

Abstract
Progressive rod-cone degeneration (prcd) is a late-onset, autosomal recessive photoreceptor degeneration of dogs, and a homolog for some forms of human retinitis pigmentosa (RP). Previously, the disease relevant interval was reduced to a 106 Kb region on CFA9, and a common phenotype-specific haplotype was identified in all affected dogs from several different breeds, and breed varieties. Screening of a canine retinal EST library identified partial cDNAs for novel candidate genes in the disease relevant interval. The complete cDNA of one of these, PRCD, was cloned in dog, human and mouse. The gene codes for a 54 amino acid (aa) protein in dog and human, and 53 aa protein in the mouse; the first 24 aa, coded for by exon 1, are highly conserved in 14 vertebrate species. A homozygous mutation (TGC → TAC) in the second codon shows complete concordance with the disorder in 18 different dog breeds/breed varieties tested. The same homozygous mutation was identified in a human patient from Bangladesh with autosomal recessive (ar) RP. Expression studies support the predominant expression of this gene in the retina, with equal expression in the retinal pigment epithelium (RPE), photoreceptors and ganglion cell layers. This study provides strong evidence that a mutation in the novel gene, PRCD, is the cause of autosomal recessive retinal degeneration in both dogs and man.
Keywords: Dogs; Disease Models, Animal; Genetic diversity; Genetic linkage; Genetic markers; Genetic predisposition to disease; Genetic variation; Mutation; Retinal Degeneration; Retinitis Pigmentosa
INTRODUCTION
Progressive rod-cone degeneration (prcd) is a canine, late-onset, autosomal recessive photoreceptor degeneration of unknown molecular etiology [1]. This disorder is one of several inherited diseases grouped under the progressive retinal atrophy (PRA) rubric. These serve as animal models for human retinitis pigmentosa (RP) due to shared molecular defects, and/or phenotypic similarities (see [2] for review). Together with the rd mouse and RCS rat, prcd represents one of the best characterized models of inherited retinal degeneration (see [3; 4; 5; 6; 7; 8; 9; 10; 11; 12]). Previously, the disease was mapped to the centromeric end of canine chromosome 9 (CFA9), a region orthologous to the telomeric end of human chromosome 17 (HSA17q22; [13; 14]). A physical and radiation hybrid map of the region confirmed the syntenic organization of the homologous chromosomal segments, but demonstrated also that the order of gene clusters, and genes within clusters, differed between man and dog [15].
In a companion paper, we reported the use of linkage disequilibrium (LD) analysis across several dog breeds affected with prcd [2; 9] to narrow the candidate region, and direct the positional cloning of prcd [15]. As individual dog breeds represent genetic isolates, this powerful technique reduced the critical interval from ~6.4 Mb to 106 Kb, and identified a chromosomal haplotype shared by 14 different dog breeds or breed varieties [16]. This suggested that a common ancestral chromosome containing the mutant gene was widely distributed as modern dog breeds developed. The LD region, flanked by AANAT and ST6GalNac1, is completely contained in a single RPC181 BAC clone (10-M13; 21; http://bacpac.chori.org), and includes three known genes: CYGB, RHBDL, and STHM (ST6GalNac2). All the genes in the interval were evaluated and excluded from causative association with disease [16].
In a complementary effort we created a normalized canine retinal EST library that was catalogued by suggested gene homology and chromosomal location [17]. Intended as a means to identify and evaluate positional candidate genes within the prcd interval, the combination of sequence and expression data yielded several potential candidate genes in the original disease relevant interval. The application of this approach on the final shared haplotype identified a mutation in a novel gene of unknown function, referred to as Progressive Rod-Cone Degeneration (PRCD). In this study, we report the preliminary characterization of PRCD. Furthermore, we analyzed samples from 1,863 human patients with RP and other inherited retinal disorders, whose diseases had been previously excluded from causal association with genes known to cause retinal degeneration (RetNet-Retinal Information System; http://www.sph.uth.tmc.edu/RetNet/), and demonstrate that the identical mutation that causes prcd in dogs is homozygously present in a human RP patient.
RESULTS
Identification and characterization of PRCD
Identification of new candidate genes from a retinal EST library
Although linkage analysis of prcd-informative pedigrees mapped the disease locus to CFA9 [14], the original canine genome sequence draft (http://www.genome.gov/11007358; July 2004) suggested a translocation of the centromeric CFA9 region to CFA18. To establish the correct chromosomal position of the disease interval, FISH hybridization was performed with clones isolated from an 8× redundant dog genomic RPCI81 BAC library ([18]; http://bacpac.chori.org/) that are specific for CFA9 (189-B1), or are included in the physical map of the prcd interval (429-O8; [15]). Both probes co-localized on the same canine chromosome that was identified as CFA9 by the chromosome specific probe and G-banding pattern (data not shown). The assignment of the prcd-critical interval to CFA9 was confirmed in the new canine genome draft assembly ([19], May 2005 v2.0).
As presented in a companion paper [16], LD mapping identified a common 106 Kb candidate region on CFA9 flanked by AANAT and ST6GalNac1. This interval, and flanking genes, are contained in RPCI81 BAC 10-M13, and includes RefSeq genes AANAT, RHBDL6, CYGB, ST6GalNac1, and ST6GalNac2, all of which were previously excluded from causative association with disease. To complement the positional cloning of prcd, and begin characterization of the canine retinome, we developed a normalized canine retinal cDNA library [17] containing a total of 6,316 retinal expressed clones that were assembled into 3,980 unique sequences, termed “contig”, with unique identifier numbers (www.bioinformatics.upenn.edu/canine_retinalESTs/). BLASTN of those clones against the 3.2× low-pass genomic RPCI81 BAC 10-M13 sequence produced significant alignment for three contig sequences, 1131, 1365, and 3041 (Figure 1A). Contig 3041 represents five exons of a previously deposited, unpublished gene (DN904971, CF407776, CF407775, DN37851), and we found no sequence changes in this gene between prcd-affected and normal dogs.
Figure 1.

Display of the prcd candidate region. (A) LD was used to reduce the candidate region to 106 Kb [16]. This interval harbors three canine retinal ESTs (contigs 1131, 1365 and 3041). PRCD was cloned within orthologous regions in the dog (B), human (C) and mouse (D), and showed a high degree of conservation in gene structure.
Cloning of PRCD
Contig sequences 1131 and 1365 are 1,219 bp apart, and were assumed to represent a single gene (Figure 1A). They did not have a recognizable exon/intron structure, and, as both are adjacent to poly A stretches in the genomic DNA, could have represented potential genomic contamination of the EST library. Comparison between the canine, human and mouse genomic and cDNA sequences identified potential orthologous genes conserved in these species, and a suggested translation site approximately 6 Kb upstream of contig 1131 (Figure 1B–D). This was confirmed by RT-PCR from retinal RNA, and resulted in cloning of the full length ORF in all three species.
Canine PRCD (cPRCD) was first partially retrieved utilizing primers designed from contig 1131 and predicted exons 1 and 2, extended by 5’ and 3’ RACE, and the most common retinal transcript was confirmed by RT-PCR (695 bp, 4 exons; accession number: DQ390330). Additional splice variants were obtained during the cloning process (accession numbers: DQ390331, DQ390332, DQ390333, DQ390334, DQ390335, DQ390336). RT-PCR from human retina yielded a 417 bp product spanning four exons (accession number: DQ390338, Figure 1C); the sequence confirms hypothetical LOC400621, for which 20% of the total expression has been estimated to be present in the eye (http://genome-www5.stanford.edu/cgi-bin/source/sourceResult?choice=Gene&option=CLUSTER&criteria=Hs.434271). While this putative gene is supported by a variety of clones and splice variants, only two of the currently posted ESTs (BX646189, BN413679) contain parts of the proposed first exon, with the ATG site present only in the first of the two. Our clone locates the ATG start codon of the human ORF 84 bp upstream of the splice site at the end of exon 1, exactly as in the dog. The human ORF ends with a TAA stop codon at position 20 of exon 3, as in the dog. In mouse a TAG is present at the same position.
The murine 596 bp transcript (accession number: DQ390337; Figure 1D) spans five exons, and is partially homologous to a full length mouse RIKEN cDNA clone isolated from 16 day cerebellum (AK137302). Our clone has a different profile in the 5’ and 3’ ends; it is missing a putative 5’ exon, and 200 bp of the AK137302 3’UTR. We have not been able to amplify this putative 5' exon from mouse retinal cDNA, and the exon 1 we have characterized is in agreement with GeneScan prediction for the mouse genome draft, placing the ATG site 81 bp upstream the end of exon 1. We have identified an additional splice variant that is missing part of exon 1 (data not shown), indicating potential alternative splicing in the mouse. These sites are not conserved in the dog or human.
Structure and conservation of PRCD
The overall nucleotide conservation of PRCD between the dog, man, and mouse (dog-man=81%; dog-mouse=78%; human-mouse=78%) is slightly lower than previously observed for comparisons between mammalian gene orthologs [20]. However, the conservation profile at the protein level suggests functional importance, particularly for the first 24 aa coded for by exon 1 (Figure 2A). SignalP 3.0 Sever (http://www.cbs.dtu.dk/services/SignalP/) predicts a likely cleavage site between aa 20 and 21, suggesting function as a signal peptide with a C-terminal transmembrane domain; this conclusion is supported by other prediction software (e.g. “DAS”-transmembrane prediction server). We identified the ortholog of cPRCD exon 1 in 14 different vertebrate species for which genomic sequence is available (Figure 2B), confirming the evolutionary constraint of this peptide which is predicted to form an α-helix structure in a 3D model (Figure 2C). According to the secondary structure prediction methods used by the BIOINFO metaserver, the C2Y mutation in the human and canine sequences (see below) does not appear to change the predicted α-helical conformation of the initial 15 aa fragment. PRCD could not be identified in non-vertebrate species (e.g. D. melanogaster, A. thaliana or any published insect genome).
Figure 2.

(A) Alignment of translated PRCD ORF producing a 54 amino acid protein for human and dog; the mouse is missing a single amino acid (aa 10). The first 28 aa are completely conserved between the first two species, while 4 aa acids are replaced in the predicted mouse protein. Replacements are highlighted in gray if non-conservative, and the identical (.), strongly (*) and weakly (+) conserved positions are indicated underneath the sequence. Percentage of homology between species is shown on the right. (B) Alignment of exon1 coded protein sequence in 11 additional species demonstrating absolute conservation of the first 4 aa. (C) Three dimensional modeling of PRCD suggests the formation of an α-helix of approximately 20 aa at the N-terminal end (ball and stick). The C2Y mutation (highlighted in gray) does not appear to influence the secondary structure.
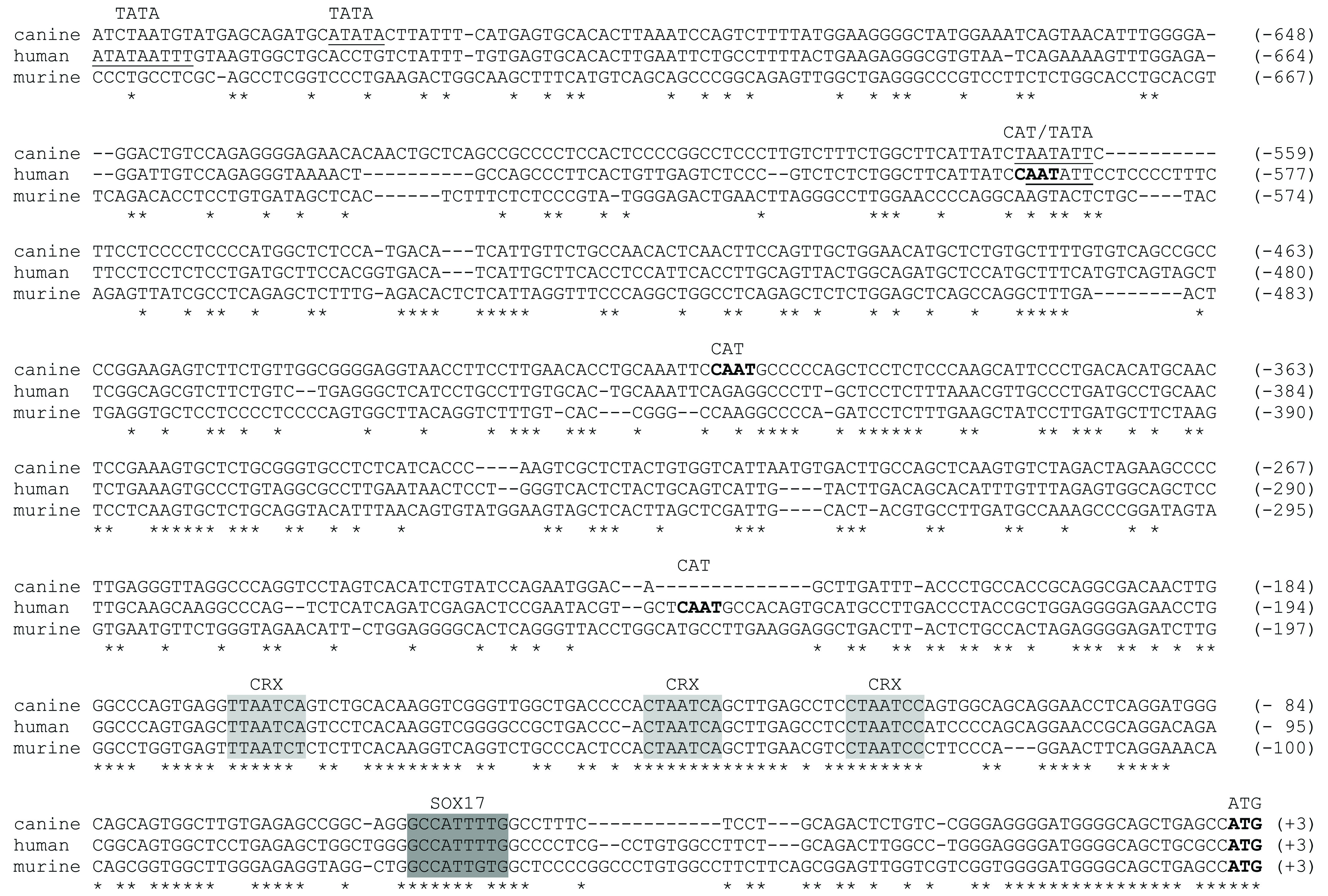
We screened for potential transcription binding sites 1,000 bp upstream to the ATG using the consite algorithm [score threshold of 80% (http://mordor.cgb.ki.se/cgi-bin/CONSITE/consite)] or manually (CRX, NRL). Overall, 86, 74 and 74 motifs are located on the positive strand only in the dog, human and mouse sequence, and represent binding site for 32, 36 and 23 different factors, respectively. 13 transcription factors are common to all three species (AML-1, ARNT, bZIP910, E74A, Hen-1, Max, NF-Y, n-MYC, Snail, Sox5, Sox17, Thing1-E47, USF). Pair-wise analysis of alignments, however, identified only one Sox17 site, located approximately 50 bp in front of the ATG, and CRX binding sites that were spatially conserved (Supplementary Figure 1, dark and light gray boxes, respectively). The same comparative analysis suggests potential involvement of CFI-USP and Snail as regulators through binding sites in intron 1 of PRCD (data not shown). CRX binding sites were also recognized in the potential 5’UTR of the additional 11 species for which exon 1 sequence is available, strongly suggesting the importance of this regulatory site. A potential binding motif for another retinal regulator, NRL, was found in the putative promoter region of the chicken, but could not be identified in the other species.
Putative TATA boxes are located at position −565 in dog and −593 in human (AATATT; Supplementary Figure 1); a second box could be present at −723 and −759 in the dog and human, respectively, but is less conserved between species (common motif ATATA), and the human might have a third box at −803 (not shown in Supplementary Figure 1 alignment). Putative CAAT motifs are observed at −408 in the dog, and at −242 and −595 in man. However, none of these transcription initiation sites could be identified in the mouse sequence.
Mutation in PRCD causes retinal degeneration in dog and man
Identification and segregation of the cPRCD mutation
Retinal cDNA sequence comparison of the cPRCD ORF (Supplementary Table 1, primers 3 and 4) from 8.6 week prcd-affected and 10.4 week normal dogs identified a single G to A transition at nucleotide 5 of the coding sequence. This mutation causes a cysteine to tyrosine change (C2Y) at the second aa of the protein in the affected animal. The causal association between this mutation and prcd was established by: (a) consistent and stable segregation of the mutation in the prcd reference colony; (b) segregation of the mutation in well maintained pedigrees of breeds included in previous prcd breeding studies; (c) segregation of the mutation in dog breeds for which the diagnosis of prcd was obtained by the investigators (GDA, GMA), and pedigrees were followed over several generations; (d) observation of the prcd mutation in breeds with a clinical diagnosis of retinal degeneration compatibly with prcd; (e) absence of the mutated allele from breeds not segregating the prcd phenotype. Details of these studies are presented below:
(a) We have maintained a reference colony segregating prcd for more than 10 generations. The founder chromosomes were from affected Toy and Miniature poodles (TP, MP [9]). Affected chromosomes originating from Labrador retriever (LR), Portuguese water dog (PWD), Nova Scotia duck tolling retriever (NSDTR), Australian cattle dog (ACD) and English cocker spaniels (ECS) were introduced into the colony subsequently, and segregate the same phenotype. The affected haplotype present in this colony has been identified [16], and shows complete concordance with the phenotype. The prcd mutation is included in this haplotype described by 98 polymorphisms in Supplementary Table 1 of the companion paper [16], and does not show any discordance from inheritance within the reference colony.
(b) We also have access to outside pedigrees from these breeds in which prcd segregates. These animals were screened for the disease-specific haplotype and cPRCD mutation. Complete concordance between affected phenotype and genotype was confirmed for affected and obligate carrier in ACD, ECS, LR, NSDTR, MP, PWD, and TP using a restriction enzyme analysis (see Figure 3A and B for illustration). In addition, we have selected a small subset of dogs from some breeds (ACD, LR, MP, TP) that had a clinical retinal disease atypical for the breed-specific form of retinal degeneration. For example, these included dogs showing clinical signs much earlier or later than the characteristic age of onset of prcd in the breed, inflammatory retinopathy, or other clinical disease features not considered characteristic of prcd. In all cases, these dogs were found to be heterozygous or homozygous normal at the disease locus. Sequence analysis using a subset of markers from the prcd LD region confirmed exclusion of prcd, and suggested that the clinical disease(s) present in these dogs were geno- or phenocopies.
Figure 3.

(A) Example of a prcd-informative ECS pedigree showing co-segregation of haplotypes with the disease. GRB2, AANAT and SEC14L are outside the candidate region, but a benign AANAT SNP is in complete linkage disequilibrium with the disease in this breed [16]. (B) Restriction enzyme test shows complete digestion of the PRCD A allele with RsaI, and of the G allele with ApaLI. (C) Assessment of the PRCD genotype by pyrosequencing.
(c) Pedigrees were available for fours breeds of which three were not included in the breeding studies [9]: American cocker spaniel (ACS), American Eskimo (AE [21]), Chesapeake Bay retriever (CBR), and Entlebucher mountain dog (EMD [22]). With the exception of one dog, there was complete concordance between the cPRCD mutation and affected and obligate carrier status. The exception was an AE that was homozygous affected (A/A), but clinically normal at 13 years. As the disease in this and some other breeds, e.g. ECS, can occur at a much later age [2; 9], definite phenotypic diagnosis will require histologic assessment after death of the animal.
(d) Selected samples of representative dog breeds that showed a retinal disease that appeared to be clinically compatible with prcd were collected by the investigators, or provided by colleagues. These included the following breeds: Chinese crested (CC), Silky terrier (ST), Finnish lapphund (FL), Kuvasz (KU), Lapponian herder (LH), and Swedish lapphund (SL). The homozygous G →A mutation in cPRCD was found in all 5 breeds. Analysis for these breeds is currently in progress.
(e) To establish that the A allele is not just a rare SNP in the population that has been concentrated in a few selected breeds, high throughput pyrosequencing analysis (Figure 3C) was used to screen 1,120 samples from unrelated dogs from 2 groups: (i) 1,007 samples from 37 breeds with non-prcd form of retinal degeneration where dogs of known phenotype were tested. In several of these breeds, the molecular basis of the retinal disease was known, and the genotype at the disease locus established (Supplementary Table 2). (ii) 113 samples of unrelated dogs from 54 breeds without known retinal disease (Supplementary Table 3). All 1,120 dogs tested from 91 different breeds were homozygous normal (G/G) at nucleotide 5 of cPRCD. Furthermore, 24 red and 6 grey wolves also were tested, and all were homozygous normal (G/G) at the mutation locus.
In summary, the cPRCD mutation has been observed in 18 individual dog breeds or breed varieties (Table 1) to date, and was confirmed to cause disease in the homozygous state. The disease causing allele was never observed in dogs or breeds confirmed not to segregate prcd.
Table 1.
Breeds affected by prcd
| Abbreviation | Breed name |
|---|---|
| ACS | American cocker spaniel |
| AE | American Eskimo |
| ACD | Australian cattle dog |
| CBR | Chesapeake Bay retriever |
| CC | Chinese crested |
| ECS | English cocker spaniels |
| EMD | Entlebucher mountain dog |
| FL | Finnish lapphund |
| KU | Kuvasz |
| LR | Labrador retriever |
| LH | Lapponian herder |
| MP | Miniature poodle |
| NSDTR | Nova Scotia duck tolling retriever |
| PWD | Portuguese water dog |
| ST | Silky terrier |
| STCD | Australian stumpy tail cattle dog |
| SL | Swedish lapphund |
| TP | Toy poodle |
Mutation identification and screening in human populations
We have found seven different hPRCD sequence variations in a total of 1,863 patients, most of whom were clinically diagnosed with RP or allied retinal digenerations (Table 2). Of these, 4 were found in the heterozygous state in the non-coding regions of the gene, and appeared unlikely to represent disease-causing variations. One patient with autosomal recessive RP (arRP) showed a G→A transition in the heterozygous state at 88 bp of the coding sequence. This variation would be expected to cause a conservative substitution of methionine for valine at aa 30 of the PRCD protein (V30M). DNA sequencing of the entire hPRCD coding sequence in this patient failed to detect a second sequence variation. The heterozygosity of the variation in this patient, coupled with the conservative nature of the predicted change in the protein, make it difficult to assert that this variation causes human retinal disease even though the change was not observed in the control population (215 individuals; Table 2).
Table 2.
hPRCD sequence changes observed in heterozygous (het) or homozygous (hom) state in patients with RP, Leber Congenital Amaurosis (LCA), cone-rod dystrophy (CRD), Usher syndrome (USH), and normal Caucasian (NML) and India/Pakistan (I/P NML) control populations.
| Disorder | RP | LCA | CRD | USH | NML | I/P NML | ||
|---|---|---|---|---|---|---|---|---|
| Patient/control numbers | 1,241 | 283 | 224 | 115 | 166 | 49 | ||
| Position | Sequence change | aa change | ||||||
| −106 het | C→T | 1 | 0 | 0 | 0 | 0 | 0 | |
| −40U het | T→C | 1 | 0 | 0 | 0 | 0 | 0 | |
| 5 hom | TGC→TAC | Cys2Tyr | 1 | 0 | 0 | 0 | 0 | 0 |
| 49 het | CGC→TGC | Arg17Cys | 1 | 0 | 0 | 0 | 0 | 5 |
| 88 het | GTG→ATG | Val30Met | 1 | 0 | 0 | 0 | 0 | 0 |
| *57U het | C→G | 1 | 0 | 0 | 0 | 0 | 0 | |
| *80 het | G→A | 1 | 0 | 0 | 0 | 0 | 0 | |
Another transition (C→T in the 49th nucleotide of the coding sequence, predicting the substitution of Cysteine for Arginine in codon 17, R17C) was observed in the heterozygous state in one RP patient of Indian origin, but was also observed in the heterozygous state in five members of the Indian/Pakistani control group. Bi-directional DNA sequencing was used to examine the entirety of the hPRCD coding sequence in the RP patient with the R17C change, and no other variations were detected. This result, combined with the high frequency of occurrence (5% of all alleles) of the variation in the ethnic control samples, make it likely that it represents a non-disease-causing ethnic-specific polymorphism.
In contrast, a woman from Bangladesh with arRP was found to harbor a G→A transition in the fifth nucleotide of the hPRCD coding sequence which would be predicted to result in a substitution of tyrosine for cysteine (C2Y) in the second amino acid of the protein. This is the identical mutation at both the DNA and protein levels that was observed in prcd-affected dogs. The patient was 32 years old when examined at the University of Iowa. She was born in Bangladesh, and first noticed difficulty seeing at night as a small child. Her family history was significant for four unaffected siblings, one brother who became blind at a young age, and two other siblings still living in Bangladesh with an unknown vision problem. Her parents were first cousins and had normal vision. The patient’s best corrected visual acuity was 20/70 in the right eye, and hand motions in the left eye with a manifest refraction of −3.75+1.00×180 in the right eye and −2.00 sphere in the left eye. Slit lamp examination revealed clear corneas and anterior chambers that were deep, clear and quiet. The lenses were clear, and there were no vitreous cells. Analysis of the papillary response revealed a 0.9 log unit relative afferent pupillary defect. Goldmann perimetry showed a greater loss of sensitivity in the left eye than the right (Figure 4A). Fundus examination revealed optic discs with fairly normal color, but markedly attenuated arterioles (Figure 4B). There was extensive bone-spicule-like pigmentation in all four quadrants of both eyes, densest at the equator, and this intra-retinal pigment was admixed with lighter colored deposits at the level of the retinal pigment epithelium. There were also small patches of geographic atrophy near fixation in both eyes. Ganzfeld electroretinography was performed, but no responses could be recorded from either eye under any stimulus condition.
Figure 4.

Clinical assessment of an arRP patient of Bangladesh birth at age 32. (A) The Goldmann visual field of the right eye shows complete loss of the I2e isopter. The I4e isopter (shown in blue) is limited to less than 5 degrees. There is persistence of a temporal island that is sensitive only to the V4e target (shown in purple). (B) Fundus photograph of the right eye of the patient centered on the inferotemporal vascular arcade. The optic disk is of fairly normal color, but the retinal arterioles are extensively attenuated. Typical bone-spicule-like pigmentation can be seen admixed with pale deposits at the level of retinal pigment epithelium. Small patches of atrophy of the retinal pigment epithelium can be seen adjacent to the fovea (the fovea can be recognized as the region of darker pigmentation to the left of the optic nerve head).
Expression and function of the PRCD gene
Expression profile of cPRCD in normal and mutant animals
Transcription of cPRCD was examined by northern analysis using probes specific for either exons 1 or 2–4 (Figure 5A). The same level of expression is observed in retinas from normal and prcd affected dogs, and show two distinct retinal transcripts of 2 Kb and 6.5 Kb. Experiments with a probe specific for the 3’UTR located in contig 1131 sequence yielded the same results (data not shown). The same transcripts were observed in RPE/choroid, but the level of expression in other tissues is too low for detection. RT-PCR amplification using primers located in exons 1 and 4 (Supplementary Table 1, primers 1 and 31) showed low levels of cPRCD expression in a variety of tissues (data not shown).
Figure 5.

Expression profile of cPRCD. (A) Northern analysis with probes specific for exon 1 and exons 2–4 of cPRCD identifying two distinct transcripts (2 and 6.5 Kb) expressed in the normal and prcd-affected retinas (R=10.4 weeks; R prcd=8.6 weeks). No signal is present in normal or affected brain (B=15.7 weeks; B prcd=7.7 weeks), or normal spleen (S=22.1 weeks). The same transcripts are expressed at lower levels in the RPE/choroid (RPE) when compared to retina. β-Actin was used as loading control. (B) qRT-PCR results show high expression in the retina compared to other tissues. Expression profile (shown as relative expression against 18S) has been established for a normal dog at 16 weeks of age (center) and is similar to the expression levels found in affected retinas of different ages (left). Relative expression normalized against retina provides fold reduction in the expression between tissues (right). (C) Individual retinal layers (RPE, photoreceptor and ganglion cell layer) from a normal eye were isolated with a laser capture microscope (respective regions are illustrated by octagons below black bars), and analyzed by qRT-PCR. No significant differences in expression was observed between the layers.
To quantify the expression profile of cPRCD, we performed qRT-PCR on different tissues of a 16 wk old normal dog for two probes: one specific for exon 1, and the second probe spanning parts of exons 2 and 3. Results are shown for the exon1 probe only, as no significant differences between the two probe sets were found. Relative expression was calculated against an 18S probe as difference of cycles leading to amplification. In addition, retinal samples from prcd-affected dogs of various ages (10, 16, and 23 weeks; 2 and 3 years) also were analyzed by qRT-PCR. Comparison of cPRCD expression in the normal vs. prcd-affected retina showed no significant changes in expression with age or disease stage (Figure 5B). Expression in spleen, liver, and pancreas was insufficient for quantification, and no expression was observed in the lung (data not shown). In comparison to normal retina, there is significantly lower expression in all other tissues tested (Figure 5B); there was ~6.5 fold lower expression in RPE/choroid, 16 fold decrease in kidney, and 100–1000 fold decreases in other tissues. Even though expression levels for cerebrum represented a pooled sample of frontal, occipital and temporal lobes, analysis of individual samples showed comparable low levels of expression (data not shown).
Laser capture microscopy was used in the normal retina to specifically compare cPRCD expression in the RPE, photoreceptor and ganglion cell layers. Comparable expression in all three layers suggests that this gene has uniform expression in many retinal cell types, and is not photoreceptor-specific or enriched (Figure 5C).
Expression of wildtype and mutant hPRCD in cos cells
His tag localization was performed on COS cells transfected with either wild type or mutant (C2Y) hPRCD. Whereas the wild type fusion protein was distributed diffusely throughout the cytoplasm (Figure 6A, D; upper panels), in the majority of transfected cells the mutant protein was present in spherical aggregates within the cytoplasm (Figure 6E, H; lower panels). The distribution of the wild type protein did not substantially overlap with that of actin filaments in the cytoplasm, but did extend into cell processes, and showed greatest labeling around the nucleus.
Figure 6.

Immunocytochemical localization of wild type (A) and mutant (E) PRCD in COS-7 cells. No co-localization was observed with phalloidin labeled actin (B, F), or nuclei stained with DAPI (C, G) in the merged images (D, H). Localization of the His tag was present in a diffuse web-like distribution in most cells transfected with the wild type construct (A, D), whereas labeling was generally concentrated in vesicular profiles in the cytoplasm in cells transfected with the mutant construct (E, H). Scale bar: 25µm.
DISCUSSION
A combination of LD mapping [16] and the characterization the canine retinome [17] identified the 3' end of a novel retinal gene. This gene was subsequently cloned in dog, human, and mouse, and a mutation found that causes canine and human retinal degeneration. The gene has been given the preliminarily name of “progressive rod-cone degeneration” with the gene symbol “PRCD” (HUGO gene nomenclature committee, approved on February 22, 2006 by Dr. Elspeth Bruford).
PRCD has a predominant transcript of 671 bp in the dog retina, and encodes a conserved ORF (78–81% conservation). GenBank entries of human and murine transcripts, as well as potential alternative splicing identified in the dog, suggest alternative splice variants for this gene, and some of these have been characterized in dogs (data not shown). However, we could not identify usage of additional 5' exons in any of the retinal transcripts of dog or mouse by RT-PCR or 5' RACE. The correct sequence was confirmed, and the final ORF chosen based on conservation between species. PRCD encodes a 54 aa protein in dog and human; in mouse it is 53 aa as 3 in frame coding nucleotides (#28–30) are missing from exon 1. The conservation of potential regulatory elements and binding sites further suggests the correct positioning of the proposed ORF (Supplementary Figure 1). Three CRX binding sites are located just upstream of the ATG site in dog, human and mouse; this factor is thought to drive photoreceptor expression [23; 24]. A large number of potential transcription factor binding sites could be identified within the 5’UTR and intron 1, and 13 of these were common to the three species investigated. However, only the Sox17 site showed high positional conservation, suggesting that this might be one of the PRCD interacting genes even though this transcription factor predominantly regulates the transcription of endodermal genes [25; 26]. Additional studies currently are underway to further assess the regulation and function of PRCD.
Comparison of the cDNA between prcd-affected and normal dogs identified G to A substitution at position 5 of the cPRCD ORF that causes a C2Y change in the translated protein. No other change within the available RPCI81 BAC 10-M13 sequence was found that segregated with the disease phenotype. Subsequent screening of the cPRCD mutation indicated the absence of the mutated allele (A) in 1,120 dogs from 91 different breeds not diagnosed with the prcd form of PRA as well as 24 red and 6 grey wolves (Supplementary Tables 2 and 3). As well, breeds identified to segregate the prcd haplotype [16] showed complete concordance between the mutated allele in the homozygous state and the clinical diagnosis of prcd. The high number of dogs investigated supports the association between mutation and disease phenotype, and this is particularly important when identifying a mutation in a novel gene of unknown function.
Some exceptions in the investigated breeds, however, were noted, and bear mentioning. We have identified dogs with retinal degeneration that were homozygous for the normal (G) allele. Labrador retrievers with advanced retinal degeneration at < 1 yr rather than 6–8 years represented age-outliers for the breed-specific disease [2]. Others, e.g. CC, ST, ACD, had retinal degeneration that was clinically similar to prcd, either in age of onset or clinical appearance, but were homozygous normal for cPRCD. It is likely that these retinal degenerations, if not acquired phenocopies, are caused by mutations in different retinal disease genes, an issue that we have previously reported for the Norwegian elkhound and miniature schnauzer breeds (see [2] for review). For all of the discordant individuals for which haplotype analysis was carried out, we were able to exclude the segregating chromosomes from association with prcd [16].
An AE dog was of particular interest because she typed homozygous affected for prcd (A/A), but did not show clinical signs at 13 years. Both chromosomes were confirmed to produce the disease phenotype in affected relatives through extended haplotype analysis. Such a delay in onset of disease is not surprising given the variation in disease onset and progression which occurs in some breeds with prcd [9]. For example, in the ECS we have found the age of early diagnosis to vary from 3.1–13.5 years in dogs having the same mutation in cPRCD (OptiGen LLC, unpublished information). Comparative analysis of the genetic background between different phenotypic variants of prcd has been proposed to identify the genetic modifiers. Such studies will contribute to our understanding of PRCD function, and the molecular mechanisms of disease.
The cloning and characterization of the hPRCD ortholog, and the subsequent screening of patients clinically diagnosed with various forms of retinal disease, led to the identification of a patient that is homozygous for the same sequence variation at, both the nucleotide and amino acid levels, as found in pcrd dogs. This coincidence may suggest that function-altering sequence variations that occur elsewhere in this gene are likely to have a different phenotype from the patients that were screened in this study. The one RP patient identified with a hPRCD mutation had advanced disease in the fourth decade of life with marked constriction of the visual fields, a non-recordable electroretinogram, and extensive bone-spicule-like pigmentation. Such advanced disease is similar to what is observed in most canines affected with prcd at a comparable age [2]. Although the patient has a family history of vision problems, the remaining family members still live in Bangladesh, and the acquisition of additional samples and phenotype characterization of family members has been difficult. This data hopefully will become available soon. Although this study shows that hPRCD mutations are an extremely rare cause of retinitis pigmentosa in patients ascertained in North America, it will require further study to determine whether mutations in this gene are responsible for a larger fraction of this disease in Bangladesh or other Asian populations.
Expression of PRCD indicates two main retinal transcripts of 2 and 6.5 Kb using northern analysis, both of which contain exons 1–4, as well as the originally identified 3’UTR region. The expression of cPRCD was determined by qRT-PCR, and showed that that highest level is in retina followed by RPE/choroid, and that other tissues have very low or negligible levels. However, the use of laser microscopy to specifically isolate different retinal cell layers allowed more precisely to quantify the contribution of each to the total cPRCD mRNA pool. This showed that there was approximately equal expression in the RPE, photoreceptor and ganglion cell layers, thus explaining why there is no decrease in cPRCD expression as the photoreceptor cells are lost with disease progression. Data from immunocytochemical subcellular localization experiments suggest that the mutant form of the protein exhibits a distinct pattern of localization into spherical profiles within the cytoplasm, and we posit that this altered localization may impair function and/or its interactions with other protein in the affected human and canine retina.
As can be appreciated, it is extremely difficult to asses the function of a protein that has not been known thus far, and does not belong to any known gene family. To date, we have shown that PRCD is causally associated with retinal degeneration in dog and man. As well, in silico analysis strongly suggests the functional importance of this gene based on the high conservation in vertebrate species. Our working hypothesis includes the formation of an α-helix involving the first 24 aa of PRCD that might contain a transmembrane segment; partial homology of this region to other confirmed or predicted bacterial proteins (e.g. AAS62120; ABD02230) highly supports this hypothesis. The first four aa, totally conserved in 14 vertebrate species analyzed, are not predicted to be part of the transmembrane domain. They are likely to play an important role as the recognition site for interacting proteins. Therefore, the described mutation in the highly conserved second aa would significantly influence the function of PRCD. The nature of the mutation, and observed change in the localization, are concordant with changes in the long-term viability of the retina rather than a fast acting developmental disorder. However, the exact disease mechanism will require a series of further studies.
The importance of this novel gene, PRCD, for maintenance of rod photoreceptor structure, function, and/or survival is attested to by its high level of conservation in vertebrate species. The significance of this conservation, absolute for the first 4 codons, is underscored by the causal association of the C2T mutation reported here with hereditary retinal degeneration in both dogs and man. The apparent absence of a homologous gene in invertebrates suggests that the function of the PRCD protein is critically important to those aspects of photoreceptor structure or metabolism unique to vertebrate visual cells. As these issues yield to further studies, they should provide new insights into vertebrate photoreceptor evolution, as well as to the mechanisms of photoreceptor health and disease.
MATERIALS AND METHODS
FISH mapping
Four ml of peripheral blood containing 0.2 ml preservative-free heparin was incubated with 31 ml of complete medium for 96 hr at 37°C [27]. After incubation, 5ml of medium was aspirated, and cells arrested with 0.2ml colcemid (10µ/ml) during a 1.5 hr incubation. Chromosomal spreads were prepared on slides using standard hypotonic solution and methanol/acetic acid fixation, and chromosome painting performed [28] using RPCI81 BAC clones specific to CFA9 (#189-B1), or included in the physical map (#429-O8) of the prcd interval [15]. FISH images of probes hybridized onto G-banded metaphases were captured using the ISIS software (In situ Imaging System; Metasystems) with a Paco CCD camera mounted on an Aristoplan (Leitz) microscope [28].
Protein structural analysis
Three-dimensional (3D) models for PRCD sequences were built by using the MODELLER program [29; 30; 31] with (a) experimentally-determined structures for the templates extracted from the Protein Data Bank, and (b) sequence alignments obtained from the BIOINFO metaserver [32]. The pairwise alignments produced by the metaserver were assigned relatively-low scores (low level of certainty) by the 3-D-Jury tool. However, the set of highest-score alignments points to structures having a short α-helical fragment at the N-terminal end of the protein. 3D models for all the sequences were generated by comparative protein modeling through satisfaction of spatial constraints with the MODELLER program, minimizing infringements of distance and dihedral-angle restraints from the template structures.
Study samples
Research dogs were maintained at the Retinal Disease Studies Facility (RDSF) in Kennett Square, Pennsylvania under cyclic light conditions, and prcd status was established using morphological, ERG or genetic criteria [14]. The dogs represented several breeds in which prcd allelism had been established through interbreed crosses [2; 9]: ECS, LR, MP and TP. Allelism with prcd has also been established for ACD, NSDTR, and PWD [16]. In addition to the colony derived research dogs, we obtained blood samples and clinical records of dogs submitted by their owners or other scientists directly to our lab, or through OptiGen, LLC. In all animals maintained in the research colony, and many outside cases, two of the investigators (GMA, GDA) carried out the clinical evaluations.
Euthanasia with a barbiturate overdose was performed at a single time of the day (10AM) to prevent fluctuation the abundance of retinal expressed genes, and retinas were isolated from the eyecup within 1–2 minutes of death. Other tissues were obtained within 5–10 minutes, and all tissues were flash frozen in liquid nitrogen and stored at −80°C until used. The human retina was isolated from a 76year old female donor 8 hours post mortem, and obtained through the National Disease Research Interchange. After isolation, it was snap frozen in liquid nitrogen, and processed as described. Neural retina from C57B1/6 mice were pooled to obtain sufficient tissue for cDNA amplification of the murine PRCD.
A total of 1,863 unrelated human patients with a clinical diagnosis of heritable photoreceptor disease were studied. All individuals provided written informed consent. Approximately a third of these patients were ascertained in the Retina Clinic of the University of Iowa, while the remainder was ascertained by inherited retinal disease specialists elsewhere in the United States. 1,241 patients were diagnosed with RP, 283 with Leber Congenital Amaurosis (LCA), 224 with cone-rod dystrophy (CRD), and 115 with Usher syndrome (USH). All of these patients had been screened previously for mutations in one or more genes known to cause their diseases (RetNet-Retinal Information System; http://www.sph.uth.tmc.edu/RetNet/), and none of them had been found to harbor a plausible disease-causing variation in a known retinal disease gene. Normal controls from the University of Iowa consisted of 166 subjects (general population controls) of mixed ages who had no personal or family history of heritable retinal disease. Over 80% of this group described themselves as ‘Caucasian’. The second group consisted of 49 unrelated control subjects from India and Pakistan.
Sample preparation
Total RNA was isolated using trizol extraction followed by treatment with DNAse (1 unit RQ1 DNAse; Promega) if used for qRT-PCR, and protection with RNase Inhibitor (Roche Applied Science). Double-stranded cDNA was synthesized using the Thermoscript RT-PCR kit (Invitrogen) with random hexamer primers. Amplification was obtained at standard conditions (Supplementary Table 1, primers 1, 2 and 11; accession numbers:DQ390332, DQ390333), and extension of cDNA sequence by RACE was obtained from a 10 week normal retina through the SMART™RACE cDNA amplification kit (Clontech) following the manufacturer’s protocol (Supplementary Table 1, primers 5–8). RT-PCR using primers from exons 1 and 4 (Supplementary Table 1, primers 9 and 10) confirmed the main transcript (695 bp, 4 exons; accession number: DQ390330), but suggested a second transcript, which does not splice out intron 1 (accession number: DQ390331). Additionally, primers located specifically in exon 2 and the predicted exon 5 of cPRCD (Supplementary Table 1, primers 12 and 13) yielded three splice variants (796 bp, 763 bp, 509 bp), utilizing different potential splicing sites between exons 4 and 5 (accession numbers: DQ390334, DQ390335, DQ390336).All amplicons were cloned into pCR®2.1-TOPO®vector (Invitrogen) following the manufacturer’s protocol, and stored in glycerol. Selected clones were sequenced for insert verification. DNA was extracted from spleen or blood using the QIAmp DNA Mini Kit (Qiagen). All RNA and DNA samples were evaluated on a 1% denaturing agarose-formaldehyde or agarose gel prior to use, and level of purity and concentration was measured with a spectrophotometer.
Restriction enzyme analysis
Amplification from genomic DNA with primers 3 and 17 (Supplementary Table 1) at an annealing temperature of 58°C under conditions described above resulted in a 512 bp PCR product. Restriction enzyme digestion performed in a 25µl reaction on 500ng PCR product with RsaI resulted in 396 and 116 bp bands in the presence of the mutated (A) allele; digestion with ApaLI resulted in 397 and 115 bp bands in the presence of the normal (G) allele. The genotypes resulting from the digestion were visualized on 6% polyacrylamide gels.
Pyrosequencing
High throughput screening for the mutant (A) and normal G) PRCD alleles was done by Pyrosequencing™ (Biotage). PCR was performed on isolated DNA with primers (Supplementary Table 1; primer 18 and biotin labeled primer 19) under standard conditions with 1.5mM MgCl2 at 55 oC annealing temperature in 35 cylces, checked on a 2% agarose gel, and purified with Streptavidin Sepharose High Performance Beads (Amersham). Primer 20 was added to initiate the reaction, and primer elongation was achieved and recorded on a Pyrosequencer PSQ™ 96MA using the PSQ™ 96 SNP Reagent Kit (Biotage).
Human PRCD screen
DNA was extracted from peripheral blood according to a previously described protocol [33]. Samples from 1,863 patients and 215 controls were screened for variations in the whole coding sequence of PRCD using single strand conformational polymorphism (SSCP) analysis as previously described [34]. To conserve DNA, the 49 Indian/Pakistani control samples were screened for variations in exon 1 only. All samples exhibiting an abnormal electrophoretic pattern by SSCP were further characterized using automated bi-directional sequencing on an ABI 3730 model using the BigDye version 3 chemistry (Applied Biosystems).
Northern analysis
Samples were loaded on a 1% agarose-formaldehyde denaturing gel, and 3µg of 0.24–9.5 Kb RNA ladder was used as a size marker (Invitrogen). The gel ran with continuously circulating 1×MOPS running buffer (Ambion) for 16 hours at 21 volts. After three 5 min washes in DEPC treated water, 20 min in 0.05N NaOH, and a 15 min soak in 10×SSC, transfer to a nylon-based membrane (GeneScreen Plus, NEN Life Science) was done with 10×SSC buffer using a standard protocol. Full transfer was confirmed by exposing the gel to UV light. The membrane was washed in 2×SSC for 2 min, and RNA was cross-linked to the membrane (exposure=0.12 joules per cm2; Stratalinker UV Crosslinker).
Northern probes were amplified from cDNA clones and labeled with alpha-dCTP- P32 using RadPrime DNA labeling System (Invitrogen). Pre-hybridization and hybridization were carried out with ExpressHyb solution (Clontech) at 68°C for 30 minutes. Hybridization was carried out at 68°C for 16–18 hours, and the blots rinsed several times with 2×SSC, 0.05%SDS; the washes with the same solution were done twice with continuous agitation for 40 min. Then the blot was washed with 0.1×SSC and 0.1% SDS with continuous shaking at 50°C for 40 min with one change of fresh solution. Blots were exposed to x-ray film at −70°C for 24–96 hours with two intensifying screens. Loading control was achieved by hybridizing canine specific β-actin (Z70044) probe to the membranes under the same conditions, and exposure to x-ray film for 4 hours.
Quantitative real time PCR (qRT-PCR)
Expression profiling of PRCD was done on a 7500 Real Time PCR System (Applied Biosystems) with two TaqMan® assays. Probes and primers were designed using the Primer Express® Software (Applied Biosystems) resulting in two primer sets. One produced a 55 bp product specific for exon 1 (Supplementary Table 1, primers 24 and 25, probe 26), and the second one a 63 bp product spanning the exon 2 and 3 junction (Supplementary Table 1, primers 21 and 22, probe 23). TaqMan PCR amplification was performed in a two-step RT-PCR reaction, reverse transcribing the RNA first with the TaqMan® Reverse Transcription Reagents (Applied Biosystems) followed by amplification of the target sequence using TaqMan® Universal Master Mix with the specific probe sets. Gene expression was evaluated through relative quantification against an 18S endogenous control (Hs99999901_s1, Applied Biosystems).
Laser capture microscopy
Glass slides with 10 µm frozen sections of normal canine retina (16 wk) fixed in 4% paraformaldehyde and embedded in OCT [35] were thawed for 20 seconds, and subsequently dehydrated through serial immersion in 70%, 95%, and 100% EtOH for 1 min each, followed by 2×5min in xylene. Laser capture was performed with a PixCell II Laser Capture Microdissection apparatus (Arcturus) on CapSure HS LCM Caps (Arcturus) at 50mW, capturing a total of 3,024 ganglion, 2,525 RPE, and 1,850 photoreceptor shots. RNA was isolated with the PicoPure RNA Isolation kit (Arcturus), and cDNA amplified with the RiboAmp OA RNA Amplification Kit (Arcturus), both following manufacturer’s protocols. A final cDNA concentration of ~15ng/µl was obtained based on spectrophotometer measurement for each cell class isolated, and 1µl was used for qRT-PCR as described.
Protein expression and localization
COS-7 cells (American Type Culture Collection, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium (with 10% (v/v) fetal calf serum, 1% penicillin/streptomycin, L-glutamine, sodium pyruvate, non-essential amino acids, HEPES and gentamycin). Prior to transfection, cells were split and grown on cover-slips in 6-well plates. Transfection of expression vectors (pCDNA3.1v5His see above) containing either the wild-type hPCRD gene or the C2Y mutant were mediated using Lipofectamine 2000 (Invitrogen) in Opti-MEM 1 (Gibco) for 48 hours according to manufacturer’s instructions.
Cells were fixed for 5 min in 4% formaldehyde (prepared from paraformaldehyde) in 10mM phosphate buffered saline (PBS), pH 7.4. Coverslips were then washed in PBS, and their top surfaces were covered with blocking solution (1mg/mL bovine serum albumin in PBS) for 15 min. Following a brief rinsing step in PBS, coverslips were then incubated with anti-His tag primary antibody (Invitrogen) at a concentration of 6µg/mL in PBS with 0.1% Triton X-100 for one hour. After rinsing with PBS, coverslips were then incubated with an anti-mouse secondary antibody conjugated to Alexa-546 (Invitrogen) at a 1 to 200 dilution for 30 min. Coverslips were washed, and then counterstained with 4’-6-diamidino-2-phenylindole (DAPI) and with Alexa-488-conjugated phalloidin, an actin binding dye, diluted 1 to 200 (Invitrogen). Coverslips were then placed on glass slides, mounted with Aquamount (Ted Pella), and photographed with a BX41 fluorescence microscope (Olympus).
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
We are grateful to Dr. Charles Aquadro, Department of Molecular Biology and Genetics, Cornell University for help with pyrosequencing, Dr. Peter Felsburg in the Section of Medical Genetics, University of Pennsylvania for access to the Applied Biosystems 7500 Real Time PCR System, and Dr. Neal Rubinstein, Department of Cell and Developmental Biology and Pennsylvania Muscle Institute, University of Pennsylvania for access to and help with laser capture microscopy. Some blood samples with pedigrees and clinical reports were provided by Drs. David Sargan (FL), Keith Murphy (AE subset) Simon Petersen-Jones (EMD), The Seeing Eye Inc. (LR subset), Dr. Ron Riis, Elaine Ostrander (many non-affected breeds), and many interested dog breeders and breed clubs. Dr. Jean Bennett provided normal mouse retinas for RNA extraction, and Ulana Prociuk assisted with chromosomal spreads. The authors especially wish to acknowledge Jennifer Johnson, Julie Jordan and Amy Antosh for technical assistance, Jean Andorf and Louisa Affatigato for sequencing of human samples, Jaroslaw Pillardy and Qi Sun for bioinformatics support, and Amanda Nickle, Gerri Antonini and staff of RDS facility for care and supervision of the research dog colony. This manuscript is dedicated to Sue Van Sloun for steadfast support, encouragement and help.
Supported by: Foundation Fighting Blindness (FFB), Morris Animal Foundation/The Seeing Eye, Inc., NIH grants EY06855, EY13132, Van Sloun Fund for Canine Genetic Research, L.J. Niles Foundation, ONCE International Prize for Biomedicine and R&D for New Technologies for the Blind, targeted donations from many individuals and breed clubs, HHMI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The discovery of the PRCD gene and potential tests identifying the mutation in the dog has been patented. Authors B. Zangerl, O. Goldstein, S.E. Pearce-Kelling, J.S. Felix, G.M. Acland and G.D. Aguirre are holding rights to this patent through Cornell University, and authors J.S. Felix, G.M. Acland and G.D. Aguirre are co-owners of the company (OptiGen®, LLC, Ithaca, NY) that has licensed the technology from Cornell University.
REFERENCES
- 1.Aguirre G, Alligood J, O'Brien P, Buyukmihci N. Pathogenesis of progressive rod-cone degeneration in miniature poodles. Invest Ophthalmol Vis Sci. 1982;23:610–630. [PubMed] [Google Scholar]
- 2.Aguirre GD, Acland GM. Models, Mutants and Man: Searching for Unique Phenotypes and genes in the Dog Model of Inherited Retinal Degeneration. In: Ostrander EA, Giger U, Lindblad-Toh K, editors. The Dog and Its Genome. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2006. pp. 291–325. [Google Scholar]
- 3.Wiggert B, Kutty G, Long KO, Inouye L, Gery I, Chader GJ, Aguirre GD. Interphotoreceptor retinoid-binding protein (IRBP) in progressive rod-cone degeneration (prcd)-biochemical, immunocytochemical and immunologic studies. Exp Eye Res. 1991;53:389–398. doi: 10.1016/0014-4835(91)90245-a. [DOI] [PubMed] [Google Scholar]
- 4.Anderson RE, Maude MB, Acland GM, Aguirre GD. Plasma lipid changes in PRCD-affected and normal miniature poodles given oral supplements of linseed oil. Indications for the involvement of n-3 fatty acids in inherited retinal degenerations. Exp Eye Res. 1994;58:129–137. doi: 10.1006/exer.1994.1001. [DOI] [PubMed] [Google Scholar]
- 5.Mieziewska K, van Veen T, Aguirre GD. Structural changes of the interphotoreceptor matrix in an inherited retinal degeneration: a lectin cytochemical study of progressive rod-cone degeneration. Invest Ophthalmol Vis Sci. 1993;34:3056–3067. [PubMed] [Google Scholar]
- Aguirre GD, Andrews L. Nomarski evaluation of rod outer segment renewal in a hereditary retinal degeneration. Comparison with autoradiographic evaluation. Invest Ophthalmol Vis Sci. 1987;28:1049–1058. [PubMed] [Google Scholar]
- 7.Aguirre G, O'Brien P. Morphological and biochemical studies of canine progressive rod-cone degeneration. 3H-fucose autoradiography. Invest Ophthalmol Vis Sci. 1986;27:635–655. [PubMed] [Google Scholar]
- 8.Parkes JH, Aguirre GD, Rockey JH, Liebman PA. Progressive rod-cone degeneration in the dog: characterization of the visual pigment. Invest Ophthalmol Vis Sci. 1982;23:674–678. [PubMed] [Google Scholar]
- 9.Aguirre GD, Acland GM. Variation in retinal degeneration phenotype inherited at the prcd locus. Exp Eye Res. 1988;46:663–687. doi: 10.1016/s0014-4835(88)80055-1. [DOI] [PubMed] [Google Scholar]
- 10.Huang JC, Chesselet MF, Aguirre GD. Decreased opsin mRNA and immunoreactivity in progressive rod-cone degeneration (prcd): cytochemical studies of early disease and degeneration. Exp Eye Res. 1994;58:17–30. doi: 10.1006/exer.1994.1191. [DOI] [PubMed] [Google Scholar]
- 11.Aguirre GD, Acland GM, Maude MB, Anderson RE. Diets enriched in docosahexaenoic acid fail to correct progressive rod-cone degeneration (prcd) phenotype. Invest Ophthalmol Vis Sci. 1997;38:2387–2407. [PubMed] [Google Scholar]
- 12.Gropp KE, Huang JC, Aguirre GD. Differential expression of photoreceptor-specific proteins during disease and degeneration in the progressive rod-cone degeneration (prcd) retina. Exp. Eye Res. 1997;64:875–886. doi: 10.1006/exer.1996.0257. [DOI] [PubMed] [Google Scholar]
- 13.Bardien S, Ramesar R, Bhattacharya S, Greenberg J. Retinitis pigmentosa locus on 17q (RP17): fine localization to 17q22 and exclusion of the PDEG and TIMP2 genes. Hum Genet. 1997;101:13–17. doi: 10.1007/s004390050577. [DOI] [PubMed] [Google Scholar]
- Acland GM, Ray K, Mellersh CS, Gu W, Langston AA, Rine J, Ostrander EA, Aguirre GD. Linkage analysis and comparative mapping of canine progressive rod-cone degeneration (prcd) establishes potential locus homology with retinitis pigmentosa (RP17) in humans. Proceedings National Academy of Sciences, USA. 1998;95:3048–3053. doi: 10.1073/pnas.95.6.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sidjanin DJ, Miller B, Kijas J, McElwee J, Pillardy J, Malek J, Pai G, Feldblyum T, Fraser C, Acland GM, Aguirre GD. Radiation hybrid map, physical map, and low-pass genomic sequence of the canine prcd region on Cfa9 and comparative mapping with the syntenic retion on human chromosome 17. Genomics. 2003;81:138–148. doi: 10.1016/s0888-7543(02)00028-9. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein O, Zangerl B, Sidjanin DJ, Kijas JW, Pearce-Kelling SE, McElwee J, Nelson JL, Felix JS, Acland GM, Aguiree GD. Linkage disequilibrium mapping in domestic dog breeds identifies the ancestral disease chromosome transmitting progressive rod-cone degeneration (prcd) 2006 doi: 10.1016/j.ygeno.2006.05.013. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zangerl B, Sun Q, Pillardy J, Johnson JL, Schweitzer PA, Hernandez AG, Liu L, Acland GM, Aguirre GD. Development and characterization of a normalized canine retinal cDNA library for genomic and expression studies. Invest Ophthalmol Vis Sci. 2006;47:2632–2638. doi: 10.1167/iovs.05-1463. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Mignot E, Faraco J, Kadotani H, Cantanese J, Zhao B, Lin X, Hinton L, Ostrander EA, Patterson DF, de Jong JP. Construction and characterization of an eightfold redundant dog genomic bacterial artificial chromosome library. Genomics. 1999;58:9–17. doi: 10.1006/geno.1999.5772. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, Mauceli E, Xie X, Breen M, Wayne RK, Ostrander EA, Ponting CP, Galibert F, Smith DR, Dejong PJ, Kirkness E, Alvarez P, Biagi T, Brockman W, Butler J, Chin CW, Cook A, Cuff J, Daly MJ, Decaprio D, Gnerre S, Grabherr M, Kellis M, Kleber M, Bardeleben C, Goodstadt L, Heger A, Hitte C, Kim L, Koepfli KP, Parker HG, Pollinger JP, Searle SM, Sutter NB, Thomas R, Webber C, Baldwin J, Abebe A, Abouelleil A, Aftuck L, Ait-Zahra M, Aldredge T, Allen N, An P, Anderson S, Antoine C, Arachchi H, Aslam A, Ayotte L, Bachantsang P, Barry A, Bayul T, Benamara M, Berlin A, Bessette D, Blitshteyn B, Bloom T, Blye J, Boguslavskiy L, Bonnet C, Boukhgalter B, Brown A, Cahill P, Calixte N, Camarata J, Cheshatsang Y, Chu J, Citroen M, Collymore A, Cooke P, Dawoe T, Daza R, Decktor K, Degray S, Dhargay N, Dooley K, Dooley K, Dorje P, Dorjee K, Dorris L, Duffey N, Dupes A, Egbiremolen O, Elong R, Falk J, Farina A, Faro S, Ferguson D, Ferreira P, Fisher S, Fitzgerald M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 20.Makalowski W, Boguski MS. Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proc Natl Acad Sci U S A. 1998;95:9407–9412. doi: 10.1073/pnas.95.16.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moody JA, Famula TR, Sampson RC, Murphy KE. Identification of microsatellite markers linked to progressive retinal atrophy in American Eskimo Dogs. Am J Vet Res. 2005;66:1900–1902. doi: 10.2460/ajvr.2005.66.1900. [DOI] [PubMed] [Google Scholar]
- Heitmann M, Hamann H, Brahm R, Grussendorf H, Rosenhagen CU, Distl O. Analysis of prevalence of presumed inherited eye diseases in Entlebucher Mountain Dogs. Vet Ophthalmol. 2005;8:145–151. doi: 10.1111/j.1463-5224.2005.00339.x. [DOI] [PubMed] [Google Scholar]
- 23.Furukawa T, Morrow EM, Cepko CL. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997;91:531–541. doi: 10.1016/s0092-8674(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 24.Furukawa T, Morrow EM, Li T, Davis FC, Cepko CL. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat Genet. 1999;23:466–470. doi: 10.1038/70591. [DOI] [PubMed] [Google Scholar]
- 25.Sinner D, Rankin S, Lee M, Zorn AM. Sox17 and beta-catenin cooperate to regulate the transcription of endodermal genes. Development. 2004;131:3069–3080. doi: 10.1242/dev.01176. [DOI] [PubMed] [Google Scholar]
- 26.Niimi T, Hayashi Y, Futaki S, Sekiguchi K. SOX7 and SOX17 regulate the parietal endoderm-specific enhancer activity of mouse laminin alpha1 gene. J Biol Chem. 2004;279:38055–38061. doi: 10.1074/jbc.M403724200. [DOI] [PubMed] [Google Scholar]
- 27.Werner P, Raducha MG, Prociuk U, Henthorn PS, Patterson DF. Physical and linkage mapping of human chromosome 17 loci to dog chromosomes 9 and 5. Genomics. 1997;42:74–82. doi: 10.1006/geno.1997.4723. [DOI] [PubMed] [Google Scholar]
- 28.Graphodatsky AS, Yang F, O'Brien PC, Serdukova N, Milne BS, Trifonov V, Ferguson-Smith MA. A comparative chromosome map of the Arctic fox, red fox and dog defined by chromosome painting and high resolution G-banding. Chromosome Res. 2000;8:253–263. doi: 10.1023/a:1009217400140. [DOI] [PubMed] [Google Scholar]
- Sanchez R, Sali A. Comparative protein structure modeling. Introduction and practical examples with modeller. Methods Mol Biol. 2000;143:97–129. doi: 10.1385/1-59259-368-2:97. [DOI] [PubMed] [Google Scholar]
- 30.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 31.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 32.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 33.Buffone GJ, Darlington GJ. Isolation of DNA from biological specimens without extraction with phenol. Clin Chem. 1985;31:164–165. [PubMed] [Google Scholar]
- Webster AR, Heon E, Lotery AJ, Vandenburgh K, Casavant TL, Oh KT, Beck G, Fishman GA, Lam BL, Levin A, Heckenlively JR, Jacobson SG, Weleber RG, Sheffield VC, Stone EM. An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2001;42:1179–1189. [PubMed] [Google Scholar]
- 35.Beltran WA, Rohrer H, Aguirre GD. Immunolocalization of ciliary neurotrophic factor receptor alpha (cntfralpha) in mammalian photoreceptor cells. Mol Vis. 2005;11:232–244. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.