at early and late time points. As shown in Fig. 2B, transforming growth factor β-induced (TGFβi), insulin-like growth factor 1 (IGF1), and matrix metalloproteinase-10 (MMP10) transcript levels are unchanged in TGF-α transgenic mice 4 days after Dox induction compared with control but are significantly upregulated in the lungs at 6 wk on Dox. TGFβi is known to be directly induced by TGF-β, serves as a ligand recognition sequence for several integrins, and has been demonstrated to directly increase primary fibroblast production of collagen (43). In sum, the β6 integrin and active TGF-β are expressed at later time points following the induction of TGF-α after fibrosis is already established and progressing.
Fig. 2.

Activation of the TGF-β pathway after TGF-α-induced fibrosis is established. A: active TGF-β levels in the bronchoalveolar lavage fluid (BALF) from TGF-α mice are significantly elevated from control mice following 6 wk of Dox (n = 4–7 for each group). B: αvβ6/TGF-β pathway specific gene transcripts transforming growth factor β-induced (TGFβi), insulin-like growth factor 1 (IGF1), and matrix metalloproteinase-10 (MMP10) are unchanged in TGF-α transgenic mice 4 days after Dox induction compared with control but are significantly upregulated in the lungs at 6 wk on Dox. Real-time PCR analysis was performed on total lung RNA isolated from the lungs.
Therapeutic neutralization of β6 integrin attenuates TGF-α-induced fibrosis.
To determine the contribution of the αvβ6/TGF-β pathway on the progression of established fibrotic disease, TGF-α transgenic mice already administered Dox for 4 wk were treated with function-blocking anti-αvβ6 antibody (6.3G9) while remaining on Dox for an additional 4 wk (8 wk total). Controls included single transgenic mice (CCSP/-) and TGF-α transgenic mice treated with isotype control antibody (1E6) following 4 wk of administration while also continuing on Dox an additional 4 wk. Body weights in TGF-α transgenic mice treated with isotype control antibody decreased 25% from baseline following 8 wk of Dox administration (Fig. 3A). Body weight loss was attenuated in mice treated with anti-αvβ6 antibody, with an average weight loss of 13% from baseline at 8 wk on Dox; however, body weights remained lower than CCSP/- controls (Fig. 3A). There were no differences in the total lung hydroxyproline levels between TGF-α transgenic mice treated with control or anti-αvβ6 antibodies (Fig. 3B). Masson's trichrome staining of lung sections of TGF-α transgenic mice treated with isotype control antibody demonstrate marked pleural thickening with advanced perivascular and peribronchial fibrosis affecting both large and small vessels and airways (Fig. 4A). TGF-α mice treated with anti-αvβ6 antibody displayed reduced pleural thickening compared with isotype control-treated TGF-α transgenic mice. To quantify the overall changes in the fibrotic lesions of the lungs, we established a scoring system based on the distribution and severity of fibrotic lesions in perivascular, peribronchial, alveolar, and pleural areas of the lung. Compared with CCSP/- control mice, overexpression of TGF-α for 8 wk caused a significant increase in the fibrosis score that was not significantly reduced in TGF-α mice treated with anti-αvβ6 antibody (Fig. 4B). To quantify the fibrotic changes specifically in the pleura, the pleural thickening of lung sections was measured. Mice treated with anti-αvβ6 antibody demonstrate significantly reduced pleural thickening compared with isotype control antibody-treated TGF-α transgenic mice (Fig. 4C). Changes in lung mechanic measurements of airway resistance, tissue elastance, and lung compliance were significantly attenuated in TGF-α transgenic mice treated with anti-αvβ6 antibody compared with TGF-α transgenic mice treated with isotype control antibody but values remained altered compared with nonfibrotic CCSP/- controls (Fig. 5). Together, increases in pleural thickening and alterations in lung mechanics in TGF-α mice were significantly attenuated when αvβ6 was inhibited after disease was already established, supporting a role for the αvβ6 pathway in mediating a component of EGFR/TGF-α-induced pulmonary/pleural fibrosis.
Fig. 3.
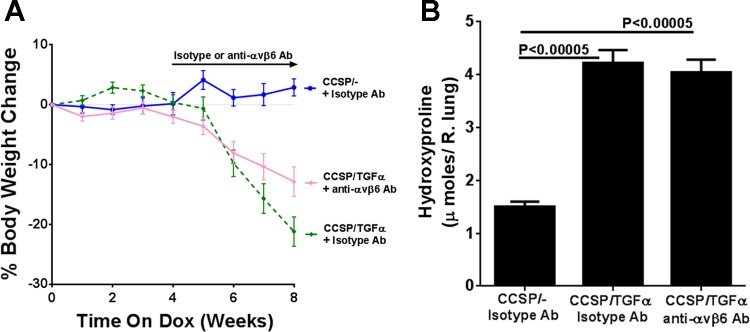
Therapeutic neutralization of αvβ6 integrin attenuates the loss of body weight but does not change total lung collagen production. To assess the efficacy of αvβ6 inhibition in established fibrosis, TGF-α transgenic mice were treated with the anti-αvβ6 monoclonal antibody 6.3G9 and the isotype control antibody 1E6 after 4 wk of Dox, while remaining on Dox for an additional 4 wk. A: Dox-induced expression of TGF-α for 8 wk caused progressive weight loss in isotype control-treated mice (green line), whereas mice treated with anti-αvβ6 4 wk after TGF-α induction demonstrated an attenuation in the fall in body weight (pink line), but weights remained below CCSP/- controls (blue line). Mice were weighed weekly during treatments and no change in bodyweights of mice is represented as 0%. Data are means ± SE (n = 14–20 for each group). B: total lung hydroxyproline levels were elevated in TGF-α transgenic mice and unchanged in mice treated with anti-αvβ6 antibodies.
Fig. 4.

Therapeutic neutralization of αvβ6 integrin attenuates progression of lung fibrosis. A: representative photomicrographs of lung tissues for each group stained with Masson's trichrome. Top, pleural regions of the lung; bottom, adventitia. Scale bar, 100 μm. B: TGF-α mice treated with anti-αvβ6 antibodies administered 4 wk after the initiation of TGF-α induction did not show a significant attenuation in the lung fibrosis score. C: mean pleural thickness was significantly attenuated in anti-αvβ6-treated mice. Data are means ± SE (n = 14–20 for each group). Micrographs in A are representative of sections from 10 mice in each group.
Fig. 5.

Therapeutic neutralization of αvβ6 integrin attenuates progression of TGF-α-dependent changes in lung mechanics. TGF-α transgenic mice administered anti-αvβ6 antibody 4 wk after treatment with Dox demonstrated attenuated increases in airway resistance and tissue elastance and decreases in compliance compared with isotype control-treated CCSP/TGF-α transgenic mice receiving 8 wk of Dox. Data are means ± SE (n = 14–20 for each group).
Lack of β6 integrin attenuates TGF-α-induced fibrosis. Genetic ablation of the β6 integrin has been shown to impair TGF-β signaling in the lung and protect mice from developing pulmonary fibrosis (23, 42). To test the effects of genetic β6 integrin inhibition at the initiation of lung fibrosis, CCSP and TGF-α transgenic mice were mated with β6-null mice to generate CCSP/TGF-α/β6+/+ and CCSP/TGF-α/β6−/− mice and the degree of fibrosis compared in adult mice 8 wk following administration of Dox. Active TGF-β levels, as assessed by ELISA from the BAL, were significantly increased in CCSP/TGF-α/β6+/+ compared with CCSP/-/β6+/+ controls, whereas levels were unchanged between CCSP/TGF-α/β6−/− and CCSP/-/β6−/− controls (data not shown, n = 5–7 mice in each group). These findings support that the β6 integrin mice prevented the TGF-α-induced increase of active TGF-β. Masson's trichrome staining of lung sections of CCSP/TGF-α/β6−/− mice demonstrated mild attenuation of adventitial and pleural fibrosis compared with CCSP/TGF-α/β6+/+ (Fig. 6). The lung fibrosis score was significantly reduced in CCSP/TGF-α/β6−/− mice compared with CCSP/TGF-α/β6+/+ (Fig. 7A), and there was a trend toward reduced pleural thickening in CCSP/TGF-α/β6−/− mice compared with CCSP/TGF-α/β6+/+ mice, but differences were not statistically significant (Fig. 7B). There were no differences in soluble lung collagen levels between CCSP/TGF-α/β6+/+ and CCSP/TGF-α/β6−/− mice (Fig. 7C). Lung mechanic measurements of airway resistance and tissue elastance were significantly reduced in CCSP/TGF-α/β6−/− mice compared with CCSP/TGF-α/β6+/+ mice with no differences in lung compliance (Fig. 8). In sum, ablation of the β6 integrin modestly attenuated histological and physiological changes in the lungs of TGF-α transgenic mice following 8 wk of Dox.
Fig. 6.

Genetic loss of β6 integrin attenuates progression of lung fibrosis. TGF-α transgenic mice were mated with β6-null mice to test the effects of genetic β6 integrin inhibition at the initiation of lung fibrosis. Representative photomicrographs of lung tissues stained with Masson's trichrome comparing TGF-α transgenic mice in the presence or absence of the β6 integrin following 8 wk of Dox. Top, pleural regions of the lung; bottom, adventitia. Scale bar, 100 μm. Presented sections are representative of sections from 6–8 mice in each group.
Fig. 7.

Genetic loss of β6 integrin attenuates progression of lung fibrosis. A: lung fibrosis score is significantly attenuated in TGF-α transgenic mice in the β6 knockout mice. B: mean pleural thickness was not significantly attenuated in TGF-α transgenic mice in the β6-nulls. C: total lung soluble collagen levels were elevated in TGF-α transgenic mice and unchanged in TGF-α transgenic mice in the β6 knockout mice. Data are means ± SE (n = 4–8 for each group).
Fig. 8.

Genetic loss of β6 integrin attenuates progression of TGF-α-dependent changes in lung mechanics. Increases in airway resistance and tissue elastance were significantly attenuated in TGF-α transgenic mice in the β6 knockout mice compared with wild-type TGF-α transgenic mice; there were no differences lung compliance. Data are means ± SE (n = 6–8 for each group).
DISCUSSION
TGF-β is a multifunctional cytokine prominently involved in the pathogenesis of fibrosis through its potent effects on fibroblast differentiation, extracellular matrix formation, and epithelial mesenchymal transformation (14). Increased TGF-β activation is demonstrated in multiple mouse models of injury-induced pulmonary fibrosis and progressive fibrotic disease develops in adenoviral or transgenic TGF-β overexpression models (33, 49). Here we demonstrated upregulation of active TGF-β in the BAL of TGF-α transgenic mice along with increased mRNA levels of the TGF-β target genes, TGFβi, IGF1, and MMP10, but only after TGF-α-induced fibrosis is already established. Minimal activation of TGF-β proximately following TGF-α induction is consistent with our earlier findings in which we did not detect activity of TGF-β in lung homogenates 1 or 4 days following TGF-α induction (21). Together these findings support TGF-β activation as a late, indirect, and partial contributor to fibrogenesis in the TGF-α transgenic model.
One of the primary mechanisms of activation of TGF-β in the lung is through the αvβ6 integrin (2). To specifically determine the role for TGF-β in the TGF-α fibrosis model we inhibited the αvβ6 integrin after fibrosis was established by administering mice a function blocking anti-αvβ6 antibody. TGF-α transgenic mice administered αvβ6 integrin inhibitor displayed modest attenuation in the progression of physiological and histological end points of lung fibrosis compared with controls. One of the prominent pathological features of pulmonary fibrosis in the TGF-α transgenic model is the progressive thickening of the lung pleura. This feature is also found in patients with several human fibrotic diseases. Previous reports demonstrate that transient transfer of the active TGF-β1 gene by adenoviral vectors into the pleural cavity and mesothelium induces homogenous, prolonged, and progressive pleural fibrosis associated with severe impairment of pulmonary function (13). In the present study, pharmacological neutralization of the αvβ6 integrin reduced the pleural thickening by almost 50%, showing that pleural fibrosis observed in TGF-α transgenic mice is partly dependent on the TGF-β pathway.
To further validate the anti-αvβ6 antibody data and to examine the efficacy of inhibiting the αvβ6 integrin at the initiation of fibrosis, TGF-α transgenic mice were mated with β6-null mice and fibrosis end points were compared between wild-type and knockout mice. In agreement with the pharmacological studies, genetic ablation of the β6 integrin mildly attenuated selected physiological and histological end points, yet a significant degree of fibrosis remained evident. Since the increases in TGF-β following TGF-α overexpression were prevented in β6 integrin knockout mice, these findings support a limited role for the TGF-β pathway in the progression of lung fibrosis in the TGF-α model. However, our assessment of TGF-β was limited to only the BAL and does not conclusively demonstrate fibrosis is independent of the TGF-β pathway at a cellular or basal level.
TGF-β induces fibrogenesis through both SMAD-mediated canonical and noncanonical pathways. The SMAD-independent pathways include the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK) cascade and the phosphatidylinositol 3′-kinase (PI3K) pathways (48). The relative contribution from both the canonical and noncanonical TGF-β pathways in mediating pulmonary fibrosis remains under investigation; however, recent studies in both fibroblasts and in the TGF-β transgenic mouse model demonstrate that TGF-β induces fibrogenesis extensively through MAPK and PI3K activation independent of Smad activity (3, 63). The EGFR also activates multiple downstream effector pathways, and previous data from our laboratory demonstrate that TGF-α-induced pulmonary fibrosis is directly mediated through activation of the MAPK and PI3K pathways (32, 37). These reports thus suggest that both the EGFR and SMAD-independent TGF-β pathways overlap significantly through the MAPK and PI3K in mediating fibrogenesis in vivo. In the present study, the attenuation of fibrosis from β6 inhibition likely reflects the TGF-β canonical pathways contribution to TGF-α fibrosis, since MAPK and PI3K pathways would remain activated through the intact EGFR signaling.
A number of reports in animal models of lung fibrosis demonstrate that strategies to inhibit TGF-β signaling markedly prevent pulmonary fibrosis (9, 11, 12, 34). However, incomplete resolution of lung fibrosis progression by targeting the TGF-β pathway is not unique to the TGF-α model (27). Several studies also report that antagonizing the TGF-β pathway in bleomycin-induced pulmonary fibrosis does not completely prevent fibrosis (17, 44, 55), and inhibition of TGF-β had no effect on airway remodeling in the mouse model of house dust mite-induced chronic allergic airway disease (15). In addition to the lung, models of renal fibrosis also demonstrate fibrosis progression despite inhibition of the TGF-β pathway (56, 62). And liver fibrosis in mice caused by Schistosoma mansoni infection is completely independent of the TGF-β1 pathway but is mediated through interleukin-13 (27). The variability in the effects of targeting TGF-β likely reflect differences in the relative contribution of non-TGF-β fibrogenic proteins and pathways in mediating fibrogenesis among the fibrosis models and strains of mice studied.
In summary, β6 integrin inhibition attenuated pulmonary fibrosis in the TGF-α transgenic fibrosis model, with the most prominent effects noted in the pleural regions of the lung. These findings support that bidirectional cross talk exists between EGFR and TGF-β in vivo in the propagation of lung/pleural fibrosis. However, the results from the present study suggest that TGF-β pathway inhibition alone is not effective to sufficiently reverse ongoing fibrosis following EGFR activation. These findings are also consistent with our previous work in the TGF-α model in which specific pharmacological inhibitors to the MAPK and PI3K pathways were only modestly effective in reversing fibrosis when administered after extensive fibrotic disease was already present (32, 35). Together these data implicate heterogeneity of multiple active pathways in the maintenance of pulmonary fibrotic lesions. The translational implications from these findings suggest that effective pharmacological therapy to reverse disease or prevent progression may require simultaneous inhibition of multiple pathways. In IPF, upregulation of the signaling intermediates from several fibrogenic pathways have been identified including TGF-β, EGFR, MAPK, and PI3K (6, 8, 29, 58, 60, 61). Many cell signaling pathways ultimately converge onto common pathways that regulate the cellular processes associated with pulmonary fibrosis, including fibroblast proliferation, differentiation, migration, and reduced apoptosis. Continued studies in preclinical models such as the TGF-α transgenic model will identify the mechanistic role of these pathways involved in the initiation, maintenance, and distribution of fibrotic lesions in the pleura and adventitia and will be important in designing future therapeutic strategies.
GRANTS
This work was supported by National Institutes of Health Grant, HL8655598 (W. D. Hardie), American Heart Association Scientist Development Grant, 12SDG9130040 (S. K. Madala), 1R03AR062832 (S. K. Madala), and Parker B. Francis Fellowship (S. K. Madala).
DISCLOSURES
S. M. Violette and P. H. Weinreb are employed by Biogen, the maker of the inhibitor we tested in this manuscript.
AUTHOR CONTRIBUTIONS
S.K.M., T.R.K., S.S., C.D., S.M.V., D.S., and W.D.H. conception and design of research; S.K.M., S.S., C.D., R.E., and W.D.H. performed experiments; S.K.M., R.E., M.I., S.M.V., P.H.W., D.S., and W.D.H. analyzed data; S.K.M., T.R.K., M.I., S.M.V., and W.D.H. interpreted results of experiments; S.K.M. and W.D.H. prepared figures; S.K.M. and W.D.H. drafted manuscript; S.K.M., T.R.K., R.E., M.I., S.M.V., P.H.W., D.S., and W.D.H. edited and revised manuscript; S.K.M., T.R.K., S.S., C.D., R.E., M.I., S.M.V., P.H.W., D.S., and W.D.H. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the veterinary services at Cincinnati Children's Hospital for expert care of mice used in this study and Angelica Schehr for technical assistance in measure lung mechanics.
REFERENCES
- 1.Absher M, Sjostrand M, Baldor LC, Hemenway DR, Kelley J. Patterns of secretion of transforming growth factor-alpha (TGF-alpha) in experimental silicosis. Acute and subacute effects of cristobalite exposure in the rat. Reg Immunol 5: 225–231, 1993 [PubMed] [Google Scholar]
- 2.Aluwihare P, Munger JS. What the lung has taught us about latent TGF-beta activation. Am J Respir Cell Mol Biol 39: 499–502, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrianifahanana M, Wilkes MC, Gupta SK, Rahimi RA, Repellin CE, Edens M, Wittenberger J, Yin X, Maidl E, Becker J, Leof EB. Profibrotic TGFβ responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases. FASEB J 27: 4444–4454, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrianifahanana M, Wilkes MC, Repellin CE, Edens M, Kottom TJ, Rahimi RA, Leof EB. ERBB receptor activation is required for profibrotic responses to transforming growth factor beta. Cancer Res 70: 7421–7430, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 116: 217–224, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Antoniou KM, Margaritopoulos GA, Soufla G, Symvoulakis E, Vassalou E, Lymbouridou R, Samara KD, Kappou D, Spandidos DA, Siafakas NM. Expression analysis of Akt and MAPK signaling pathways in lung tissue of patients with idiopathic pulmonary fibrosis (IPF). J Recept Signal Transduct Res 30: 262–269, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Ask K, Bonniaud P, Maass K, Eickelberg O, Margetts PJ, Warburton D, Groffen J, Gauldie J, Kolb M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta3. Int J Biochem Cell Biol 40: 484–495, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baughman RP, Lower EE, Miller MA, Bejarano PA, Heffelfinger SC. Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 16: 57–61, 1999 [PubMed] [Google Scholar]
- 9.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, Murphy A, Chakravarty S, Dugar S, Higgins L, Protter AA, Gauldie J. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med 171: 889–898, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Breuss JM, Gillett N, Lu L, Sheppard D, Pytela R. Restricted distribution of integrin beta 6 mRNA in primate epithelial tissues. J Histochem Cytochem 41: 1521–1527, 1993 [DOI] [PubMed] [Google Scholar]
- 11.D'Alessandro-Gabazza CN, Kobayashi T, Boveda-Ruiz D, Takagi T, Toda M, Gil-Bernabe P, Miyake Y, Yasukawa A, Matsuda Y, Suzuki N, Saito H, Yano Y, Fukuda A, Hasegawa T, Toyobuku H, Rennard SI, Wagner PD, Morser J, Takei Y, Taguchi O, Gabazza EC. Development and preclinical efficacy of novel transforming growth factor-beta1 short interfering RNAs for pulmonary fibrosis. Am J Respir Cell Mol Biol 46: 397–406, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest 114: 1308–1316, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decologne N, Kolb M, Margetts PJ, Menetrier F, Artur Y, Garrido C, Gauldie J, Camus P, Bonniaud P. TGF-beta1 induces progressive pleural scarring and subpleural fibrosis. J Immunol 179: 6043–6051, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Eickelberg O. Endless healing: TGFbeta SMADs, fibrosis. FEBS Lett 506: 11–14, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Fattouh R, Midence NG, Arias K, Johnson JR, Walker TD, Goncharova S, Souza KP, Gregory RC, Jr, Lonning S, Gauldie J, Jordana M. Transforming growth factor-beta regulates house dust mite-induced allergic airway inflammation but not airway remodeling. Am J Respir Crit Care Med 177: 593–603, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med 5: 167sr161, 2013 [DOI] [PubMed] [Google Scholar]
- 17.Giri SN, Hyde DM, Hollinger MA. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax 48: 959–966, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, Simon KJ, Chun Wang L, Leone DR, Lobb RR, McCrann DJ, Allaire NE, Horan GS, Fogo A, Kalluri R, Shield CF, 3rd, Sheppard D, Gardner HA, Violette SM. αvβ6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol 170: 110–125, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardie WD, Bejarano PA, Miller MA, Yankaskas JR, Ritter JH, Whitsett JA, Korfhagen TR. Immunolocalization of transforming growth factor alpha and epidermal growth factor receptor in lungs of patients with cystic fibrosis. Pediatr Dev Pathol 2: 415–423, 1999 [DOI] [PubMed] [Google Scholar]
- 20.Hardie WD, Davidson C, Ikegami M, Leikauf GD, Le Cras TD, Prestridge A, Whitsett JA, Korfhagen TR. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-α-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 294: L1217–L1225, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 286: L741–L749, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Hardie WD, Prows DR, Piljan-Gentle A, Dunlavy MR, Wesselkamper SC, Leikauf GD, Korfhagen TR. Dose-related protection from nickel-induced lung injury in transgenic mice expressing human transforming growth factor-alpha. Am J Respir Cell Mol Biol 26: 430–437, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, Simon KJ, Hahm K, Allaire NE, Rinaldi NJ, Goyal J, Feghali-Bostwick CA, Matteson EL, O'Hara C, Lafyatis R, Davis GS, Huang X, Sheppard D, Violette SM. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med 177: 56–65, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RV, Jr, Sheppard D. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol 133: 921–928, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishii Y, Fujimoto S, Fukuda T. Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am J Respir Crit Care Med 174: 550–556, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Katsumoto TR, Violette SM, Sheppard D. Blocking TGFβ via inhibition of the αvβ6 integrin: a possible therapy for systemic sclerosis interstitial lung disease. Int J Rheumatol 2011: 208219, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, Wakefield LM, Letterio JJ, Wynn TA. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol 173: 4020–4029, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Kawakami T, Ihn H, Xu W, Smith E, LeRoy C, Trojanowska M. Increased expression of TGF-beta receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-beta signaling to scleroderma phenotype. J Invest Dermatol 110: 47–51, 1998 [DOI] [PubMed] [Google Scholar]
- 29.Khalil N, O'Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, Bereznay OH, Greenberg AH. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 5: 155–162, 1991 [DOI] [PubMed] [Google Scholar]
- 30.Khalil N, Parekh TV, O'Connor R, Antman N, Kepron W, Yehaulaeshet T, Xu YD, Gold LI. Regulation of the effects of TGF-beta 1 by activation of latent TGF-beta 1 and differential expression of TGF-beta receptors (T beta R-I and T beta R-II) in idiopathic pulmonary fibrosis. Thorax 56: 907–915, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korfhagen TR, Swantz RJ, Wert SE, McCarty JM, Kerlakian CB, Glasser SW, Whitsett JA. Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J Clin Invest 93: 1691–1699, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Cras TD, Korfhagen TR, Davidson C, Schmidt S, Fenchel M, Ikegami M, Whitsett JA, Hardie WD. Inhibition of PI3K by PX-866 prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Pathol 176: 679–686, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CG, Kang HR, Homer RJ, Chupp G, Elias JA. Transgenic modeling of transforming growth factor-β1: role of apoptosis in fibrosis and alveolar remodeling. Proc Am Thorac Soc 3: 418–423, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, Minoo P. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 121: 277–287, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madala SK, Edukulla R, Davis KR, Schmidt S, Davidson C, Kitzmiller JA, Hardie WD, Korfhagen TR. Resistin-like molecule α1 (Fizz1) recruits lung dendritic cells without causing pulmonary fibrosis. Respir Res 13: 51, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol 46: 380–388, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol 46: 380–388, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madtes DK, Busby HK, Strandjord TP, Clark JG. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am J Respir Cell Mol Biol 11: 540–551, 1994 [DOI] [PubMed] [Google Scholar]
- 39.Madtes DK, Elston AL, Hackman RC, Dunn AR, Clark JG. Transforming growth factor-alpha deficiency reduces pulmonary fibrosis in transgenic mice. Am J Respir Cell Mol Biol 20: 924–934, 1999 [DOI] [PubMed] [Google Scholar]
- 40.Madtes DK, Rubenfeld G, Klima LD, Milberg JA, Steinberg KP, Martin TR, Raghu G, Hudson LD, Clark JG. Elevated transforming growth factor-alpha levels in bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 158: 424–430, 1998 [DOI] [PubMed] [Google Scholar]
- 41.Manning CB, Cummins AB, Jung MW, Berlanger I, Timblin CR, Palmer C, Taatjes DJ, Hemenway D, Vacek P, Mossman BT. A mutant epidermal growth factor receptor targeted to lung epithelium inhibits asbestos-induced proliferation and proto-oncogene expression. Cancer Res 62: 4169–4175, 2002 [PubMed] [Google Scholar]
- 42.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96: 319–328, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Nacu N, Luzina IG, Highsmith K, Lockatell V, Pochetuhen K, Cooper ZA, Gillmeister MP, Todd NW, Atamas SP. Macrophages produce TGF-beta-induced (beta-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts. J Immunol 180: 5036–5044, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oga T, Matsuoka T, Yao C, Nonomura K, Kitaoka S, Sakata D, Kita Y, Tanizawa K, Taguchi Y, Chin K, Mishima M, Shimizu T, Narumiya S. Prostaglandin F (2alpha) receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-beta. Nat Med 15: 1426–1430, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Polosa R, Puddicombe SM, Krishna MT, Tuck AB, Howarth PH, Holgate ST, Davies DE. Expression of c-erbB receptors and ligands in the bronchial epithelium of asthmatic subjects. J Allergy Clin Immunol 109: 75–81, 2002 [DOI] [PubMed] [Google Scholar]
- 46.Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, Davies DE. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J 14: 1362–1374, 2000 [DOI] [PubMed] [Google Scholar]
- 47.Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, Devitt ML, Horan GS, Weinreb PH, Lukashev ME, Violette SM, Grant KS, Colarossi C, Formenti SC, Munger JS. Inhibition of integrin αvβ6, an activator of latent transforming growth factor-β, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med 177: 82–90, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. J Cell Biochem 102: 593–608, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stahlman MT, Orth DN, Gray ME. Immunocytochemical localization of epidermal growth factor in the developing human respiratory system and in acute and chronic lung disease in the neonate. Lab Invest 60: 539–547, 1989 [PubMed] [Google Scholar]
- 51.Strandjord TP, Clark JG, Guralnick DE, Madtes DK. Immunolocalization of transforming growth factor-alpha, epidermal growth factor (EGF), and EGF-receptor in normal and injured developing human lung. Pediatr Res 38: 851–856, 1995 [DOI] [PubMed] [Google Scholar]
- 52.Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem 275: 11858–11864, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Van Winkle LS, Isaac JM, Plopper CG. Distribution of epidermal growth factor receptor and ligands during bronchiolar epithelial repair from naphthalene-induced Clara cell injury in the mouse. Am J Pathol 151: 443–459, 1997 [PMC free article] [PubMed] [Google Scholar]
- 54.Waheed S, D'Angio CT, Wagner CL, Madtes DK, Finkelstein JN, Paxhia A, Ryan RM. Transforming growth factor alpha (TGFα) is increased during hyperoxia and fibrosis. Exp Lung Res 28: 361–372, 2002 [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, Wang Y, Hyde DM, Gotwals PJ, Koteliansky VE, Ryan ST, Giri SN. Reduction of bleomycin induced lung fibrosis by transforming growth factor beta soluble receptor in hamsters. Thorax 54: 805–812, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang S, Wilkes MC, Leof EB, Hirschberg R. Noncanonical TGF-β pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal Physiol 298: F142–F149, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weinreb PH, Simon KJ, Rayhorn P, Yang WJ, Leone DR, Dolinski BM, Pearse BR, Yokota Y, Kawakatsu H, Atakilit A, Sheppard D, Violette SM. Function-blocking integrin αvβ6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J Biol Chem 279: 17875–17887, 2004 [DOI] [PubMed] [Google Scholar]
- 58.White ES, Thannickal VJ, Carskadon SL, Dickie EG, Livant DL, Markwart S, Toews GB, Arenberg DA. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med 168: 436–442, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Worthington JJ, Klementowicz JE, Travis MA. TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci 36: 47–54, 2011 [DOI] [PubMed] [Google Scholar]
- 60.Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med 205: 1659–1672, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshida K, Kuwano K, Hagimoto N, Watanabe K, Matsuba T, Fujita M, Inoshima I, Hara N. MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J Pathol 198: 388–396, 2002 [DOI] [PubMed] [Google Scholar]
- 62.Zhou G, Li C, Cai L. Advanced glycation end-products induce connective tissue growth factor-mediated renal fibrosis predominantly through transforming growth factor beta-independent pathway. Am J Pathol 165: 2033–2043, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou Y, Lee JY, Lee CM, Cho WK, Kang MJ, Koff JL, Yoon PO, Chae J, Park HO, Elias JA, Lee CG. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-beta-induced pulmonary fibrosis. J Biol Chem 287: 41991–42000, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]