

Abstract
Hyperhomocysteinemia (HHcy) is associated with elderly frailty, skeletal muscle injury and malfunction, reduced vascular integrity and function, and mortality. Although HHcy has been implicated in the impairment of angiogenesis after hindlimb ischemia in murine models, the underlying mechanisms are still unclear. We hypothesized that HHcy compromises skeletal muscle perfusion, collateral formation, and arteriogenesis by diminishing postischemic vasculogenic responses in muscle fibers. To test this hypothesis, we created femoral artery ligation in wild-type and heterozygous cystathionine β-synthase (CBS+/−) mice (a model for HHcy) and assessed tissue perfusion, collateral vessel formation, and skeletal muscle function using laser-Doppler perfusion imaging, barium angiography, and fatigue tests. In addition, we assessed postischemic levels of VEGF and levels of its muscle-specific regulators: hypoxia-inducible factor (HIF)-1α and peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α. The observations indicated dysregulation of VEGF, HIF-1α, and PGC-1α levels in ischemic skeletal muscles of CBS+/− mice. Concomitant with the reduced ischemic angiogenic responses, we also observed diminished leptin expression and attenuated Akt signaling in ischemic muscle fibers of CBS+/− mice. Moreover, there was enhanced atrogene, ubiquitin ligases that conjugate proteins for degradation during muscle atrophy, transcription, and reduced muscle function after ischemia in CBS+/− mice. These results suggest that HHcy adversely affects muscle-specific ischemic responses and contributes to muscle frailty.
Keywords: hyperhomocysteinemia, skeletal muscle, vascular endothelial growth factor, hypoxia-inducible factor, peroxisome proliferator-activated receptor-γ coactivator-1α, atrogin-1, muscle ring finger-1, ischemia, angiogenesis, CD31, leptin, Akt
although the vital functions of skeletal muscles have been extensively studied, little is known about the mechanistic role of the factors that either promote or inhibit skeletal muscle adaptability in response to chronic ischemia. Poor vascularity and diminished revascularization potential are the hallmarks of loss of adaptive responses. These factors limit exercise performance and disrupt the vital functions of skeletal muscles chronically. The development of novel therapeutic strategies that can either reverse the detrimental effects or promote skeletal muscle adaptability and sustain long-term skeletal muscle integrity and function are necessary. Given that the lack of revascularization potential causes elderly frailty, dependence, and metabolic disorders such as diabetes (36), it is vital to define the metabolic factors and associated signaling pathways that are detrimental to long-term skeletal muscle function.
The metabolic factors that regulate VEGF expression are currently under intense investigation to decipher its regulation during poor exercise tolerance and elderly frailty. Deletion of muscle-specific VEGF expression has been reported to reduce exercise endurance, capillary-to-fiber ratio, and capillary density (33). Expression of VEGF is crucial for ischemic angiogenesis and is highly upregulated in skeletal muscles after ischemia (15, 23, 46, 49). Furthermore, the extent of VEGF expression in muscle fibers determines arteriole formation over capillary formation and thus helps in faster recovery (43). In addition, muscle-specific VEGF expression levels could contribute to strain-specific ischemic responses (30), underscoring the consequences of differential VEGF expression regulation. The transcription factors hypoxia-inducible factor (HIF)-1α and peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α are important regulators of VEGF expression in the hindlimb ischemic model and have been shown to regulate skeletal muscle angiogenesis in response to tissue hypoxia and nutrient deprivation (2, 34, 44, 46). In addition to these physiological regulators, abnormal accumulation of certain metabolites has been shown to regulate VEGF levels. Reports from the literature vary on the correlation between VEGF and HHcy from positive in some studies to negative or no correlation in other studies in relation to ischemia and other disease conditions (5, 25, 28, 42, 52). These reports indicate that homocysteine (Hcy), a sulfur-containing intermediary amino acid produced from methionine during demethylation process that supplies methyl donors for the cellular methylation process, could be a potential regulator of VEGF levels and angiogenesis. In this study, we examined muscle-specific regulators of angiogenesis during postischemic angiogenesis in the HHcy condition.
Disorders in methionine metabolism are the primary cause for HHcy and cause multiorgan damage (17, 21, 22, 48). Genetic mutations of enzymes involved in synthesis of methionine from Hcy (remethylation pathway) and the synthesis of cysteine from Hcy (trans-sulfuration pathway) have been shown to cause HHcy in both humans and mice (3, 19, 29, 50). Mutations of methylene tetrahydrofolate reductase involved in remethylation of Hcy to methionine have been reported to cause HHcy. Other mutations include cystathionine β-synthase (CBS), an enzyme involved in irreversibly removing Hcy by conversion to cystathionine. Homozygous CBS knockout mice die shortly after birth due to multiple defects (19, 50). However, heterozygous CBS (CBS+/−) mice live a normal lifespan and therefore serve as a suitable model for moderate HHcy exhibiting similar cardiovascular pathology as in humans (3). Another enzyme, cystathionine γ-lyase, functions downstream of CBS to convert cystathionine into beneficial cysteine and has been shown to cause muscle atrophy after cysteine supply is limited (19, 50). Nutritional imbalance, age, and type of physical activity have also been reported to raise plasma Hcy levels and have been shown to be associated with poor skeletal muscle performance (48). HHcy has been associated with skeletal muscle injury and malfunction, elderly frailty, and mortality (17, 21, 48, 51). Severe HHcy has been associated with below normal body weights and musculoskeletal abnormalities (22). The studies above suggest that HHcy causes muscle malfunctions that are aggravated during nutritional imbalance, specifically with vitamin B6, B9, and B12 deficiencies, methionine excess, and a low cysteine supply in methylene tetrahydrofolate reductase and CBS mutant backgrounds. However, how HHcy causes poor skeletal muscle function and frailty remain elusive. A deficiency in CBS protein levels has been proposed to compromise angiogenesis in humans and mice and elevate Hcy levels (3, 10, 53). Although HHcy has been reported to compromise tissue perfusion rates and angiogenesis in murine hindlimb ischemia models (5, 7), the molecular mechanisms of HHcy-mediated suppression of angiogenesis and the expression of provasculogenic molecules in muscle fibers after ischemia remain incompletely understood. Specifically, the expression of different endothelial cell proliferation regulators at different time points after ischemia and their regulation in specific muscle type has not been evaluated.
In a previous report (5) involving hindlimb ischemia in wild-type (WT) and CBS+/− mice, there were no differences in thigh muscle VEGF levels after 7 days of ischemia. However, a significant reduction in capillary density was observed using CD31 labeling (5). Given that muscle fiber-specific expression of VEGF levels plays a crucial role in the regulation of endothelial cell proliferation (CD31 positive) and the consequent angiogenesis, in the present study, we hypothesized that postischemic angiogenic regulators in muscle fibers are induced differentially at different time points between WT and CBS+/− mice after hindlimb ischemia. In addition, it has been demonstrated that VEGF levels taper after acute induction after muscle ischemia, suggesting the significance of time-sensitive VEGF expression (46). The present study aimed to unravel the molecular mechanisms that contribute to differential HHcy-mediated postischemic muscle responses and poor revascularization and to test the impact of these changes on physical performance.
MATERIALS AND METHODS
Animal maintenance and genotyping.
Male WT (C57BL/6J) mice and CBS+/− (B6.129P2-Cbstm1Unc/J 002853) mice at ∼2 mo old were used in this study. Mice were procured from the Jackson Laboratory (Bar Harbor, ME) and housed in the animal care facility at the University of Louisville with ad libitum access to standard chow and water. Pups were genotyped as recommended by the Jackson Laboratory and as previously reported (47). Unless otherwise mentioned, four to five mice were used in each group for physiological function experiments. For molecular experiments, we used a minimum of three different mice per group.
Femoral artery ligation.
All protocols were approved by the Institutional Animal Care and Use Committee. Femoral artery ligation (unilateral or bilateral) was performed under intraperitoneal pentobarbital sodium (50 mg/kg) anesthesia as previously described (26, 32). Briefly, strict aseptic conditions were implemented before the skin was opened at the medial thigh region. The underneath tissue/fascia were separated to expose the femoral artery, vein, and nerve. After careful separation of the femoral artery from the bundle, the artery was ligated using 6-0 silk at proximal and distal places (before bifurcation) and was transected. The skin opening was sutured (interrupted sutures) using 6-0 silk suture. Betadine antiseptic was applied after closure of the skin. Mice were allowed to recover on a warm pad before being returned to their cages. Postoperative care, such as separate caging and continuous monitoring, was provided. Tissue harvesting was performed for immunohistochemistry and for Western blot quantification on days 5 and 7 of postischemia using 2,2,2-tribromoethanol as an anesthetic. Barium angiograms were performed at different days (0, 7, and 28 days) after the creation of femoral artery disruption. In the case of bilateral hindlimb ischemia, the procedure was repeated on both limbs.
Barium angiograms.
At different time points after hindlimb ischemia (0, 7, 28 days), mice were injected with 10% barium sulfate in normal saline through carotid artery catheterization after pentobarbital anesthesia. The injection rate was ∼1 ml/min. Heparin (20 U/ml) was used along with barium sulfate to visualize the nascent angiogenesis in day 7 postischemic limbs. Angiograms were captured and analyzed using the Carestream whole animal X-ray imaging system (Carestream Molecular Imaging, Woodbridge, CT) as previously described (12). Vessel density was quantified using VesSeg tool (Institute for Signal Processing, University of Luebeck, Lübeck, Germany) as previously reported (27). Collateral vessels were counted manually from the postischemic angiograms from day 28. Unilateral hindlimb ischemia was used for day 0 and 7 postischemic angiograms and bilateral hindlimb ischemia was used for day 28 postischemic angiograms. A minimum of four ischemic legs were used for angiogram quantification.
Laser-Doppler tissue perfusion imaging and flowmetry.
MoorLDI (Moor Instruments) was used to measure tissue perfusion intensity and blood flow rates. We assessed both tissue perfusion and mean blood flow rates compared with the nonischemic limb from the same animal to minimize positional and uncontrollable field differences. Relative mean plantar foot tissue perfusion for the ischemic limb was measured and is presented as a percentage of recovery to the nonischemic limb. All measurements were performed on medial aspects of the feet distal to the site of ligation. The focus was beneath the skin surface (tissue perfusion).
Immunohistochemistry.
Tissue harvesting was done on days 5 and 7 from animals subjected to unilateral hindlimb ischemia. Both ischemic and contralateral nonischemic gastrocnemius muscles were collected and frozen in liquid nitrogen using tissue freezing medium (Triangle Biomedical Sciences, Durham, NC). Tissues were stored at −80°C. Cryosections of 8 μm thickness were prepared and sectioned using a Leica CM1850 cryostat (Leica Microsystems). Sections were probed with specific antibodies by following a previously described protocol (11). Briefly, sections were fixed with 4% paraformaldehyde for 15 min, blocked and permeabilized with PBS containing 3% BSA and 0.3% Triton X-100 for 1 h, incubated overnight with primary antibody, incubated for 1 h with secondary antibody, and stained for 15 min with 4′,6-diamidino-2-phenylindole. Slides were washed in between steps for three times each with 5-min duration. Stained slides were mounted, and images were obtained with a laser scanning microscope (Olympus FluoView1000, B&B Microscope).
Western blot analysis.
Homogenized tissues were lysed using RIPA lysis buffer with protease and phosphatase inhibitors and brief sonication. The protein concentration was estimated using the Bio-Rad Bradford reagent. Equal amounts of total proteins (30 μg) were resolved using SDS-PAGE and transferred to polyvinylidene membranes. Membranes were probed with overnight primary antibody and 1 h of secondary antibody. The peroxidase signal was developed using SuperSignal West Pico (Pierce), and signal capturing was obtained with the Bio-Rad ChemiDoc XRS+ system and Image Lab software (Bio-Rad).
Quantitative PCR.
Total RNA was isolated using TRIzol reagent. The quantity of RNA was assayed using a nano drop spectrophotometer. Equal amounts of total RNA were used for cDNA synthesis with a Promega kit (Improm-II RT system, A3800) using oligo-dT primers. Real-time PCR was performed using cyber green chemistry (FastStart Essential DNA Green Master, no. 06402712001, Roche) and a light cycler (Roche). The following primer pairs were used to amplify specific gene expression: muscle ring finger (MuRF)-1, forward 5′-GTGTGAGGTGCCTACTTGCTC-3′ and reverse 5′-GCTCAGTCTTCTGTCCTTGGA-3′; GAPDH, forward 5′-TGGATTTGGACGCATTGGTC-3′ and reverse 5′-TTTGCACTGGTACGTGTTGAT-3′; atrogin-1, forward 5′-GCAAACACTGCCACATTCTCTC-3′ and reverse 5′-CTTGAGGGGAAAGTGAGACG-3′; and leptin, forward 5′-ATGTGCCCTTCCGATATACAACC-3′ and reverse 5′-CGTGTCATCCACTAATCTTCTGG-3′.
Statistical analysis.
Densitometry analysis was performed using ImageJ software. P values of <0.05 were considered significant. Student's t-test was used to enumerate the levels of significance between two different groups.
Antibodies and reagents.
Antibody sources were as follows: anti-VEGF antibody from Santa Cruz Biotechnology (Santa Cruz, CA); anti-HIF-1α antibody from Thermo Scientific (Rockford, IL); anti-PGC-1α, anti-phosphorylated (p)Akt1 (p473), and anti-dystrophin antibodies from Abcam (Cambridge, MA); anti-GAPDH antibody and anti-CD31 antibody from Millipore (Billerica, MA). Horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology, and Alexa fluor-conjugated secondary antibodies were from Life Technologies (Grand Island, NY).
Swim test.
The live animal behavior recording and analysis system (Topscan) from Clever Systems (Reston, VA) was used to collect swim parameters such as the speed of movement and total distance moved by tracking the animal while swimming in a water tub with second-to-second data (live video recording). The swim test was performed at ∼3 wk postischemia using bilateral hindlimb ischemic animals. Data from the first 20 min were analyzed, and the level of significance was enumerated. Baseline performance was calculated from the same age group (the nonischemic group of matching age) and was used to measure relative functional performance after ischemia.
RESULTS
Tissue perfusion is limited in CBS+/− mice after hindlimb ischemia.
To compare the ischemic responses in skeletal muscles of CBS+/− mice (a model for HHcy) with those of WT control mice, we performed either unilateral or bilateral femoral artery ligation. Tissue perfusion and flow rates were measured using laser-Doppler imaging on days 0, 7, and 28 after ischemia. The results suggested an effective cut in the supply of nutrients and other growth-promoting factors distal to the ligation site in all ischemic limbs (Fig. 1). On day 0, we could not observe any significant differences in tissue perfusion between CBS+/− and WT control mice. However, on day 7, there were observable differences in limb tissue perfusion between CBS+/− and WT mice (Fig. 1, A and B). The difference was prominent on day 28 after ischemia as observed by the differential tissue perfusion between the groups (Fig. 1, C–E). In addition, Duane et al. (7), using diet-induced HHcy in rats, also observed near-complete tissue perfusion recovery after ischemia in 28 days in WT control animals but not in HHcy animals. Significant changes in tissue perfusion on day 7 after ischemia between WT and CBS+/− mice were observed in the present study.
Fig. 1.

Limited recovery in tissue perfusion of hindlimbs from heterozygous cystathionine β-synthase (CBS+/−) mice after femoral artery ligation. A: laser-Doppler perfusion imaging showing the intensity of limb perfusion on days 0 and 7 after ischemia in both nonligated (NL) and ligated (L) limbs. Mean perfusion rates of ischemic limbs compared with NL limbs (P < 0.05) are shown in the graph. WT, wild-type control mice. B: quantification of perfusion rates on days 0 and 7 and presented as percentages from baseline rates (100%) in the NL limb. *P < 0.05. C: live perfusion units (PUs) recorded at the foot region after 28 days of bilateral ischemia. D: tissue perfusion intensity map on day 28 after ischemia. E: quantification of PUs between WT and CBS+/− mice (at 28 days after ischemia). *Significant difference (P < 0.05).
Barium sulfate angiograms after hindlimb ischemia reveal attenuated angiogenesis in CBS+/− mice.
To determine whether revascularization occurred in skeletal muscles after ischemia and to quantify the vessel architecture, we performed barium sulfate infusion at different time points after ischemia (days 0, 7, and 28) in both CBS+/− and WT groups. To rule out any preexisting difference in the vascular density, we performed barium angiograms immediately after femoral artery ligation (Fig. 2A). The results suggested that there were no significant differences in vessel density on day 0 after femoral artery ligation. To further confirm the differences in vessel density after ischemia and to determine the differences in collateral arteriogenesis, we performed bilateral femoral ligation and day 28 postischemic barium angiograms (Fig. 2B). Based on vessel density quantifications from the day 28 angiograms, postischemic vessel density was significantly lower in CBS+/− mice compared with WT control mine on day 28 (Fig. 2C). In addition, we observed inconsistent collateral vessel formation and angiogenesis with reduced postischemic vessel length in CBS+/− mice (Fig. 2, B–E). In contrast with CBS+/− mouse tissues, WT ischemic tissues exhibited highly branched, prominent, and longer neovasculatures in the ischemic tissues. The vessel density on day 7 was also found to be defective in CBS+/− mice compared with WT control mice (Fig. 2F). These observations suggest that HHcy mice have defective revascularization (as measured by vessel density) and reduced collateral (the directional vessels that originate near the site of ligation to reestablish the nutrient supply) formation. These observations also confirm the attenuated ischemic vasculogenic events that were observed in previous reports (5, 7) with HHcy in murine models of hindlimb ischemia.
Fig. 2.

Attenuated angiogenesis in CBS+/− mice after hindlimb ischemia. A: day 0 postischemic angiograms showing complete femoral artery ligation. B: barium sulfate angiograms showing postischemic angiogenesis, which was severely compromised in CBS+/− mice at 28 days after bilateral femoral artery ligation. Blue arrows represent the collateral vessel formed after ischemia. The red asterisks represent sites of ligation. C: quantification of vessel density at 4 wk after ischemia by the VesSeg tool. *P < 0.05. D: vessel length determined using the ImageJ program. **P < 0.01. E: collateral vessels were manually counted from the angiograms. Av number, average number. *P < 0.05. F: quantification of vessel density at 1 wk after ischemia by the VesSeg tool. AU, arbitrary units. *P < 0.05.
Attenuated VEGF expression and endothelial proliferation in muscle fibers of CBS+/− mice.
To further understand the mechanism causing the reduced vessel density in CBS+/− mice after ischemia, we assayed for VEGF levels and patterns in ischemic tissue sections on days 5 and 7 after ischemia. We observed postischemic VEGF expression specifically in gastrocnemius muscle tissue sections. VEGF expression levels were significantly reduced in CBS+/− mice but were highly expressed in muscle fibers of WT ischemic mice (Figs. 3 and 4). VEGF expression was highly localized in ischemic muscle fibers, which has also been observed by others (46). The difference was more pronounced on day 5 after ischemia in gastrocnemius muscles (Fig. 3C). This could be due to delayed ischemic upregulation of VEGF levels in muscle fibers of ischemic CBS+/− mice. To further confirm the differences in VEGF levels in ischemic tissue sections, we assayed VEGF levels in protein lysates using Western blot analysis (Fig. 4). The Western blot analysis results further confirmed the differences in VEGF levels between WT and CBS+/− ischemic muscle fibers. Taken together, these results suggest that there is differential VEGF induction after ischemia in muscle fibers between WT and CBS+/− mice.
Fig. 3.

Reduced muscle fiber VEGF expression in CBS+/− mice after hindlimb ischemia. A: confocal image (×60) showing reduced expression levels of VEGF (green) in ischemic gastrocnemius muscles of CBS+/− mouse sections compared with WT mouse sections after 7 days of ischemia. Contralateral nonischemic gastrocnemius muscles from CBS+/− and WT mice were used as controls. Dystrophin (red) was used to locate the muscle fibers. 4′,6-Diamidino-2-phenylindole (DAPI) staining was done to locate nuclei. B: relative VEGF fluorescence intensity (from day 7 images in A) was quantified using imageJ and is presented in fluorescence units (FUs). ***P < 0.001. C: confocal images (×20) showing VEGF expression (green) in day 5 ischemic gastrocnemius muscle sections from WT and CBS+/− mice.
Fig. 4.

Differential regulation of proangiogenic regulators [hypoxia-inducible factor (HIF)-1α and peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α] in CBS+/− mice after hindlimb ischemia. A: levels of proangiogenic molecules in total protein lysates from day 7 ischemic and nonischemic muscles of CBS+/− and WT mice determined with representative Western blot analysis. B: band intensities quantified from 3 Western blots. *P < 0.05.
Next, we assayed for the CD31 glycoprotein marker (a marker for endothelial cells) in ischemic tissue sections and found that the levels were induced in WT muscle sections after ischemia (Fig. 5). In contrast with nonischemic control and ischemic CBS+/− sections, there were multiple endothelial cells surround the muscle fibers and there was an enhanced capillary-to-muscle fiber ratio in ischemic sections of WT mice (Fig. 5). In conjunction with a weak recovery of tissue perfusion, CD31 levels were significantly attenuated in ischemic muscles from CBS+/− mice, including the capillary-to-muscle fiber ratio, compared with WT mice after ischemia (Fig. 5, B and C).
Fig. 5.

Attenuated endothelial cell proliferation in CBS+/− mice after hindlimb ischemia. A: endothelial proliferation was visualized using CD31 antibody (magenta), and nuclei were stained with DAPI. Dystrophin (green) was used to locate muscle fibers. Images are shown at ×60 magnification. Gastrocnemius muscle sections were obtained at day 7 of ischemia. B: relative CD31 fluorescence intensity was quantified using ImageJ and is presented in FUs. ***P < 0.001. C: the capillary-to-fiber ratio was enumerated from 10 sections belonging to 3 different mice. Although there was significant (**P < 0.01) proliferation of endothelial cells in CBS+/− mice after ischemia, the levels were significantly less than those from WT ischemic sections (***P < 0.005).
Interestingly, nonischemic sections from CBS+/− mice showed a nonsignificant reduction in basal levels of CD31 compared with WT nonischemic sections (P = 0.053).
Differential expression of VEGF regulators in muscle fibers of ischemic CBS+/− mice.
Next, to determine if regulators of VEGF expression in muscle fibers are differentially expressed between WT and CBS+/− skeletal muscle fibers after ischemia, we assayed for HIF-1α and PGC-1α levels by immunohistochemistry and Western blot analysis. There was a significant reduction in the levels of both regulators of VEGF induction (HIF-1α and PGC-1α) in ischemic CBS+/− skeletal muscle fibers (Figs. 4 and 6). These results imply that there is differential regulation of postischemic levels of HIF-1α and PGC-1α between WT and CBS+/− mice that could be responsible for the delayed/improper induction of VEGF levels after ischemia in muscle fibers of CBS+/− mice.
Fig. 6.

Diminished expression of VEGF regulators in CBS+/− mice after hindlimb ischemia. A: confocal images (×60) showing the expression levels of PGC-1α (green) and HIF-1α (red) in ischemic gastrocnemius muscles of CBS+/− and WT mice on day 7 of ischemia. Contralateral nonischemic gastrocnemius muscles from CBS+/− and WT mice were used as controls. Nuclei were stained with DAPI. B: relative quantification of fluorescence intensity of PGC-1α (in FUs) measured using ImageJ and presented in graphical form. **P < 0.01; ***P < 0.005. C: quantification of HIF-1α fluorescence intensities across sections. **P < 0.01.
Diminished Akt activation and reduced proangiogenic gene expression contribute to the attenuated vasculogenesis in CBS+/− mice.
The Akt/mammalian target of rapamycin (mTOR) pathway has been shown to regulate HIF-1α expression in various cell types (6, 14, 18). To determine if there was reduced activation of survival signaling concomitant with attenuated angiogenesis in ischemic CBS+/− mouse muscles, we assayed for levels of activation-specific Akt phosphorylation (Ser473) in tissue lysates. As shown in Fig. 7, A and B, there was a marginal but significant reduction in Akt activation, as evidenced by reduced levels of pAkt1 (Ser473) in ischemic muscles from CBS+/− mice compared with ischemic muscles from WT mice. These results suggested that diminished Akt signaling in skeletal muscle fibers might contribute to the derailed ischemic angiogenesis in CBS+/− mice.
Fig. 7.
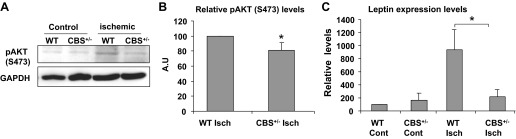
Attenuated Akt signaling and diminished leptin levels contribute to the derailed ischemic angiogenesis. A: representative Western blot images. Equal amounts of protein lysates obtained from normal (WT and CBS+/−) and ischemic (WT and CBS+/−) muscle fibers were probed with phosphorylated (p)Akt (Ser473)-specific antibody. GAPDH levels were used to visualize equal loading. B: quantification of relative pAkt intensities (from 3 different blots of ischemic WT and CBS+/− muscle fibers). *P < 0.05. C: enumeration of relative expression levels of leptin transcripts by quantitative PCR from three different total RNA samples obtained from normal (WT and CBS+/−) and ischemic (WT and CBS+/−) muscle fibers. *P < 0.05.
In addition to VEGF, many other growth factors, cytokines, and signal regulators also regulate angiogenesis (16, 35, 41). To determine additional alterations in angiogenic regulators in ischemic tissues of WT and CBS+/− mice, we assayed gene expression levels of leptin, a molecule known to be expressed in muscle fibers that can regulate endothelial proliferation (35). As shown in Fig. 7C, relative quantification of leptin expression levels revealed that leptin levels were attenuated in ischemic muscles from CBS+/− mice, whereas leptin levels showed a robust upregulation in WT ischemic muscles. These results further suggested that decreased proangiogenic molecules such as leptin, in addition to VEGF, in ischemic muscles contribute to the reduced angiogenic potential in CBS+/− mice.
Enhanced atrogene expression and limited fatigue resistance in CBS+/− mice after ischemia.
Muscle atrophy is associated with various muscle pathologies and involves the degradation of muscle proteins through the ubiquitin proteasome pathway. The atrogenes E3 ubiquitin ligases atrogin-1 and MuRF-1 are upregulated during muscle atrophy and conjugate various muscle proteins with ubiquitin for proteosomal degradation (9). To further evaluate the consequences of limited angiogenesis and attenuated ischemic responses, we assayed for levels of atrogin-1 and MuRF-1 (atrogenes) by quantitative PCR in ischemic and contralateral nonischemic tissues of WT and CBS+/− mice on day 7 of ischemia. In WT ischemic muscles, there were no difference in the levels of these genes (Fig. 8A). However, both atrogin-1 and MuRF-1 mRNA were upregulated significantly in ischemic muscle fibers from CBS+/− mice. These results suggested that the lack of prompt revascularization might direct postischemic muscles toward muscle atrophy through atrogene expression in CBS+/− mice. To further understand whether the limited nutrient supply and enhanced atrogene expression might confer fatigue and limit exercise performance, WT and CBS+/− mice were subjected to a swim capability test after ∼3 wk of bilateral femoral artery ligation. CBS+/− mice showed a reduced speed of motion and total distance travelled (potential indicators of fatigue) compared with WT mice (Fig. 8, B–D). These results further corroborate the findings that suggest limited vascularity in CBS+/− mice after ischemia and imply that a delayed and/or abnormal regulation of postischemic muscle responses might promote muscle weakness and cause compromised physical performance.
Fig. 8.

Limited postischemic endurance capacity and enhanced atrogene expression in CBS+/− mice. A: all measurements were enumerated from mice with bilateral femoral artery ligation (∼3 wk postischemia). Trace diagrams were obtained from TOPSCAN live animal movement recordings while swimming for the first 10 min. B: the duration of time that mice were moving with a speed of <5 mm/s was calculated and is presented as the percentage below the baseline performance. The slower the velocity of movement, the lower the performance. Swim performance parameters were enumerated from a 20-min swim duration and are presented as a bar graph. *P < 0.05. C: the total distance moved in the first 20 min was calculated and is presented as the percentage below the baseline distance moved. *P < 0.05. D: total RNA from tissues of ischemic and contralateral nonischemic muscles were obtained at 7 days after ischemia and assayed for atrogin-1 and muscle ring finger (MuRF)-1 levels by quantitative PCR. The fold difference was calculated from baseline expression levels (WT nonischemic expression levels). *P < 0.05.
DISCUSSION
HHcy has been implicated in multiorgan damage and compromised angiogenesis and is an independent risk factor for peripheral arterial disease (7, 24). In severe HHcy and homocystinuria, musculoskeletal deformities have been reported along with reduced muscle mass (17, 21, 22, 31, 48, 51). In addition, HHcy results in muscle damage, exercise intolerance, and elderly frailty (17, 21, 22, 31, 48, 51). In mouse models of HHcy, reduced body weight and muscle deformities have been reported that were contingent upon cysteine deficiency (19). However, the molecular mechanisms that regulate muscular defects and muscle frailty are not completely known.
Previous studies have examined the role of defective ischemic responses as a cause for poor vascularity in murine models of HHcy. Duan et al. (7) observed reduced ischemic perfusion recovery on day 7 in a rat model of HHcy, and the reduction in postischemic neoangiogenesis was attributed to reduced bioavailability of nitric oxide. Bosch-Marcé et al. (5) proposed that reduced Akt signaling in endothelial cells but not reduced VEGF levels or its associated signaling in thigh muscles on 7 days after ischemia as the cause for defective endothelial proliferation and compromised revascularization. Given that muscle-specific VEGF levels play a crucial role in endothelial cell proliferation and potentially contribute to VEGF receptor-stimulated Akt activation in endothelial cell proliferation and survival, it is not known how the differences in Akt activation are produced that culminate in defective endothelial cell proliferation in the presence of HHcy. In skeletal muscles, it has been shown that muscle-specific VEGF levels not only regulate endothelial proliferation but also determine arteriole formation over capillaries formation and potentially contribute to exercise endurance and postischemic muscle integrity.
In the present study, we observed differences in angiogenesis as early as 7 days after ischemia, suggesting that the molecular changes might precede the vasculogenic events. Consistent with these observations, pronounced differences in VEGF levels on day 5 after ischemia were observed in gastrocnemius muscles (Fig. 3C). Nonetheless, the differences in VEGF levels were significant between ischemic gastrocnemius muscles of WT and CBS+/− mice on day 7 after femoral artery ligation (Figs. 3 and 4). These findings suggest that HHcy mediates reduced and delayed VEGF level expression in ischemic muscle fibers. The apparent differences could be due to time point (5 days after ischemia) and/or muscles distal to the site of ligation, thus exhibiting pronounced ischemic responses (gastrocnemius muscle). VEGF levels have been reported to vary between different muscle types and in response to physical activity (4, 37, 38). Moreover, variations in VEGF expression in the presence of HHcy have been reported in different tissues (5, 25, 28, 42, 52). However, the sources for all such variations mentioned above are unknown. The duration of HHcy, concentration of Hcy, nutritional status, age, sex, and amount of physical activity could be considered as confounding factors that modulate HHcy-mediated VEGF expression patterns. The results of the present study suggest that dysregulation of postischemic VEGF levels in skeletal muscles contribute to the observed decrease in vasculogenic events in CBS+/− mice. These observations further corroborate findings from other studies (8, 33) that suggest the significance of muscle fibers in the regulation of endothelial proliferation through VEGF expression after ischemia and subsequent functional and perfusion recovery. In addition to VEGF, there are other cytokines or growth factors that are known to regulate endothelial proliferation and are produced in muscle fibers. Leptin has been demonstrated to cause endothelial proliferation as well as the tissue remodeling necessary for angiogenesis (35). Moreover, HIF-1α has been reported to induce leptin gene expression (13). These findings, along the present finding of attenuated leptin transcript induction after ischemia in muscle fibers from CBS+/− mice, suggest that HIF-1α regulation in ischemic muscle fibers is important for proper angiogenesis during HHcy.
Multiple studies (34, 40, 45) have demonstrated the contribution of muscle-specific HIF-1α and PGC-1α in the postischemic revascularization potential in a dose-dependent manner. The present findings from CBS+/− mice indicate that under nutrient and O2-deprived conditions, Hcy exerts an attenuation of HIF-1α and PGC-1α levels. Moreover, there was reduced postischemic Akt signaling, as evidenced by decreased pAkt levels. It is plausible that the reduced Akt signaling, to some extent, is responsible for the reduced HIF-1α levels, as previously observed (6, 14, 18). Based on these findings, we propose that HHcy-mediated postischemic suppression of HIF-1α and PGC-1α levels as a contributing factor for the attenuated VEGF expression and diminished ischemic endothelial cell proliferation. Future studies are necessary to examine the detailed mechanisms of diminished HIF-1α and PGC-1α levels in ischemic conditions with HHcy. Other potential mechanisms involve an attenuation of the AMP-activated protein kinase (AMPK) pathway with the HHcy condition after ischemia. Hypoxia-induced AMPK activation has been demonstrated to enhance VEGF levels in skeletal muscles and increase capillary density (54, 55). Furthermore, AMPK activation enhances expression levels of PGC-1α and also the transcriptional function of PGC-1α in skeletal muscles and heart tissue (20, 54).
The association of peripheral arterial disease and vessel occlusive disorders with HHcy has been documented in clinical studies (1, 24). Lack of revascularization blocks the recovery process after vascular occlusive events and leads to eventual aggravation of the disease. Results from the present study demonstrate that attenuated vasculogenic events are correlated with an enhancement in atrogene expression and diminished physical activity during swim performance. Absence of a prompt recovery in tissue perfusion and reestablishment of nutrient supply contribute to the muscle atrophy and may lead to HHcy-associated elderly frailty and exercise intolerance after local ischemic events. Given that forkhead box protein O (FOXO) transcription factors are important in the induction of atrogene expression and the Akt pathway inhibits FOXO transcription (39), it is possible that the attenuation of postischemic Akt signaling in HHcy conditions might not only reduce HIF-1α levels but also might enhance muscle atrophy through lack of FOXO inhibition. In addition, it is also notable that HHcy-mediated suppression of PGC-1α levels might contribute to FOXO transcriptional activation and the eventual muscle atrophy. PGC-1α-driven inhibition of FOXO transcription has been reported in the context of skeletal muscles (1).
In conclusion, we have shown that diminished Akt signaling, VEGF, leptin, and CD31 levels were accompanied with reduced tissue perfusion and collateral formation in muscle fibers of CBS+/− mice after ischemia. Furthermore, there was an attenuation of ischemic HIF-1α and PGC-1α levels in CBS+/− mice. Consistent with a poor ischemic response, CBS+/− mice also exhibited higher levels of atrogene expression and decreased swim performance. These results suggest that HHcy is involved in muscle frailty through reduced postischemic molecular responses, collateral formation, and angiogenesis, especially when there is damage or occlusion to major arteries, such as the femoral artery. It is plausible that prompt enhancement of VEGF and leptin expression in skeletal muscle fibers might benefit in the recovery process during ischemic occlusions and peripheral arteriole disease associated with HHcy. In addition, promotion of muscle-specific VEGF and/or leptin levels and/or local expression of HIF-1α and PGC-1α or activation of the Akt pathway might improve the elderly frailty associated with HHcy.
GRANTS
This work was supported in part by National Institutes of Health Grants HL-74185, NS-84823, and HL-108621 (to S. Tyagi).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.V. and S.C.T. conception and design of research; S.V. performed experiments; S.V. analyzed data; S.V., S.G., S.P., and S.C.T. interpreted results of experiments; S.V. prepared figures; S.V. drafted manuscript; S.V. edited and revised manuscript; S.V., S.G., S.P., and S.C.T. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Irving Gilbert Joshua (Department of Physiology and Biophysics, University of Louisville) for constructive criticism.
REFERENCES
- 1.Andras A, Stansby G, Hansrani M. Homocysteine lowering interventions for peripheral arterial disease and bypass grafts. Cochrane Database Syst Rev 7: CD003285, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 451: 1008–1012, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Beard RS, Jr, Bearden SE. Vascular complications of cystathionine β-synthase deficiency: future directions for homocysteine-to-hydrogen sulfide research. Am J Physiol Heart Circ Physiol 300: H13–H26, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birot OJ, Koulmann N, Peinnequin A, Bigard XA. Exercise-induced expression of vascular endothelial growth factor mRNA in rat skeletal muscle is dependent on fibre type. J Physiol 552: 213–221, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosch-Marce M, Pola R, Wecker AB, Silver M, Weber A, Luedemann C, Curry C, Murayama T, Kearney M, Yoon YS, Malinow MR, Asahara T, Isner JM, Losordo DW. Hyperhomocyst(e)inemia impairs angiogenesis in a murine model of limb ischemia. Vasc Med 10: 15–22, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Conde E, Alegre L, Blanco-Sanchez I, Saenz-Morales D, Aguado-Fraile E, Ponte B, Ramos E, Saiz A, Jimenez C, Ordonez A, Lopez-Cabrera M, del Peso L, de Landazuri MO, Liano F, Selgas R, Sanchez-Tomero JA, Garcia-Bermejo ML. Hypoxia inducible factor 1-alpha (HIF-1 alpha) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS One 7: e33258, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duan J, Murohara T, Ikeda H, Sasaki K, Shintani S, Akita T, Shimada T, Imaizumi T. Hyperhomocysteinemia impairs angiogenesis in response to hindlimb ischemia. Arterioscler Thromb Vasc Biol 20: 2579–2585, 2000 [DOI] [PubMed] [Google Scholar]
- 8.Frey SP, Jansen H, Raschke MJ, Meffert RH, Ochman S. VEGF improves skeletal muscle regeneration after acute trauma and reconstruction of the limb in a rabbit model. Clin Orthop Relat Res 470: 3607–3614, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaugler M, Brown A, Merrell E, DiSanto-Rose M, Rathmacher JA, Reynolds TH. PKB signaling and atrogene expression in skeletal muscle of aged mice. J Appl Physiol 111: 192–199, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson JB, Carson NA, Neill DW. Pathological findings in homocystinuria. J Clin Pathol 17: 427–437, 1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS One 7: e32388, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Givvimani S, Sen U, Tyagi N, Munjal C, Tyagi SC. X-ray imaging of differential vascular density in MMP-9−/−, PAR-1−/+, hyperhomocysteinemic (CBS−/+) and diabetic (Ins2−/+) mice. Arch Physiol Biochem 117: 1–7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grosfeld A, Andre J, Hauguel-De Mouzon S, Berra E, Pouyssegur J, Guerre-Millo M. Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. J Biol Chem 277: 42953–42957, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Harada H, Itasaka S, Kizaka-Kondoh S, Shibuya K, Morinibu A, Shinomiya K, Hiraoka M. The Akt/mTOR pathway assures the synthesis of HIF-1α protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J Biol Chem 284: 5332–5342, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Hiasa K, Ishibashi M, Ohtani K, Inoue S, Zhao Q, Kitamoto S, Sata M, Ichiki T, Takeshita A, Egashira K. Gene transfer of stromal cell-derived factor-1α enhances ischemic vasculogenesis and angiogenesis via vascular endothelial growth factor/endothelial nitric oxide synthase-related pathway: next-generation chemokine therapy for therapeutic neovascularization. Circulation 109: 2454–2461, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol 59: 15–26, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Hoogeveen EK, Kostense PJ, Jakobs C, Dekker JM, Nijpels G, Heine RJ, Bouter LM, Stehouwer CD. Hyperhomocysteinemia increases risk of death, especially in type 2 diabetes: 5-year follow-up of the Hoorn Study. Circulation 101: 1506–1511, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol Cell Biol 22: 7004–7014, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine γ-lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 285: 26358–26368, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci USA 104: 12017–12022, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kado DM, Bucur A, Selhub J, Rowe JW, Seeman T. Homocysteine levels and decline in physical function: MacArthur Studies of Successful Aging. Am J Med 113: 537–542, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Kalra BR, Ghose S, Sood NN. Homocystinuria with bilateral absolute glaucoma. Ind J Ophthalmol 33: 195–197, 1985 [PubMed] [Google Scholar]
- 23.Karkkainen AM, Kotimaa A, Huusko J, Kholova I, Heinonen SE, Stefanska A, Dijkstra MH, Purhonen H, Hamalainen E, Makinen PI, Turunen MP, Yla-Herttuala S. Vascular endothelial growth factor-D transgenic mice show enhanced blood capillary density, improved postischemic muscle regeneration, and increased susceptibility to tumor formation. Blood 113: 4468–4475, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg 38: 316–322, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Lee I, Lee H, Kim JM, Chae EH, Kim SJ, Chang N. Short-term hyperhomocysteinemia-induced oxidative stress activates retinal glial cells and increases vascular endothelial growth factor expression in rat retina. Biosci Biotechnol Biochem 71: 1203–1210, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat Prot 4: 1737–1746, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Machens HG, Grzybowski S, Bucsky B, Spanholtz T, Niedworok C, Maichle A, Stockelhuber B, Condurache A, Liu F, Egana JT, Kaun M, Mailander P, Aach T. A technique to detect and to quantify fasciocutaneous blood vessels in small laboratory animals ex vivo. J Surg Res 131: 91–96, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Maeda M, Yamamoto I, Fujio Y, Azuma J. Homocysteine induces vascular endothelial growth factor expression in differentiated THP-1 macrophages. Biochim Biophys Acta 1623: 41–46, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Maron BA, Loscalzo J. The treatment of hyperhomocysteinemia. Annu Rev Med 60: 39–54, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McClung JM, McCord TJ, Keum S, Johnson S, Annex BH, Marchuk DA, Kontos CD. Skeletal muscle-specific genetic determinants contribute to the differential strain-dependent effects of hindlimb ischemia in mice. Am J Pathol 180: 2156–2169, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller A, Mujumdar V, Shek E, Guillot J, Angelo M, Palmer L, Tyagi SC. Hyperhomocyst(e)inemia induces multiorgan damage. Heart Vessels 15: 135–143, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Niiyama H, Huang NF, Rollins MD, Cooke JP. Murine model of hindlimb ischemia. J Vis Exp: 1035, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olfert IM, Howlett RA, Tang K, Dalton ND, Gu Y, Peterson KL, Wagner PD, Breen EC. Muscle-specific VEGF deficiency greatly reduces exercise endurance in mice. J Physiol 587: 1755–1767, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pajusola K, Kunnapuu J, Vuorikoski S, Soronen J, Andre H, Pereira T, Korpisalo P, Yla-Herttuala S, Poellinger L, Alitalo K. Stabilized HIF-1α is superior to VEGF for angiogenesis in skeletal muscle via adeno-associated virus gene transfer. FASEB J 19: 1365–1367, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, Jang Y, Cho SY, Kim HS. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med 33: 95–102, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Raschke S, Eckardt K, Bjorklund Holven K, Jensen J, Eckel J. Identification and validation of novel contraction-regulated myokines released from primary human skeletal muscle cells. PLoS One 8: e62008, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson RS, Wagner H, Mudaliar SR, Henry R, Noyszewski EA, Wagner PD. Human VEGF gene expression in skeletal muscle: effect of acute normoxic and hypoxic exercise. Am J Physiol Heart Circ Physiol 277: H2247–H2252, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Richardson RS, Wagner H, Mudaliar SR, Saucedo E, Henry R, Wagner PD. Exercise adaptation attenuates VEGF gene expression in human skeletal muscle. Am J Physiol Heart Circ Physiol 279: H772–H778, 2000 [DOI] [PubMed] [Google Scholar]
- 39.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarkar K, Fox-Talbot K, Steenbergen C, Bosch-Marce M, Semenza GL. Adenoviral transfer of HIF-1α enhances vascular responses to critical limb ischemia in diabetic mice. Proc Natl Acad Sci USA 106: 18769–18774, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semenza GL. Pulmonary vascular responses to chronic hypoxia mediated by hypoxia-inducible factor 1. Proc Am Thorac Soc 2: 68–70, 2005 [DOI] [PubMed] [Google Scholar]
- 42.Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N, Tyagi SC. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol 303: C41–C51, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Springer ML, Ozawa CR, Banfi A, Kraft PE, Ip TK, Brazelton TR, Blau HM. Localized arteriole formation directly adjacent to the site of VEGF-induced angiogenesis in muscle. Mol Ther 7: 441–449, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, Gassmann M, Candinas D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J 15: 2445–2453, 2001 [DOI] [PubMed] [Google Scholar]
- 45.Tadaishi M, Miura S, Kai Y, Kano Y, Oishi Y, Ezaki O. Skeletal muscle-specific expression of PGC-1α-b, an exercise-responsive isoform, increases exercise capacity and peak oxygen uptake. PLoS One 6: e28290, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuomisto TT, Rissanen TT, Vajanto I, Korkeela A, Rutanen J, Yla-Herttuala S. HIF-VEGF-VEGFR-2, TNF-α and IGF pathways are upregulated in critical human skeletal muscle ischemia as studied with DNA array. Atherosclerosis 174: 111–120, 2004 [DOI] [PubMed] [Google Scholar]
- 47.Tyagi N, Qipshidze N, Sen U, Rodriguez W, Ovechkin A, Tyagi SC. Cystathionine β synthase gene dose dependent vascular remodeling in murine model of hyperhomocysteinemia. Int J Physiol Pathophysiol Pharmacol 3: 210–222, 2011 [PMC free article] [PubMed] [Google Scholar]
- 48.Veeranki S, Tyagi SC. Defective homocysteine metabolism: potential implications for skeletal muscle malfunction. Int J Mol Sci 14: 15074–15091, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner PD, Olfert IM, Tang K, Breen EC. Muscle-targeted deletion of VEGF and exercise capacity in mice. Respir Physiol Neurobiol 151: 159–166, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine β-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci USA 92: 1585–1589, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong YY, Almeida OP, McCaul KA, Yeap BB, Hankey GJ, Flicker L. Homocysteine, frailty, and all-cause mortality in older men: the health in men study. J Gerontol A Biol Sci Med Sci 68: 590–598, 2013 [DOI] [PubMed] [Google Scholar]
- 52.Yan TT, Li Q, Zhang XH, Wu WK, Sun J, Li L, Zhang Q, Tan HM. Homocysteine impaired endothelial function through compromised vascular endothelial growth factor/Akt/endothelial nitric oxide synthase signalling. Clin Exp Pharmacol Physiol 37: 1071–1077, 2010 [DOI] [PubMed] [Google Scholar]
- 53.Yap S, Naughten ER, Wilcken B, Wilcken DE, Boers GH. Vascular complications of severe hyperhomocysteinemia in patients with homocystinuria due to cystathionine beta-synthase deficiency: effects of homocysteine-lowering therapy. Semin Thromb Hemost 26: 335–340, 2000 [DOI] [PubMed] [Google Scholar]
- 54.Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv Z, Gu Z, Zhang J, Wang J, Zen K, Xiang Y, Wang D, Zhang CY. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res 20: 676–687, 2010 [DOI] [PubMed] [Google Scholar]
- 55.Zwetsloot KA, Westerkamp LM, Holmes BF, Gavin TP. AMPK regulates basal skeletal muscle capillarization and VEGF expression, but is not necessary for the angiogenic response to exercise. J Physiol 586: 6021–6035, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]