

Abstract
Bronchopulmonary dysplasia (BPD), a lung disease of prematurely born infants, is characterized in part by arrested development of pulmonary alveolae. We hypothesized that heme oxygenase (HO-1) and its byproduct carbon monoxide (CO), which are thought to be cytoprotective against redox stress, mitigate lung injury and alveolar simplification in hyperoxia-exposed neonatal mice, a model of BPD. Three-day-old C57BL/6J mice were exposed to air or hyperoxia (FiO2, 75%) in the presence or absence of inhaled CO (250 ppm for 1 h twice daily) for 21 days. Hyperoxic exposure increased mean linear intercept, a measure of alveolar simplification, whereas CO treatment attenuated hypoalveolarization, yielding a normal-appearing lung. Conversely, HO-1-null mice showed exaggerated hyperoxia-induced hypoalveolarization. CO also inhibited hyperoxia-induced pulmonary accumulation of F4/80+, CD11c+, and CD11b+ monocytes and Gr-1+ neutrophils. Furthermore, CO attenuated lung mRNA and protein expression of proinflammatory cytokines, including the monocyte chemoattractant CCL2 in vivo, and decreased hyperoxia-induced type I alveolar epithelial cell CCL2 production in vitro. Hyperoxia-exposed CCL2-null mice, like CO-treated mice, showed attenuated alveolar simplification and lung infiltration of CD11b+ monocytes, consistent with the notion that CO blocks lung epithelial cell cytokine production. We conclude that, in hyperoxia-exposed neonatal mice, inhalation of CO suppresses inflammation and alveolar simplification.
Keywords: bronchopulmonary dysplasia, CD11b, macrophage, CCL2, neonatal lung injury
an estimated 30% of premature infants with a birth weight between 500 and 1,500 g will develop respiratory distress syndrome and its consequence, bronchopulmonary dysplasia (BPD). Pathological features of BPD include interstitial fibrosis and abnormal cellular proliferation, as well as arrested alveolar development. Infants with BPD are at risk for morbidity and mortality during the first years of life, including increased medication use, hospital readmissions (14), and lung function abnormalities that persist into childhood and even early adulthood (13, 24).
Recent studies implicate lung inflammation as a contributor to BPD development. Leukocyte infiltrates, in particular neutrophils and cells of the monocyte/macrophage lineage, as well as inflammatory mediators including IL-1β, IL-6, tumor necrosis factor-α, and CXC and CC-chemokines, have been detected in the lungs of patients with BPD (3, 23, 25, 30, 33). Treatment of hyperoxia-exposed neonatal lungs with antibodies or antagonists against neutrophil (CXCL1, CXCL2) and macrophage (CCL2) chemokines prevents lung inflammation and alveolar simplification (2, 9, 50, 55).
Oxidative stress has also been implicated as a cause of pulmonary injury leading to the development of BPD. Prolonged exposure to sublethal hyperoxia in animal models recapitulates some of the processes observed in BPD (18, 40, 41), such as arrested alveolar development, a paucity of α-actin-positive myofibroblasts at the alveolar septal tips, and a corresponding increase in the number of interstitial myofibroblasts (4, 21, 22, 48). Importantly, rodents exhibit a saccular stage of lung development at birth that is only completed after 4 wk of postnatal alveolarization (53). By comparison, saccules appear in the human lung by 23 wk of gestation (11, 19). In newborn animals, hyperoxia has been shown to be a strong inducer of various proinflammatory cytokines in airway cells and pulmonary tissue (51). Reactive oxygen species may be produced by resident lung epithelial cells under hyperoxic conditions or from neutrophils and macrophages invading the lungs as part of the inflammatory process (42).
Organisms faced with hyperoxic stress have evolved sophisticated mechanisms to ensure survival. Emerging evidence highlights a crucial role for stress-inducible heme oxygenase (HO)-1 and its catalytic byproducts, carbon monoxide (CO) and biliverdin, in cytoprotection against hyperoxic and ischemic forms of tissue and cellular injury (12, 15, 31, 32, 34, 35, 38, 49). Although the precise mechanisms whereby CO modulates the inflammatory milieu remain under study, there is some evidence that it does so by tilting the milieu toward the production of the anti-inflammatory cytokine IL-10 (12, 34, 38), while at the same time blunting expression of TNF-α and IL-1β (17). In immature rodents, HO-1 expression is induced during lung maturation and following exposure to hyperoxia, consistent with adaptation to extrauterine conditions of increased oxidative stress and inflammation (10, 44). It has recently been shown that HO-1 and CO mitigate hyperoxia-induced pulmonary inflammation in neonatal mice (15). However, in this study, arrested alveolar development was minimally affected by CO administration, and the mechanisms by which CO reduced pulmonary inflammation were not explored.
We hypothesized that inhalation of low-dose episodic CO mitigates the effect of hyperoxia on alveolar development by an anti-inflammatory mechanism. We report here that inhalation of CO suppresses inflammatory cell trafficking, proinflammatory cytokine expression, and alveolar simplification in hyperoxia-exposed mice.
MATERIALS AND METHODS
Experimental animals.
All animal experiments were performed according to protocols approved by the University of Michigan Institutional Animal Care and Use Committee. Timed pregnant C57BL/6J mice were either purchased from Jackson Laboratories (Bar Harbor, ME) or bred at the University of Michigan animal facility. Newborn pups from six different litters were subdivided into four groups within 24 h of birth. At 3 days of age, pups were exposed to normoxia (21% O2), hyperoxia (75% O2), carbon monoxide (CO 250 ppm) + normoxia, or CO (250 ppm) + hyperoxia for 7, 10, 14, or 21 days. Selected experiments employed B6.129S4-Scya2tm1Rol CCL2-null mice (Jackson Laboratories).
In addition, we obtained HO-1-null mice (C57BL/6J background) from Dr. Mu-En Lee (Harvard School of Public Health) (54). Colonies were maintained by breeding HO-1−/− males with HO-1+/− females. Offspring were genotyped at the time of birth using PCR (16). For all experiments, HO-1+/+ and HO-1−/− mice were age matched at postnatal day 3. Each litter consisted of 8–10 pups per group to control for the effects of litter size on nutrition and growth. Animals were either exposed to room air or various gas mixtures for up to 3 wk.
To achieve 75% O2, 100% O2 was delivered at a flow rate of 0.5 l/min into a custom-made 1.5-m3 plastic chamber. Oxygen concentrations were monitored continuously with an oxygen analyzer (Neutronics, Exton, PA), gas lines were filtered with activated charcoal to remove excess ammonia, and calcium chloride was used to maintain CO2 levels below 0.05%. Animals were exposed to 250 ppm CO at a flow rate of 10 l/min for 1 h twice daily. A CO analyzer (Ntron model 1100; Neutronics, Exton, PA) was used to monitor CO levels continuously in the chamber. Dams were rotated between normoxia and hyperoxia conditions, or CO + normoxia and CO + hyperoxia conditions daily to minimize oxygen toxicity to the dams and to remove potential confounders of milk production induced in the various environments. Relative humidity in the chamber was maintained at 45–50%. Dams and pups were given food and water ad libitum and maintained on a 12-h:12-h dark/light cycle. Mice were euthanized with an overdose of ketamine and xylazine followed by thoracotomy after exposure for the indicated time periods and experimental conditions.
Histological analysis.
Lungs were perfused with PBS with 20 mM EDTA through the right ventricle at a constant pressure of 20 cm H2O for 3–5 min. Lungs were then intratracheally inflated with 10% formalin, fixed overnight at 4°C, then stored in 70% ethanol before being embedded in paraffin. Five-micron sections were stained with hematoxylin and eosin. Stained sections were visualized under light microscopy, and images were captured using MetaMorph version 7.0r3 software (Molecular Devices, Sunnyvale, CA) on an Eclipse TE2000-E microscope (Nikon Instruments, Melville, NY).
Morphometric analysis.
To assess alveolarization, mean alveolar diameter was determined by measurement of mean linear intercept (MLI). MLI, equal to the mean interalveolar distance, was measured by dividing the total length of a line drawn across the lung section by the number of intercepts encountered, as described (7). Briefly, three representative lung sections for each animal were photographed at ×200 magnification. A grid with parallel lines spaced at 60 μm was overlaid onto the image, and the length of each cord was defined by the intercept with the alveolar walls. Fifteen nonoverlapping fields for each section were examined. MetaMorph image analysis software was used for morphometric analysis.
Bronchoalveolar inflammatory and differential cell counts.
Bronchoalveolar lavage fluid (BAL) fluid was obtained by intratracheal instillation of 3 × 1 ml ice-cold PBS and filtered via a 70-μm cell strainer to exclude epithelial cells. Erythrocytes were lysed using RBC Lyse (BD Biosciences, San Diego, CA). BAL fluid was centrifuged at 2,000 revolution/min for 15 min, and supernatants were removed and stored in −80°C until further analysis. The cell pellet was resuspended in PBS, and total infiltrating cells (neutrophils and macrophages) were counted using a Hemavet 950FS cell hemocytometer (Drew Scientific, Dallas, TX).
Immunohistochemistry and fluorescence microscopy.
Paraffin blocks were sectioned at 500-μm intervals at a thickness of 5 μm. After deparaffinization, rehydration, and antigen retrieval, endogenous peroxidase was blocked using a rodent serum block. Serial sections were probed with fluorophore-labeled mouse anti-α-smooth muscle actin (clone 1A4, Sigma-Aldrich), Alexa Fluor (AF)-conjugated donkey anti-goat IgG (Molecular Probes, Portland OR), rat anti-mouse F4/80 (AbD Serotec, Raleigh, NC), anti-CD11b (R&D Systems, Minneapolis, MN), and rat anti-mouse Gr-1 (R&D Systems). Slides were subsequently incubated with secondary antibody, ABC reagent (Vector Laboratories, Burlingame, CA) and diaminobenzidine. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes), and immunofluorescence (DAPI). In addition, sections were incubated with AF-555-conjugated goat anti-mouse CCL2 (R&D Systems), AF633-rat anti-mouse CD68 (AbD Serotec), or AF-conjugated isotype control IgGs. To examine endogenous HO-1 staining, we used a rabbit polyclonal to rat HO-1 (StressGen; Victoria, BC, Canada). Images were visualized using an Axiovert 100M inverted fluorescent microscope (Carl Zeiss, Thornwood, NY).
Focused gene array analysis of neonatal mouse lung lysates.
Lung RNA was subjected to a targeted PCR array examining 84 mouse inflammatory cytokines (SA Biosciences, Frederick, MD). Total RNA extraction was performed using the Qiagen Animal Spin protocol (Valencia, CA). Total RNA was quantified using the Nanodrop N-1000 (Agilent Biosystems, Santa Clara, CA). After verification of RNA quality and integrity, first-strand cDNA synthesis was performed on an Eppendorf Mastercycler using a Reaction Ready First-Strand cDNA Synthesis kit (SA Biosciences). The cDNA template was combined with RT2 Real-Time SYBR Green/Rox Master Mix (SA Bioscience) and RNAse-free water. A final reaction volume of 25 μl was added to each well of PCR array. Finally, pathway-focused mRNA was amplified on a 7300 Fast Real-Time PCR system (Applied Biosystems, Foster City, CA). Genes were plotted as heat maps by hierarchical clustering using the correlation coefficient as a distance measure and the average of each cluster for cluster formation of the genes using Amap software (Bioconductor, Seattle, WA). Expression values are visualized with color ranging from red (high expression) over white (intermediate expression) to blue (low expression).
RNA isolation and real-time RT-PCR.
Quantitative RT-PCR was performed on eight genes with increased or decreased expression following the different exposure conditions in C57BL/6J mice (n = 10–18 samples/condition). Lung total RNA was extracted using an RNAeasy Mini kit (Qiagen, Gaithersburg, MD). First-strand cDNA (500 ng) was reverse transcribed with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR was performed with an ABI PRISM 7000 sequence detection system with TaqMan Universal PCR Master Mix and Assay-on-Demand gene expression probes (Applied Biosystems). We measured the following mRNAs: IL-1β, TNF-α, IL-10, IL-13, IL-6, CCL2, CXCL1, and CXCL2. β-Actin served as housekeeping gene. All primers were purchased from Applied Biosystems. All samples were analyzed in quadruplicate. mRNA expression was normalized to β-actin level by the 2−ΔΔCt method.
Protein extraction and cytokine expression.
Whole lung samples were homogenized with lysis buffer containing 1 mM Tris·HCl, pH 7.4, 0.5 M EDTA, 5 M NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 10% SDS supplemented with protease inhibitors (Complete Protease Inhibitor Cocktail; Roche, Indianapolis, IN). Supernatants were analyzed for IL-1β, IL-6, IL-10, IL-13, TNF-α, CCL2, CXCL1, and CXCL2 protein abundance with a multiplex mouse immunoassay kit (Bio-Rad, Hercules, CA) using the Luminex 200 System (Luminex, Austin, TX). Supernatants were analyzed in triplicate.
Flow cytometric analysis of infiltrating leukocyte populations.
Lung tissues were harvested, and single-cell suspensions were obtained by digestion with collagenase IV (Gibco Life Technology, Grand Island, NY) for 1 h at 25°C while being shaken. FACSlyse buffer (BD Bioscience) was used to lyse erythrocyte contaminants. Total lung cell counts were assessed by Hemavet hemocytometer (Drew Scientific) and reconfirmed by flow cytometry analysis using FITC-anti mouse CD45 antibody (BD Bioscience) and flow cytometry absolute count standard beads (BD Bioscience). Viable cells were identified by the absence of DAPI (Molecular Probes) staining. Nonspecific antibody staining was inhibited using Fc Block (BD Bioscience). Analysis of whole lung total alveolar leukocytes was performed using APC-anti-mouse CD11b and phycoerythrin (PE)-anti-mouse Ly6G/Ly-6C (Gr-1) (both from BD Bioscience). To detect infiltrating alveolar macrophages, cells were purified and identified in four stages. First, the leukocyte population was gated using FITC-conjugated anti-mouse CD45. Second, infiltrating monocyte/macrophages double positive for staining with APC-Cy7-conjugated anti-mouse F4/80high and PE-conjugated anti-mouse CD11c were then positively gated. Third, cells were gated using PerCPCy5.5-conjugated anti-mouse CD11b (all from BD Bioscience). For calculation of total cell numbers in tissue, normalization to weight of tissue was performed. Data were acquired on a BD Bioscience FACS Canto flow cytometer and analyzed with FlowJo v.9.4.11 (Tree Star, Ashland, OR).
Cell culture and ex vivo CO exposure.
Murine type II alveolar epithelial cells were isolated from C57BL/6J mice as described previously (8). Briefly, after proteolytic digestion of lung tissue, single-cell suspensions of lung tissue were depleted of macrophages by negative selection in a magnetic separator using anti-CD32 and anti-CD45 antibodies. The negative cells were plated overnight in 100-mm culture plates. The type II alveolar epithelial cells comprised the nonadherent population. These cells were recovered for cell culture and maintained in humidified incubators (5% CO2-95% air) at 37°C. Cells were grown to 60–80% confluence and rendered quiescent in medium containing serum-free medium. Selected cultures were exposed to 95% O2 or 250 ppm CO + 95% O2 in an incubator chamber (Biospherix, Lacona, NY). Control cells were cultured in standard tissue culture conditions (95% air-5% CO2).
Statistical analysis.
All results were expressed as means ± SE, with the number of experiments performed provided in the figure legends. Results were compared using a one-way ANOVA or Mann-Whitney U test, as appropriate, using GraphPad Prism software (La Jolla, CA). To pinpoint differences between specific groups, Newman-Keuls test was used. Values were considered significantly different when P ≤ 0.05.
RESULTS
CO partially mitigates the effects of hyperoxia-induced lung injury on body weight.
Prolonged neonatal exposure to hyperoxia adversely affected somatic growth (Fig. 1A). Hyperoxic exposure resulted in a significantly decreased weight at the end of the exposure period compared with air-exposed littermates. This was true for all experiments under hyperoxic conditions despite rotation of dams. Intermittent exposure to CO at 250 ppm during hyperoxic exposure tended to increase body weight, with the difference reaching statistical significance on day 21 of exposure (P < 0.05).
Fig. 1.

Low-intermittent-dose CO partially mitigates the effects of hyperoxia on body weight and alveolar arrest in neonatal mice. A: group mean body weight at 3, 7, 10, 14, and 21 days of life under conditions of normoxia (n = 167), hyperoxia (n = 106), CO + normoxia (n = 52), and CO + hyperoxia (n = 64). Hyperoxic exposure decreased body weight compared with normoxia. On day 21 of exposure, mice of the hyperoxia + CO group were significantly larger than mice of the hyperoxia group (*P < 0.05, ANOVA). B: representative images of hematoxylin and eosin-stained lungs of neonatal mice exposed to normoxia (21% oxygen), hyperoxia (75% oxygen), intermittent exposure to 250 ppm carbon monoxide plus compressed air for 1 h twice daily (normoxia + CO), or 75% oxygen and 250 ppm CO for 1 h twice daily (hyperoxia + CO). Compared with air-exposed mice, hyperoxia-exposed mice demonstrated increased alveolar size indicative of alveolar development arrest. CO-treated mice exposed to hyperoxia showed attenuated alveolar simplification compared with hyperoxia-exposed mice. CO-treated mice exposed to normoxia showed similar alveolar architecture as normoxia-treated mice. Solid bar scale = 200 μm, and all panels are ×200. C: corresponding group mean linear intercepts of all groups. Data are reported as means ± SE. n = 17–24 per group, *different from hyperoxia, P < 0.05. All data were assessed by 1-way ANOVA.
CO mitigates hypoalveolarization in hyperoxia-exposed neonatal mice.
Premature mice are born at the saccular stage of lung development. Sacculi are transformed into alveoli within the first 2 wk after birth by septal thinning and secondary septation. Histological analysis in untreated postnatal 3-day (p3) normoxia-exposed mice revealed markedly underdeveloped lungs at the saccular stage of development (Fig. 1B). After 7–21 days of exposure (postnatal days 10–24), hyperoxic mice exhibited a significant structural delay in alveolar development compared with age-matched normoxia-exposed counterparts. Alveolar simplification in hyperoxia-exposed mice was accompanied by increased collagen deposition in the alveolar walls (not shown). Lungs of mice intermittently exposed to 250 ppm CO for 1 h twice daily were structurally similar to normoxic control mice. Mice exposed to 75% oxygen and CO showed partial mitigation of hypoalveolarization compared with mice exposed to hyperoxia alone. To quantitate these differences, morphometric measurements of the MLI were compared between study groups (Fig. 1C). Compared with air-exposed mice, mean MLI for hyperoxia-exposed mice was significantly increased at 10, 14, and 21 days of exposure. When neonatal pups were exposed to 250 ppm CO in the setting of hyperoxia, we observed a significant decrease in MLI on day 10 of exposure compared with hyperoxia-exposed mice (P < 0.001). Similar decreases in MLI were observed at other time points although the differences were not statistically significant. Together, these data demonstrate that chronic neonatal hyperoxic exposure results in markedly decreased alveolarization, which is mitigated by intermittent CO treatment.
CO suppresses hyperoxia-induced lung leukocyte recruitment.
We hypothesize that CO mitigates the effect of hyperoxia on the developing lung via an anti-inflammatory mechanism. We examined the inflammatory cell response in the BAL fluid. The number of BAL polymorphonuclear cells and monocyte/macrophages was significantly increased in hyperoxia-exposed pups at 7 days (Fig. 2A), reaching a peak at 14 days of hyperoxic exposure (Fig. 2B). Macrophages from hyperoxia-exposed mice showed a larger, foamy appearance, indicative of activation (data not shown), concordant with their previously reported appearance (49). CO treatment decreased the number of neutrophils and monocyte/macrophages in the BAL fluid. These findings indicate that CO treatment may confer significant protection against hyperoxia-induced pulmonary inflammation in developing lungs.
Fig. 2.
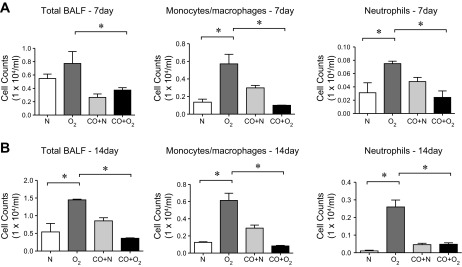
Episodic CO treatment suppresses leukocyte trafficking to the lungs of hyperoxia-exposed neonatal mice. A and B: bronchoalveolar lavage fluid (BALF) from 3-day-old pups exposed to normoxia or hyperoxia in the absence or presence of CO for 7 (A) or 14 days (B). Total BALF cells (left), monocyte/macrophages (middle), and neutrophils (right) were measured by Hemavet. n = 12–17 per group, *P < 0.05, ANOVA.
To further understand the role of HO-1/CO in neonatal lung injury, we measured lung HO-1 mRNA expression in hyperoxia-exposed mice. Hyperoxia significantly increases HO-1 expression (Fig. 3A). HO-1-null (HO-1−/−) mice exposed to hyperoxia showed exaggerated alveolar simplification compared with wild-type mice (Fig. 3B). Exaggerated hypoalveolarization was confirmed by MLI measurements (Fig. 3C). Immunohistochemistry showed that hyperoxia increases HO-1 expression in alveolar macrophages (Fig. 3D). As expected, HO-1 expression was not detectable in hyperoxia-exposed HO-1-null mice. Finally, compared with HO-1+/+ hyperoxia-exposed mice, HO-1-null mice showed a significant increase in BAL macrophages (Fig. 3E). There was no significant increase in BAL neutrophils (Fig. 3F). Together, these data demonstrate that HO-1 deficiency is sufficient to exacerbate the effects of hyperoxia on the newborn lung.
Fig. 3.

Heme oxygenase (HO-1) knockout exacerbates hypoalveolarization and increases leukocyte infiltration in hyperoxia-exposed neonatal mice. A: whole lung HO-1 mRNA expression from wild-type mice exposed to either normoxia (n = 20–26 mice per group) or hyperoxia (n = 19–23 mice per group) for up to 21 days. Hyperoxic exposure increased mRNA expression, as measured by qPCR (*P < 0.05, ANOVA). B: 3-day-old HO-1+/+ or HO-1-null (HO-1−/−) mice were exposed to air or 75% oxygen for 14 days. Hyperoxia-exposed HO-1−/− mice showed alveolar arrest with large airspaces. Hyperoxia-exposed HO-1-null mice showed exaggerated alveolar simplification compared with wild-type mice. C: group mean chord length data (n = 4–9 per group, *P < 0.05, ANOVA). D: hyperoxia-induced HO-1 expression is observed in lung alveolar macrophages (arrows). Immunohistochemistry was performed with an antibody against murine HO-1 (scale bar = 100 μm). E and F: hyperoxia-induced inflammatory cell influx in neonatal mouse lungs is exacerbated by HO-1 deletion. BALF monocytes (E) and neutrophils (F) were counted 14 days after exposure to room air (21% O2) or hyperoxia (75% O2). Compared with HO-1+/+ mice, HO-1-null mice showed a significant increase in macrophages and a trend toward increased neutrophils (n = 4–7 mice per group, means ± SE, *different from hyperoxia-exposed group, P < 0.05, ANOVA).
To further characterize the inflammatory response to hyperoxic exposure and CO treatment, lung cells were subjected to flow cytometry. More than 90% of the isolated whole lung inflammatory cells were CD45-positive cells. First, we examined cells of the monocyte/macrophage lineage. By day 7, the percentage of cells expressing the monocyte/macrophage cell surface markers F4/80+ and CD11c+ (49) significantly increased under hyperoxic exposure. The total number of F4/80+ and CD11c+ double-positive cells peaked at 14 days of hyperoxic exposure (data for day 14 are shown in Fig. 4A). CO treatment tended to decrease the accumulation of cells infiltrating in the hyperoxic-exposed lungs. On the other hand, hyperoxia-exposed HO-1-null mice showed increased F4/80+ and CD11c+ double-positive macrophages.
Fig. 4.
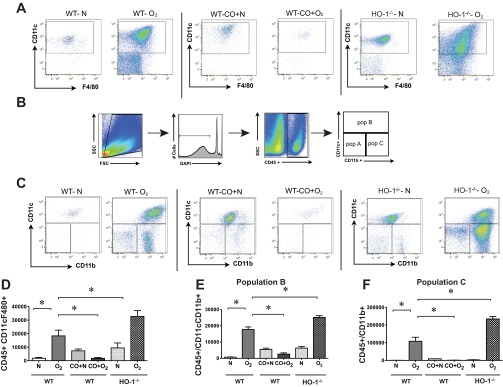
Hyperoxic exposure induces alveolar monocyte/macrophage infiltration into neonatal lungs that is ameliorated by CO-induced treatment. Representative flow cytometric scattergrams of monocyte/macrophage subpopulations within the lungs of wild-type (WT) and HO-1-null mice exposed to normoxia and hyperoxia for 14 days. Wild-type mice were also exposed to normoxia + CO or hyperoxia + CO. Each dot plot is representative of 4 independent experiments. A: CD45+, CD11c+, F4/80+ macrophages. B: gating scheme used in subsequent flow cytometric analysis. C: whole lung live CD45+ cells were analyzed for expression of CD11c and CD11b. D–F: comparison of cell counts for F4/80 and CD11c double-positive macrophages (D), CD11chigh CD11bhigh exudative macrophages (subpopulation B, E), and CD11clowCD11bhigh inflammatory monocytes (subpopulation C, F). Cell counts represent the number of cells per 106 events. Hyperoxia increased the trafficking of macrophage subpopulations into the neonatal lung, and CO treatment attenuated macrophage recruitment. HO-1−/− mice showed increases lung macrophages. n = 4–10 per group, means ± SE, *compared to hyperoxia alone, P < 0.05, ANOVA.
Recent studies suggest that, unlike resident alveolar macrophages, freshly recruited macrophages express low levels of CD11c and high levels of CD11b, which is required for their successful migration to inflamed tissues (20, 43). We therefore analyzed CD45-positive lung cells for both CD11c and CD11b (data for day 14 are shown in Fig. 4, B and C; remaining time points are not shown). We found three distinct subpopulations of cells. CD11c+ CD11blow cells (population A) increased early during hyperoxic exposure. Backgating of these cells showed them to be small, low-complexity cells, probably lymphocytes (not shown). CD11chigh CD11bhigh cells, presumably representing exudative macrophages (population B), increased significantly early during hyperoxic exposure. CD11clow CD11bhigh cells (population C), which presumably represent a transitional population of inflammatory monocytes, were also increased throughout the hyperoxic exposure period. Both CD11bhigh populations backgated to the large, high-complexity region typical of macrophages (not shown). Administration of CO dramatically reduced the number of CD11c+ F4/80+ macrophages, as well as cells in populations A, B, and C (Fig. 4, D–F). Interestingly, lungs of air/CO-exposed mice also showed smaller but significant increases in population C cells. Finally, compared with hyperoxia-exposed wild-type mice, lungs from HO-1-null mice showed increased lung macrophages, in particular CD11clow CD11b high cells.
Next, we used flow cytometry to examine lung neutrophils. We employed antibodies against Gr-1 (Ly6C/G) and CD45 to identify infiltrating neutrophils (CD45high Gr-1+). Following hyperoxic exposure, the number of CD45high Gr-1+ cells increased compared with normoxic littermates, and exogenous CO decreased infiltrating neutrophils at all time points (data from day 14 are shown in Fig. 5A). Immunofluorescent staining confirmed that hyperoxia increased lung Gr-1-positive neutrophils compared with normoxic littermates (Fig. 5B, bottom). Exogenous application of CO to hyperoxic neonatal mice attenuated the infiltration of neutrophils. Similarly, CD11b-positive (red), F4/80-positive (green; colocalization, yellow) macrophages were found in the alveolar septae and alveolar spaces of lungs from hyperoxia-exposed mice (Fig. 5B, top). Intermittent CO exposure during hyperoxia significantly decreased macrophage infiltration into the alveolar space.
Fig. 5.
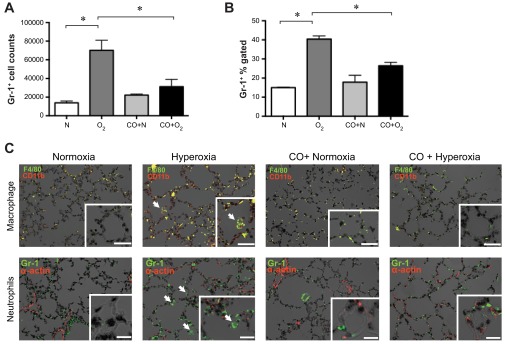
Hyperoxic exposure induces neutrophilic infiltration into neonatal lungs that is ameliorated by CO-induced treatment. A and B: high Gr-1+, CD45+ cells, representing infiltrating neutrophils, were measured by flow cytometry. Treatment of hyperoxia-exposed mice with exogenous CO decreased the number of CD45 high Gr-1+ cells. Data from 14-day exposures are shown (A, number of cells per 106 events; B, percentage of CD45+ cells). C: immunofluorescent stains of 14-day lung sections from normoxia-, hyperoxia-, CO + normoxia-, and CO + hyperoxia-exposed mice. Top: neutrophil panels show Gr-1 (green, arrows) and α-actin (red). Bottom: macrophage staining panels include F4/80 (green) and CD11b (red). Colocalization (yellow) indicates CD11b-positive macrophages (arrows). Nuclei were counterstained with DAPI (black). Original magnification is ×200, scale bar = 10 μm. Insets: high-magnification views (×400).
Exogenous CO administration regulates the expression of pro- and anti-inflammatory cytokines/chemokine under hyperoxic conditions.
To determine the cytokines responsible for hyperoxia-induced macrophage/monocyte lung infiltration, we performed a gene array focusing on inflammatory markers. Lung mRNA from day 3 normoxia-exposed mice, day 14 normoxia-exposed mice, day 14 normoxia-exposed mice, day 14 hyperoxia-exposed mice, and day 14 hyperoxia + CO-exposed mice was subjected to a targeted PCR array examining mouse inflammatory cytokines (Fig. 6). Hierarchical clustering revealed a group of 12 genes, which were downregulated during normal development, increased by hyperoxic exposure, and decreased by concomitant CO exposure. This group of genes included Ccl2, Cxcl1, and Il1b. We therefore examined the mRNA and protein levels of these and other cytokines by qPCR and multiplex immune assay, respectively. Lung samples from 14 days postexposure were studied (Fig. 7, A–F). Expression of mRNAs encoding the proinflammatory cytokines IL-1β, TNF-α, IL-6, the monocyte chemoattractant CCL-2, and the neutrophil chemoattractants CXCL1 and CXCL2 were significantly greater in the lungs of hyperoxia-treated mice compared with lungs from air-exposed mice. Hyperoxia slightly decreased β-actin copy number on days 14 and 21 of exposure, which may have led to an underestimation of cytokine induction (not shown). Similar increases were observed on the protein level. CO treatment under hyperoxia effectively suppressed all proinflammatory markers measured.
Fig. 6.
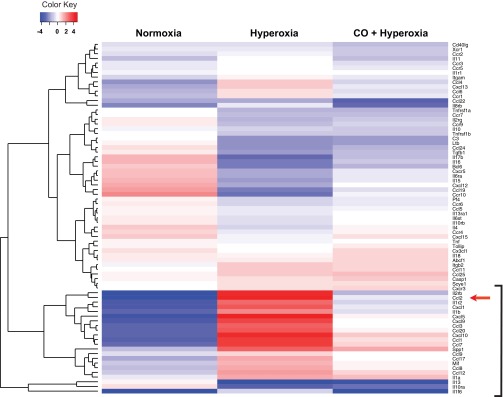
Hierarchical clustering diagram of differential inflammatory gene expression in neonatal mice lungs on 14 days after hyperoxic-induced lung injury. Gene expression was measured by focused mRNA array and analyzed by hierarchical clustering and plotted on a heat map using R software. Each lane represents the expression profile of 84 unique probe sets. Lane 1 compares gene expression from 2 P3 normoxia- and 2 14-day normoxia-exposed mice. Lane 2 compares gene expression from 2 P3 normoxia- and 2 P17 hyperoxia-exposed mice. Lane 3 compares 2 P3 normoxia- and 2 P17 hyperoxia + CO-exposed mice. Red and blue colors indicate up- and downregulated expression levels, respectively. White color indicates no change in expression level. The bracket indicates a group of genes that were downregulated with development (lane 1), increased with hyperoxic exposure (lane 2), and suppressed by CO treatment (lane 3).
Fig. 7.

Exogenous CO administration regulates the expression of pro- and anti-inflammatory cytokines under hyperoxic conditions. mRNA and protein levels were measured by quantitative PCR and multiplex immune assay at 14 days postexposure. A–F: hyperoxia increased the mRNA and protein expression of IL-1β, TNF-α, IL-6, CCL-2, IL-10, IL-13, CxCL-1, and CxCL-2. CO administration significantly reduced IL-1β, TNF-α, IL-6, CCL-2, CxCL-1, and CxCL-2 mRNA expression as well as IL-1β, CCL-2, CxCL-1, and CxCL-2 protein expression. G and H: IL-10 and IL-13 were downregulated by hyperoxic treatment and increased with CO exposure. The data pool is at least 3 independent experiments (means ± SE, n = 10–18 animals per group for mRNA analysis, n = 4 per group for protein analysis, *P <0.05, ANOVA).
In addition to the examination of proinflammatory cytokines/chemokines, experiments were also performed to determine the expression levels of two “anti-inflammatory” cytokines, IL-10 and IL-13 (Fig. 7, G and H). IL-10 and IL-13 were decreased in the lungs of hyperoxia-treated mice. Concordantly, the protein levels of both cytokines were also decreased after hyperoxic exposure. CO tended to increase IL-10 and significantly increased IL-13 mRNA and protein levels during both air and hyperoxic exposure.
CCL2 is required for hyperoxia-induced macrophage/monocyte infiltration and hypoalveolarization.
On the basis of the strong induction of CCL2 demonstrated by our microarray data, we chose to evaluate in depth the functional consequences of its expression in the development of BPD. CCL2 is a critical monocyte chemoattractant protein, which binds to its cognate ligand CCR2 expressed on monophagocytes, recruiting them to sites of inflammation. To further characterize hyperoxia-induced CCL2 expression, we performed immunofluorescence staining of lung sections for CCL2 and CD68, a macrophage marker (Fig. 8A). Hyperoxia increased the number of CD68-positive cells, indicating macrophage infiltration. CCL2 expression was also increased, primarily in CD68-negative cells of the airway and alveolar epithelium. CO administration decreased hyperoxia-induced epithelial cell CCL2 expression. We tested whether hyperoxia increases respiratory epithelial CCL2 expression in vitro. Murine type II alveolar epithelial cells were cultured in an incubator chamber containing a normoxic or hyperoxic atmosphere in the absence or presence of 250 ppm CO (Fig. 8B). Hyperoxia caused a rapid increase in CCL2 mRNA expression, which was attenuated by CO treatment. Taken together, these data suggest that CO attenuates hyperoxia-induced lung inflammation by decreasing respiratory epithelial cell CCL2 expression.
Fig. 8.

Hyperoxia exposure is associated with an increase in respiratory epithelial cell CCL2 expression. Lung sections were stained for CCL2 (red) or CD68 (green); colocalization appears yellow. Compared with normoxia-exposed mice, hyperoxia-exposed mice show an increase in CCL2 expression primarily in the epithelium as well as an increase in CD68-positive macrophages. Episodic CO treatment the presence of hyperoxic exposure suppressed CCL2 expression. Lung sections from 7, 10, 14, and 21 days of exposure are shown (line segment = 50 μm, results are typical of 3 individual experiments). B: murine type II alveolar epithelial cells were exposed to 250 ppm CO or room air in the absence or presence of high oxygen tension (>95% O2) for a duration of 2, 6, or 12 h. CCL2 mRNA expression was increased by hyperoxia and attenuated by CO. Group mean data for CCL2 mRNA expression are presented (means ± SE, n = 3, *P < 0.05, ANOVA).
To test the hypothesis that CCL2 is required for hyperoxia-induced macrophage/monocyte infiltration and hypoalveolarization, wild-type and CCL2-null mice were exposed to air or hyperoxia, as described above. As shown previously, hyperoxia blocked alveolar development, as evidenced by an increase in MLI (Fig. 9, A and B). In contrast, CCL2-null mice showed no increase in mean chord length. Hyperoxia-exposed CCL2-null mice showed a trend toward reduced BAL monocytes and, interestingly, a reduction in BAL neutrophils (Fig. 9, C and D). In CCL2-null mice, flow cytometry showed a reduction in F4/80+ CD11c+ macrophages, CD11chigh CD11bhigh (population B) exudative macrophages, and CD11clow CD11bhigh (population C) inflammatory monocytes (Fig. 10, A–E).
Fig. 9.

CCL2 deletion decreases hyperoxia-induced alveolar simplification and inflammatory cell trafficking. A: representative lung sections from CCL2+/+ and CCL2-null mice exposed to normoxia or hyperoxia for 14 days were stained with hematoxylin and eosin. B: alveolar size, as measured by mean chord length, confirmed features noted on lung histology (n = 15–18 mice per group, *P <0.05, ANOVA). B: BAL cells were measured by Hemavet. Compared with hyperoxia-exposed CCL2+/+ mice, CCL2-null mice tended to show decreased neutrophils (C) and macrophages (D). (n = 5–8 mice per group, means ± SE, *different from hyperoxia-exposed group, *P <0.05, ANOVA).
Fig. 10.

CCL2 deletion decreases alveolar monocyte/macrophage infiltration into hyperoxia-exposed neonatal lungs. Representative flow cytometric scattergrams of monocyte/macrophage subpopulations within the lungs of wild-type and CCL2-null mice exposed to normoxia and hyperoxia for 14 days. Each dot plot is representative of 4 independent experiments. A: CD45+, CD11c+, F4/80+ macrophages. B: gating scheme used for CD11c and CD11c staining was explained in Fig. 4 and is briefly shown here. Each dot plot is again representative of 4 independent experiments. Comparison of cell counts for F4/80 and CD11c double-positive macrophages (C), CD11chigh CD11bhigh exudative macrophages (subpopulation B, D), and CD11clowCD11bhigh inflammatory monocytes (subpopulation C, E). Hyperoxia increased the trafficking of macrophage subpopulations into the neonatal lung, and CCL2 deletion attenuated macrophage recruitment. (n = 8–14 mice per group, means ± SE, *different from hyperoxic-exposed group, P < 0.05, ANOVA).
DISCUSSION
In this report, we show for the first time that 1) brief episodic exposure to low-dose CO significantly mitigates the deleterious effects of hyperoxia on alveolar development, 2) low-dose CO inhibits hyperoxia-induced lung neutrophil and macrophage infiltration, and 3) HO-1 deficiency exacerbates hyperoxia-induced hypoalveolarization. Taken together, these studies indicate that, in hyperoxia-exposed neonatal mice, inhalation of the HO-1 product CO suppresses inflammatory cell trafficking, proinflammatory cytokine expression, and alveolar simplification. We also found that CO attenuates respiratory epithelial cell CCL2 expression and that CCL2 is required for hyperoxia-induced alveolar simplification, consistent with the notion that CO works in part by suppressing hyperoxia-induced CCL2.
Previous studies have examined the role of neutrophils and macrophages in the pathogenesis of neonatal hyperoxic lung injury. In neonatal rats, hyperoxia induces neutrophil influx and hypoalveolarization, which is attenuated by neutralizing antibodies against the neutrophil chemoattractants CXCL1 (2) or CXCL2 (9). Neutrophilic inflammation is also prevented by a selective chemical antagonist of CXCR2 (55). Similar to our results, neutralizing antibodies against CCL2, a monocyte chemoattractant, also decrease lung hypoalveolarization, as well as lung macrophage and neutrophil counts, in hyperoxia-exposed rats (50). However, the monocyte subpopulations involved have not previously been characterized.
In our study, we extend the above observations by showing that hyperoxia induces the accumulation of different F4/80-positive macrophage subsets into the lung, specifically CD11clow CD11bhigh and CD11chigh CD11bhigh cells. Previously, it was shown that, during influenza infection (29) and bleomycin exposure (47), CD11c− CD11b+ inflammatory monocytes are recruited to the lung and develop into a CD11chigh CD11b+ activated macrophage population known as exudative macrophages. On this basis, we conclude that the CD11clow CD11bhigh cells (our population C) represent inflammatory monocytes and that CD11chigh CD11bhigh cells (our population B) represent exudative macrophages, a major source of inflammatory cytokines and chemokines in the lung.
In addition to lung inflammation, hyperoxia increased the size of airspaces in the lung, indicating hypoalveolarization of the developing lung. Murine alveolar development begins on postnatal day 5, and saccular division is completed by the 30th postnatal day (53). This result is in agreement with previous work showing that exposure to supraphysiological levels of oxygen confers hypoalveolarization (18, 40, 41) and is consistent with the notion that oxygen exposure may be a contributing factor in the pathogenesis of BPD.
Next, we examined the effects of 1-h, twice per day CO treatment on lung development and inflammation. CO treatment during hyperoxia significantly reduced hyperoxia-induced hypoalveolarization. In addition, CO decreased the number of exudative macrophages and inflammatory monocytes in the lung. Conversely, compared with wild-type mice, HO-1-null mice exposed to hyperoxia showed increased alveolar simplification and lung infiltration with CD11b(+) cells. Overall, these data are concordant with the hypothesis that CO protects against lung injury and is facilitative for alveolar development under proinflammatory and hyperoxic stress conditions such as BPD. Unexpectedly, we found that CO treatment increased the number of lung inflammatory cells in some normoxia-treated mice. This may relate to the antiapoptotic effects of CO (27, 52), which may prevent normal clearance of leukocytes from the lung.
In our previous work, we demonstrated that HO-1-deficient mice have significantly decreased survival upon ischemic lung injury and that CO can rescue these mice (16). Recent reports have demonstrated the cytoprotective role of HO-1/CO in in vivo models of acute lung injury. Intravenous hemoglobin, a potent inducer of HO-1 (a major heme-degradative enzyme), prevented lung injury in a rat model of sepsis (37). In a separate report, intratracheal administration of hemoglobin protected rats from hyperoxia-induced lung injury (45). It was subsequently shown that adenoviral gene transfer of HO-1 conferred protection against hyperoxic-induced lung injury (35). Inhaled CO at low concentrations (50–500 ppm) protected against hyperoxia-induced lung injury in adult rats (36). Recently, the effects of HO-1 overexpression and CO administration (250 ppm for 1 h daily) were examined in a model of neonatal hyperoxic lung injury (15). HO-1 overexpression and CO each attenuated hyperoxia-induced arterial remodeling, right ventricular hypertrophy pulmonary edema, and hemosiderosis, while increasing blood vessel number. HO-1 overexpression and CO administration had modest effects on alveolar architecture and BAL, but the mechanisms driving inflammatory changes were not further explored.
To determine the mechanism of hyperoxia- and CO-mediated changes in lung inflammation, we examined the mRNA and protein levels of candidate cytokines by gene array, followed by qPCR and multiplex immune assay. Increases in lung leukocyte subpopulations with hyperoxia and mitigation of this response by CO were reflected in lung cytokine mRNA and protein levels. Lungs of hyperoxia-exposed mice showed increased levels of CCL2 mRNA and protein, which were reduced by CO treatment. We found a similar pattern for the type I cytokines IL-1β, TNF-α, CXCL1, and CXCL2. CCL2 is a chemoattractant for exudative macrophages (29). We therefore tested the hypothesis that respiratory epithelial cells produce CCL2 in response to hyperoxic exposure, leading to monocyte and macrophage infiltration of the lung. First, we found that hyperoxia increased epithelial CCL2 protein expression in vivo and that CO attenuated this response. Second, we exposed murine type II alveolar epithelial cells to hyperoxia in vitro. We found that hyperoxia increased murine type II alveolar epithelial cell CCL2 mRNA expression and that expression was inhibited by CO treatment. Finally, we exposed CCL2-null mice to hyperoxia in vivo and found that these mice were protected from alveolar simplification. In addition, the lungs of hyperoxia-exposed CCL2-null mice showed significantly reduced exudative macrophages and inflammatory monocytes, reproducing the effects of exogenous CO. Taken together, these data are consistent with the notion that inhalation of the HO-1 product CO suppresses inflammatory cell trafficking, proinflammatory cytokine expression, and alveolar simplification in part by attenuating CCL2 expression. However, other proinflammatory cytokines may also be involved. It is also possible that, in addition to targeting respiratory epithelial cells, CO has a direct inhibitory effect on leukocyte cytokine expression.
The precise mechanisms by which inflammation inhibits alveolar development are not completely known. Using a fetal lung explant model, it was recently shown that IL-1β-producing tissue macrophages mediate the inflammatory response to lipopolysaccharide and that macrophage activation inhibits airway morphogenesis (5). Conversely, macrophage depletion protected airway branching. Furthermore, tissue-specific macrophage activation induced by overexpression of IκB kinase inhibited lung development in vivo. We propose that CO protects against hyperoxia-induced alveolar simplification by blocking macrophage infiltration and activation. However, it is also possible that the neutrophilic inflammation plays a role in alveolar arrest. Both CO and the CCL2 knockout reduced neutrophil as well as macrophage infiltration, and recent studies have shown CD11b/CD11c-positive macrophages to be an important source of CXCL1 and CXCL2 (1, 39). Also, as noted above, treatment of hyperoxia-exposed neonatal lungs with antibodies or antagonists against neutrophil chemokines also prevents lung inflammation and alveolar simplification (2, 9, 55). Finally, it is also conceivable that hyperoxia retards the growth of blood vessels in the lung needed for alveolar development (4, 26, 46) and that CO promotes angiogenesis (6, 28).
In summary, we have provided a detailed in vivo analysis of lung inflammation and development after hyperoxic exposure in neonatal mice. Our studies demonstrate a direct link between CO, the CCL2-dependent control of leukocyte migration into hyperoxic lung tissue, and alveolar growth. Inhibition of inflammatory cell infiltration by either CO or CCL2 gene deletion was associated with the attenuated alveolar simplification. Improved understanding of the mechanisms regulating macrophage recruitment into the developing lung could lead to new insights into the pathogenesis of BPD. Finally, these data suggest that additional studies exploring low-dose CO as a possible therapeutic option in neonatal lung injury are warranted.
GRANTS
This work was supported by Systems in Integrative Biology predoctoral training grant T32GM008322 (to A. Anyanwu), NIH grants HL086676 (to D. Pinsky) and HL079339 (to M. Hershenson), the J. Griswold Ruth MD & Margery Hopkins Ruth Professorship (to D. Pinsky), and the A. Alfred Taubman Medical Research Institute at the University of Michigan.
DISCLOSURES
Dr. Pinsky has an equity interest in Proterris, which is developing inhaled CO for clinical use, and potential patent-related royalties from Columbia University. No other conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.C.A., M.B.H., and D.J.P. conception and design of research; A.C.A., J.K.B., O.M., H.A., and A.M.G. performed experiments; A.C.A., J.K.B., and A.P.P. analyzed data; A.C.A., J.K.B., M.B.H., and D.J.P. interpreted results of experiments; A.C.A. and J.K.B. prepared figures; A.C.A. drafted manuscript; A.P.P., M.B.H., and D.J.P. approved final version of manuscript; M.B.H. and D.J.P. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors thank Joanna D. Solarewicz, Julia Solarewicz, Martin Gruca, Joy Tsai, Saabir Kaskar, and Hussein Sheikh-Aden for technical assistance.
REFERENCES
- 1.Altmann C, Andres-Hernando A, McMahan RH, Ahuja N, He Z, Rivard CJ, Edelstein CL, Barthel L, Janssen WJ, Faubel S. Macrophages mediate lung inflammation in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol 302: F421–F432, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auten RL, Jr, Mason SN, Tanaka DT, Welty-Wolf K, Whorton MH. Anti-neutrophil chemokine preserves alveolar development in hyperoxia-exposed newborn rats. Am J Physiol Lung Cell Mol Physiol 281: L336–L344, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Baier RJ, Majid A, Parupia H, Loggins J, Kruger TE. CC chemokine concentrations increase in respiratory distress syndrome and correlate with development of bronchopulmonary dysplasia.. Pediatr Pulmonol 37: 137–148, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med 164: 1971–1980, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Blackwell TS, Hipps AN, Yamamoto Y, Han W, Barham WJ, Ostrowski MC, Yull FE, Prince LS. NF-κB signaling in fetal lung macrophages disrupts airway morphogenesis. J Immunol 187: 2740–2747, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi YK, Kim CK, Lee H, Jeoung D, Ha KS, Kwon YG, Kim KW, Kim YM. Carbon monoxide promotes VEGF Expression by increasing HIF-1α Protein level via two distinct mechanisms, translational activation and stabilization of HIF-1α protein. J Biol Chem 285: 32116–32125, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooney TP, Thurlbeck WM. The radial alveolar count method of Emery and Mithal: a reappraisal 2–intrauterine and early postnatal lung growth. Thorax 37: 580–583, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corti M, Brody AR, Harrison JH. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14: 309–315, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Deng H, Nicholas MS, Auten RL., Jr Lung inflammation in hyperoxia can be prevented by antichemokine treatment in newborn rats. Am J Respir Crit Care Med 162: 2316–2323, 2000 [DOI] [PubMed] [Google Scholar]
- 10.Dennery PA, Lee CS, Ford BS, Weng YH, Yang G, Rodgers PA. Developmental expression of heme oxygenase in the rat lung. Pediatr Res 53: 42–47, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Dilly SA. Scanning electron microscope study of the development of the human respiratory acinus. Thorax 39: 733–742, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drechsler Y, Dolganiuc A, Norkina O, Romics L, Li W, Kodys K, Bach FH, Mandrekar P, Szabo G. Heme oxygenase-1 mediates the anti-inflammatory effects of acute alcohol on IL-10 induction involving p38 MAPK activation in monocytes. J Immunol 177: 2592–2600, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Eber E, Zach MS. Paediatric origins of adult lung disease: 8. Long term sequelae of bronchopulmonary dysplasia (chronic lung disease of infancy). Thorax 56: 317–323, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehrenkranz RA, Walsh MC, Vohr BR, Jobe AH, Wright LL, Fanaroff AA, Wrage LA, Poole K, National Institutes of Child Health, Human Development Neonatal Research Network. Validation of the National Institutes of Health Consensus definition of bronchopulmonary dysplasia. Pediatrics 116: 1353–1360, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Gonzalez A, Alex Mitsialis S, Liu X, Kourembanas S. Vasculoprotective effects of heme oxygenase-1 in a murine model of hyperoxia-induced bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 302: L775–L784, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujita T, Toda K, Karimova A, Yan S, Naka Y, Yet S, Pinsky D. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 7: 598–604, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Ghosh S, Gal J, Marczin N. Carbon monoxide: Endogenous mediator, potential diagnostic and therapeutic target. Ann Med 42: 1–12 [DOI] [PubMed] [Google Scholar]
- 18.Hirakawa H, Pierce RA, Bingol-Karakoc G, Karaaslan C, Weng M, Shi GP, Saad A, Weber E, Mariani TJ, Starcher B, Shapiro SD, Cataltepe S. Cathepsin S deficiency confers protection from neonatal hyperoxia-induced lung injury. Am J Respir Crit Care Med 176: 778–785, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hislop A, Reid L. Development of the acinus in the human lung. Thorax 29: 90–94, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, Henson PM. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med 184: 547–560, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaarteenaho-Wiik R, Kinnula VL, Herva R, Soini Y, Pollanen R, Paakko P. Tenascin-C is highly expressed in respiratory distress syndrome and bronchopulmonary dysplasia. J Histochem Cytochem 50: 423–431, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Kaarteenaho-Wiik R, Pääkkö P, Herva R, Risteli J, Soini Y. Type I and III collagen protein precursors and mRNA in the developing human lung. J Pathol 203: 567–574, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Kakkera DK, Siddiq MM, Parton LA. Interleukin-1 balance in the lungs of preterm infants who develop bronchopulmonary dysplasia. Neonatology 87: 82–90, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Kennedy JD. Lung function outcome in children of premature birth. J Paediatr Child Health 35: 516–521, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Kotecha S, Wilson L, Wangoo A, Silverman M, Shaw JR. Increase in interleukin (IL)-1β and IL-6 in bronchoalveolar lavage fluid obtained from infants with chronic lung disease of prematurity. Pediatr Res 40: 250–256, 1996 [DOI] [PubMed] [Google Scholar]
- 26.Kunig AM, Balasubramaniam V, Markham NE, Seedorf G, Gien J, Abman SH. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol 291: L1068–L1078, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim HP, Choi AMK, Kim YS. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol 45: 867–873, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Volti GSD, Sangras B, Vanella A, Mezentsev A, Scapagnini G, Falck JR, Abraham NG. Carbon monoxide signaling in promoting angiogenesis in human microvessel endothelial cells. Antioxid Redox Signal 7: 704–710, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 180: 2562–2572, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Merritt TA, Stuard ID, Puccia J, Wood B, Edwards DK, Finkelstein J, Shapiro DL. Newborn tracheal aspirate cytology: Classification during respiratory distress syndrome and bronchopulmonary dysplasia. J Pediatr 98: 949–956, 1981 [DOI] [PubMed] [Google Scholar]
- 31.Minamoto K, Harada H, Lama VN, Fedarau MA, Pinsky DJ. Reciprocal regulation of airway rejection by the inducible gas-forming enzymes heme oxygenase and nitric oxide synthase. J Exp Med 202: 283–294, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mishra S, Fujita T, Lama VN, Nam D, Liao H, Okada M, Minamoto K, Yoshikawa Y, Harada H, Pinsky DJ. Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci USA 103: 5191–5196, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munshi UK, Niu JO, Siddiq MM, Parton LA. Elevation of interleukin-8 and interleukin-6 precedes the influx of neutrophils in tracheal aspirates from preterm infants who develop bronchopulmonary dysplasia. Pediatr Pulmonol 24: 331–336, 1997 [DOI] [PubMed] [Google Scholar]
- 34.Otterbein L, Bach F, Alam J, Soares M, Tao Lu H, Wysk M, Davis R, Flavell R, Choi A. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6: 422–428, 2000 [DOI] [PubMed] [Google Scholar]
- 35.Otterbein L, Kolls J, Mantell L, Cook J, Alam J, Choi A. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest 103: 1047–1054, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otterbein L, Mantell L, Choi A. Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 276: L688–L694, 1999 [DOI] [PubMed] [Google Scholar]
- 37.Otterbein LSS, Choi AM. Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am J Respir Cell Mol Biol 13: 595–601, 1995 [DOI] [PubMed] [Google Scholar]
- 38.Otterbein LE, Otterbein SL, Ifedigbo E, Liu F, Morse DE, Fearns C, Ulevitch RJ, Knickelbein R, Flavell RA, Choi AM. MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am J Pathol 163: 2555–2563, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poole JA, Gleason AM, Bauer C, West WW, Alexis N, van Rooijen N, Reynolds SJ, Romberger DJ, Kielian TL. CD11c+/CD11b+ cells are critical for organic dust-elicited murine lung inflammation. Am J Respir Cell Mol Biol 47: 652–659, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popova AP, Bentley JK, Anyanwu AC, Richardson M, Linn MJ, Lei J, Wong EJ, Goldsmith AM, Pryhuber GS, Hershenson MB. Glycogen synthase kinase-3β/β-catenin signaling regulates neonatal lung mesenchymal stromal cell myofibroblastic differentiation. Am J Physiol Lung Cell Mol Physiol 303: L439–L448, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ratner V, Starkov A, Matsiukevich D, Polin RA, Ten VS. Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia-exposed mice. Am J Respir Cell Mol Biol 40: 511–518, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rozycki HJ, Comber PG, Huff TF. Cytokines and oxygen radicals after hyperoxia in preterm and term alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 282: L1222–L1228, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Silva MFd Napimoga MH, Rodrigues DBR, Pereira SAL, Silva CL. Phenotypic and functional characterization of pulmonary macrophages subpopulations after intratracheal injection of Paracoccidioides brasiliensis cell wall components. Immunobiology 216: 821–831, 2011 [DOI] [PubMed] [Google Scholar]
- 44.Stanford SJ, Hislop AA, Oltmanns U, Nabel EG, Sang H, Haworth SG, Mitchell JA. Transition from placental to air breathing stimulates haem-oxygenase-1 expression without functional consequence for pulmonary vascular adaptation in pigs and mice. Br J Pharmacol 144: 467–476, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor J, Carraway M, Piantadosi C. Lung-specific induction of heme oxygenase-1 and hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 274: L582–L590, 1998 [DOI] [PubMed] [Google Scholar]
- 46.Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A, Korbutt G, Archer SL. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury. Circulation 112: 2477–2486, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Tighe RM, Liang J, Liu N, Jung Y, Jiang D, Gunn MD, Noble PW. Recruited exudative macrophages selectively produce CXCL10 after noninfectious lung injury. Am J Respir Cell Mol Biol 45: 781–788, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toti P, Buonocore G, Tanganelli P, Catella AM, Palmeri ML, Vatti R, Seemayer TA. Bronchopulmonary dysplasia of the premature baby: an immunohistochemical study. Pediatr Pulmonol 24: 22–28, 1997 [DOI] [PubMed] [Google Scholar]
- 49.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 123: 1986–1995, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vozzelli MA, Mason SN, Whorton MH, Auten RL. Antimacrophage chemokine treatment prevents neutrophil and macrophage influx in hyperoxia-exposed newborn rat lung. Am J Physiol Lung Cell Mol Physiol 286: L488–L493, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Wagenaar GTM, ter Horst SAJ, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, de Heer E, Hiemstra PS, Poorthuis BJHM, Walther FJ. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic Biol Med 36: 782–801, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AMK. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem 282: 1718–1726, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, Langhe SD, Kemp PJ, Riccardi D, Torday J, Bellusci S, Shi W, Lubkin SR, Jesudason E. Chapter Three: Lung Organogenesis. In: Current Topics in Developmental Biology, edited by Koopman P. New York: Academic Press, 2010, p. 73–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest 103: R23–R29, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yi M, Jankov RP, Belcastro R, Humes D, Copland I, Shek S, Sweezey NB, Post M, Albertine KH, Auten RL, Tanswell AK. Opposing effects of 60% oxygen and neutrophil influx on alveologenesis in the neonatal rat. Am J Respir Crit Care Med 170: 1188–1196, 2004 [DOI] [PubMed] [Google Scholar]