

Abstract
This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Multivariate phenotypes are frequently encountered in genetic association studies. The purpose of analyzing multivariate phenotypes usually includes discovery of novel genetic variants of pleiotropy effects, that is, affecting multiple phenotypes, and the ultimate goal of uncovering the underlying genetic mechanism. In recent years, there have been new method development and application of existing statistical methods to such phenotypes. In this paper, we provide a review of the available methods for analyzing association between a single marker and a multivariate phenotype consisting of the same type of components (e.g., all continuous or all categorical) or different types of components (e.g., some are continuous and others are categorical). We also reviewed causal inference methods designed to test whether the detected association with the multivariate phenotype is truly pleiotropy or the genetic marker exerts its effects on some phenotypes through affecting the others.
1. Introduction
Association studies, where the correlation between a genetic marker and a phenotype is assessed, are useful for mapping genes influencing complex diseases. With reduction of genotyping cost, completion of the HapMap Project [1], and more recently the 1000 Genomes Project [2], genome-wide association studies (GWAS) with several hundred thousands to tens of millions genotyped and/or imputed single nucleotide polymorphisms (SNPs) have become a common approach nowadays to search for genetic determination of complex traits.
In the study of complex diseases, several correlated phenotypes, or a multivariate (MV) phenotype with several components, may be measured to study a disorder or trait. For example, hypertension is evaluated using systolic and diastolic blood pressures; a person’s cognitive ability is usually measured by tests in domains including memory, intelligence, language, executive function, and visual-spatial function. The tests within and between domains are correlated. Most published GWAS only analyzed each individual phenotype separately, although results on related phenotypes may be reported together. Published single phenotype GWAS have successfully identified a large number of novel genetic variants predisposing to a variety of complex traits [3, 4]. However, majority of the identified genetic variants only explain a small fraction of total heritability defined as between individual phenotype variability attributable to genetic factors [4, 5]. It has been hypothesized that current GWAS may be underpowered to detect many genetic variants of moderate-to-small effects. Joint analysis of correlated phenotypes can exploit the correlation among the phenotypes, which may lead to better power to detect additional genetic variants with small effects across multiple traits or pleiotropy effects. Furthermore, joint analysis avoids multiple testing penalty incurred in analyzing each phenotype separately. Therefore, it is important to identify appropriate methods that fully utilize information in multivariate phenotypes to detect novel genetic loci in genetic association studies.
In addition to discovery of novel loci of potential pleiotropy effects, it is also important to detangle the complex relationship between phenotype components and genetic variants One of the frequently asked questions is whether a genetic variant affects multiple phenotypes simultaneously (pleiotropy) or affects one phenotype through affecting another phenotype. In this paper, we review methods for both purposes.
2. Methods for Detecting Association Using Multivariate Phenotypes
For all the methods mentioned in this section, the null hypothesis is no association between a single genetic marker and any components of a multivariate (MV) phenotype; the alternative hypothesis is the genetic maker associated with at least one phenotype component. Here we review methods for an MV phenotype consisting of all continuous, all categorical, or all time-to-event components, and methods for MV phenotypes consisting of a mixture of different types of components.
2.1. Regression Models
Regression models for clustered observations such as linear and generalized mixed effects models, generalized estimating equations, and frailty models can be used to analyze the association of a genetic marker with all continuous, categorical, or survival multivariate phenotypes.
2.1.1. Mixed Effects Models
Mixed effects models such as linear mixed effects model (LME) and generalized linear mixed effects model (GLMM) involve using fixed effects for the genetic marker effect and random effects to account for correlation among multivariate phenotypes [6, 7].
Let yjk denote the kth (k = 1,…, K) continuous component of the K-dimensional phenotype of the jth (j = 1,…, J) individual. Let gj be the genotype of a genetic marker of the jth individual, and X(gj) a score of the genotype. The linear mixed effects model takes the following form:
| (2.1) |
where β0 is the intercept or other genetic or environmental fixed effects; βk is the fixed effect size of X(gj) on the kth phenotype; ηjk(k = 1,…, K) ~ N(0, Σ) are the random effects correlated within jth person; ejk is the random errors iid. . Between any two individuals, ηjk, k = 1,…, K are independent. Within a person, ηjk, k = 1,…, K are correlated. The null hypothesis that the genetic marker is not associated with any phenotype component corresponds to H0 : β1= ,…, βK = 0. The estimation of variance parameters and fixed effect parameters can be obtained using restricted maximum likelihood method (REML) [8, 9].
When yjk is categorical, it can be modeled with generalized mixed effects model (GLMM) as follows:
| (2.2) |
where μ is a link function and μ−1 is its inverse. For Gaussian distributed traits, μ is the identity link, thus (2.2) is identical to the linear mixed effects model (2.1); for binary traits, μ is the logit link μ(x) = ln(x/1 − x). For links other than identity function, the likelihood for this model contains integrals without a close form solution. All existing algorithms for likelihood maximization are either based on theoretical or numerical approximation [10, 11].
The null hypothesis under the LME or GLMM can be tested using the likelihood ratio test or Wald chi-squared test. They can be implemented using SAS PROC Mixed or R lme4 package function lmer(). TheWald chi-squared test statistic takes the form , where β = (β1,…, βK) is estimated using (2.1) or (2.2). For example, Kraja et al. [12] have employed a model similar to (2.1) to the analyses of bivariate continuous metabolic traits. We can also fit a model assuming β1 = ··· = βK = β, that is, E(yjk | ηk) = μ−1(β0 + βX(gj) + ηjk), where a single degree-of-freedom (df) test β̂/se(β̂) can be used to test the null hypothesis. This test can be more powerful than the multi-df Wald chi-squared test if the effect sizes are in the same direction and not very different. It, however, may lack power if the β1,…, βK are very different, especially have different signs and cancel each other out.
2.1.2. Frailty Models
When the phenotypes are correlated survival times, frailty models can be used to fit the association model. Suppose the survival or censoring times are tkj for the kth (k = 1,…, K) phenotype of the jth (j = 1,…, J) individual. Let gj be the genotype of a genetic marker of the jth individual, and X(gj) a score of the genotype as follows:
| (2.3) |
where ηkj(j = 1,…, J) are subject specific random effects following N(0, Σ), and Σ is a K-dimensional correlation matrix. This is the Gaussian frailty model. There is another class of frailty models where exp(ηkj) follows a gamma distribution. A Gaussian or gamma frailty model assuming an exchangeable correlation within a person can be fitted using coxph() in the survival package of R by including a frailty() term in the regressor. In addition, including a cluster() term in coxph() fits generalized estimating equations (GEE) type of model that assumes an independent working correlation matrix [13]. Frailty models with an arbitrary prespecified Σ can be fitted with the coxme() in R coxme package for Gaussian random effects model.
Fitting a mixed effects (frailty) model requires predetermining the correlation matrix Σ of random effects ηjk within jth person. The correlation between the phenotypes yjk within a person is attributable to the random effects ηjk and the fixed effects of the genetic marker. However, since the fixed effects are unknown, it is impossible to directly infer the correlation among the random effects. Misspecifying the correlation among random effects may result in bias in the inference on fixed effects. But the bias seems to be small for genetic association studies [14, 15].
2.1.3. Generalized Estimating Equations
Different from mixed effects model is a class of models called marginal models. Instead of having random effects as regressors in addition to random errors to model correlation in multivariable response, marginal models collapse the random effects and random residual errors in the model. Generalized estimating equations (GEE) [16] solve the quasi-likelihood score function as follows:
| (2.4) |
where , and R(α) is the working correlation matrix for the residual correlation. The variance and covariance of β is estimated with the so-called robust variance estimator [16]. Similar to the LME, single- or multi-df Wald test statistic can be usually used to test that the genetic marker is not associated with any of the phenotypes.
In our experience, GEE results are inflated with low minor frequency SNPs and not as powerful as LME in general [15, 17]. However, GEE is robust to misspecification of response distribution or association model and thus can be used when the LME shows bias or inflation due to these reasons.
2.2. Variable Reduction Method
Variable reduction approaches are in general only applicable to MV phenotype consisting of all continuous phenotypes that are approximately normal distributed. It derives a single or a few new phenotypes that are linear combinations of the original phenotypes, for example,
| (2.5) |
Existing methods include principal components analysis (PCA) where for the first component, ai, i = 1,…, K are coefficients that maximize the variance of Ỹ; principal component of heritability (PCH) with coefficients maximizing the total heritability of Ỹ [18] and penalized PCH applicable to high-dimensional data [19, 20]; and principal components of heritability with coefficients maximizing the quantitative trait locus (QTL) heritability (PCQH) of Ỹ [21–24], that is, the variance explained by the genetic marker. The PCQH approaches are designed to maximize the individual phenotype variation explained by the genetic marker and thus may be more powerful than PCA and PCH in genetic association studies.
2.2.1. PCQH Approaches
The approaches proposed by Lange et al. [21, 25] and Klei et al. [23] involve using a subset of the sample to estimate the coefficients in (2.5) that maximize the correlation between Ỹ and the genetic marker. Specifically, in the estimation sample, the total phenotype variance is partitioned into QTL variance and residual variance as follows:
| (2.6) |
where Vp is the K × K total phenotype variance-covariance matrix, Vq the QTL variance matrix, and Vε the residual variance matrix. Let A = (α1,…, αK), then the variance of Ỹ = AtY explained by the genetic marker is
| (2.7) |
A that maximizes can be obtained by solving the following generalized eigen system [18]:
| (2.8) |
Vq = var(β1X,…, βKX) can be approximated by Γ11tΓ, where Γ = diag(|β1|σx,…, |βK|σx), σx is the sample standard deviation of the score of genotype X(g) across all individuals, βi is estimated using the least squared estimator of Yi = α + βiX(g) + ε, and 1 = (sign(β1),…, sign(βK)).
Lange et al. [21, 25] approaches are only applicable to family-based association design. They suggest using the noninformative families or parental genotypes to estimate A because these data will not contribute directly to the family-based association tests (FBAT). Then perform FBAT of Ỹ on X(g). However, FBAT has low power in the absence of population stratification [26] compared to population based approaches. Klei et al’s. [23] is a population-based association approach where they randomly split the sample into two subsets: one used to estimate A, the other used to test the association of Ỹ with X(g) via a linear regression model: Ỹ α + βX(g) + ε. This ensures valid P value in the association test.
2.2.2. Canonical Correlation Analysis
Canonical correlation analysis seeks coefficients so that the squared correlation between Ỹ in (2.5), and the score of genetic marker, X(g), is maximized. Here ρ̂ = corr(Ỹ,X) is called estimated canonical correlation. To obtain ρ̂, the covariance matrix of Y and X is partitioned as follows:
| (2.9) |
where ΣYY is the K × K matrix of the variance-covariance matrix of Y, ΣYX and its transpose ΣXY are K × 1 and 1 × K matrix of the covariance matrix between Y and X, ΣXX is the variance of X, a scalar. All these submatrices can be estimated using the respective sample co-variance matrix. The canonical correlation, ρ̂ = ΣXYA/(AtΣYYAΣXX)1/2, is solved as the squared root of the largest eigenvalue of , and the corresponding eigenvector A contains the coefficients for constructing Ỹ. multivariate analysis of variance (MANOVA) tests correspond to evaluating canonical correlation. Table 1 details the relationship between ρ̂ and commonly reported test statistics in MANOVA of a multivariate phenotype Y on X(g) [27].
Table 1.
Relationship between MANOVA test statistics and canonical correlation for association test of multivariate phenotype and a genetic marker.
| MANOVA test | f(ρ̂) |
|---|---|
| Roy’s largest root | ρ̂2 |
| Hotelling-Lawley trace | ρ̂2/1 − ρ̂2 |
| Wilks lambda | 1 − ρ̂2 |
| Pillai-Bartlett trace | ρ̂2 |
These tests are implemented in SAS PROC GLM and R function summary.manova(). As part of the PLINK package specifically developed for genetic analysis, Ferreira et al. [24] implemented the Wilks lambda, and its P value is obtained from F-approximation F = (ρ̂2/K)/((1 − ρ̂2)/(n − K − 1)).
Canonical correlation analysis shares similarity with PCQH [23] in that both estimate a linear combination of original phenotypes, so that the genotype score explains most of the variation (in terms of percent of total variance and squared correlation, resp.) of the new phenotype. The difference between the two approaches is that the canonical correlation analysis evaluates squared correlation using whole sample, while PCQH estimates the loadings using a subset of the sample and test the association in the rest of the sample. Extensive simulation studies performed in [28]. The author of [28] showed that MANOVA viaWilk’s lambda was substantially more powerful than PCQH [23] with K = 5 phenotypes.
2.3. Combining Test Statistics from Univariate Analysis
An alternative way to analyze multivariate phenotypes is to perform univariate phenotype-genotype association test for each phenotype individually and then combine the test statistics from the univariate analysis. The advantage of such approach is the simplicity, that is, the methods to deal with univariate phenotypes are generally simpler than methods for MV phenotypes. It is especially useful for analyzing multivariate phenotype consisting of components of different types of distributions such as continuous, dichotomous, and survival. Regression methods for analyzing such multivariate phenotype are generally complicated and not trivial to implement for MV phenotype with dimension > 2, see for example, [29, 30].
In recent years, researchers have generated large amount of univariate GWAS results for a variety of complex traits. Methods that combine the univariate results of multiple traits to detect genetic markers associated with multiple phenotypes are appealing.
2.3.1. Methods for Homogeneous Genetic Effects across Phenotypes
Assume that T = [T1, T2,…, TK]T is a vector of K test statistics obtained from association analyses of each individual component phenotype against the genetic marker. Assume that T follows a multivariate normal distribution with mean τ = (τ1, τ2,…, τK)T and a nonsingular covariance matrix Σ. For example, T can be the β coefficients from least squared regression model for individual components or the t-test statistics from the regression models. The null hypothesis of no association to any phenotypes isH0: τ = (τ1, τ2,…, τK)T = 0. O’Brien [31–33] suggested the following linear combination of T1, T2,…, TK, with weight e = (1, 1,…, 1)T of length K:
| (2.10) |
when τ1 = τ2 = ··· = τK ≠ 0 (2.10) is the most powerful test among a class of tests statistics that are linear combinations of T1, T2,…, TK. Under the null hypothesis, S follows the normal distribution with mean 0 and variance eTΣ−1e. To estimate Σ with GWAS results, Yang et al. [34] suggested using the sample covariance matrix of the statistics on a large number of SNPs genomewide with little or no linkage disequilibrium among them (say HapMap r2 < 0.1).
The power of O’Brien’s method depends on the assumption τ1 = τ2 = ··· = τK. When the means are very different or with opposite signs, O’Brien’s method may not be efficient. Yang et al. proposed a sample splitting approach that replaces the uniform weight eT by weights w estimated using a portion of the sample and only used the remaining sample to estimate T in (2.10), that is, S = wTΣ−1T. To overcome the variability introduced by a random sample splitting, Yang et al. also evaluated a cross-validation approach that averages the test statistics of 10 random splitting samples. The results showed that when τ1, τ2,…, τK are of different magnitude or in opposite directions, O’Brien’s method is less powerful than Yang et al., which indicates room for improvements for O’Brien’s method. However, the sample splitting and cross-validation methods are less powerful than O’Brien’s method with homogeneous effect sizes.
2.3.2. Methods for Heterogeneous Genetic Effects across Phenotypes
The limitation of O’Brien statistic is that it is not powerful for heterogeneous effects across multiple phenotypes, especially if some effects are of opposite directions. Another class of statistics that takes a quadratic form of the vector of the individual association statistic may overcome the limitation. For example, the following Wald chi-squared type test statistic was mentioned in Xu et al. [32].
| (2.11) |
The difference between (2.10) and (2.11) is that the vector e = (1, 1,…, 1)T is replaced by the T in (2.11). S follows a chi-squared distribution with degree of freedom equal to the number of the phenotypesK or rank of Σ if it is not full rank. Due to the “curse of dimensionality,” power of (2.11) is diminishing with the increased number of phenotypes. Similar problem has been extensively studied and discussed in high-dimensional data analysis field and most recently in the analyses of multiple rare variants. Borrowing ideas from these fields, we propose the following test statistic that may be more powerful than (2.10) and (2.11) with heterogeneous effects.
| (2.12) |
The difference between (2.12) and (2.11) is that there is no variance-covariance matrix in (2.12). This statistic was first proposed by Pan [35] to analyze multiple rare or common variants against a single phenotype, where the ti is the beta coefficient for the ith genetic variant. Different from Pan [35], here ti is the association statistic for the ith phenotype with a single marker. Based on the groundwork of Zhang [36], Pan [35] pointed out that the distribution of (2.11) is a mixture of single degree-of-freedom chi-squared variates, where cis are the eigen values of Σ, that is, the variance-covariate matrix of ti. The distribution of (2.12) can be well approximated by with
| (2.13) |
The P value is calculated as . The degree of freedom of the Ssq may be less than K with highly correlated phenotypes. In addition, (2.12) does not have the problem of instability observed for (2.10) and (2.11) when some of the components are highly correlated (in one of our applications, a correlation ~0.7 has resulted in inflated results for (2.10) and (2.11)). We have developed an R package CUMP (combining univariate results of multivariate phenotypes) that have implemented all the aforementioned combining statistics approaches. The software can be downloaded at (http://people.bu.edu/qyang/), and a short report of this software is submitted [37].
3. Identifying Pleiotropy
All the aforementioned methods can be used to detect association that is potentially due to pleiotropy. But they do not answer the question if the detected association is truly pleiotropy, that is, the marker locus affects all components of the MV phenotype directly. The detect association can affect some of the phenotypes and/or mediate through these phenotypes to affect the other phenotypes. Vansteelandt et al. [38] illustrated potential confounding mechanism between the genotype of a genetic marker and a phenotype using a causal diagram (Figure 1): the association between the genotype, denoted as G, and the response phenotype Y can occur through the paths connecting the two variables along all unbroken sequences of edges regardless of the direction of the arrows, given that there are no colliders (i.e., variables in which two arrows converge, e.g., variables K and L in Figure 1) in the sequence [39].
Figure 1.
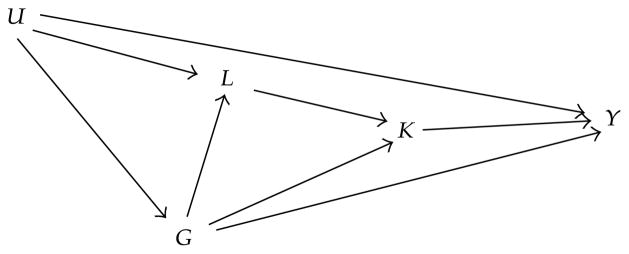
Causal diagram showing potential confounding mechanisms for the association between the genotype of a genetic marker G and the phenotype Y. The variable K denotes the intermediate phenotype, L the collection of known environmental and genetic risk factors, and U the unknown environmental and genetic risk factors such as population stratification and unknown genetic variants in linkage disequilibrium with G.
The genotype G may be associated with Y due to (1) direct causal effect, that is, G → Y; (2) through intermediate phenotype or risk factors, that is, G → K → Y or G → L → K → Y; (3) because of confounding factors, that is, G ← U → Y or G ← U → L → K → Y.
The authors showed that two commonly used approaches to detangle the complex relationship between phenotypes, genotype, and traditional risk factors are flawed. The first commonly used approach derives the residuals of Y regressing on K, say Ỹ = Y−βK, and then the association between G and Ỹ is tested. The disadvantage of this approach is that not only the direct causal effect of K on Y is removed but also any indirect effect of K on Y through G (e.g., K ← G → Y and Y ← U ← L ← G → K) and other factors (e.g., K ← L → U → Y). Therefore β may be biased in the presence of confounding factors which leads to biased test of G with Ỹ.
The second commonly used approach tests the direct effect of G on Y in a regression model including K and L as covariates. Adjustment of K removes the relationship between G and Y through G → K → Y; however, becauseK is a collider (Figure 1), the adjustment of K induces a spurious association [39, 40] along the path G → K → L ← U → Y. Additionally, adjusting for L induces spurious associated through the path G → L ← U → Y.
To overcome the limitation of the two commonly adapted approaches, Vansteelandt et al. [38] proposed a least squared regression model to estimate the direct effect size of K on Y. This regression model includes the suspected intermediate phenotype, the score of the genetic marker genotype, X(G), and other common risk factors between the two phenotypes as regressors:
| (3.1) |
The estimated effect size of the phenotype represents the direct effect of theK on Y, that is, not confounded by the effect of X mediated through any of the covariates. Then, a new phenotype is created as the residual of the response subtract the effect of K only . Then, whether the G only exerts its effect on Y through K can be tested using any standard association test statistic between the residual and the X. A negative result indicates that G only exerts its effect on Y through K while a positive result indicates that the G has a direct effect on Y and/or a spurious effect through other confounders. Extensions of the method to dichotomous and time-to-event outcomes have been proposed [41, 42].
4. Discussion
In this paper, we reviewed methods available for joint analyzing correlated phenotypes in genetic association studies. Some of these methods are designed to detect potential association with multiple phenotypes (pleiotropy), while the others are designed to test whether the detected association with the MV phenotype is truly pleiotropy or the genetic marker exerts its effects on some phenotypes through affecting the others.
For methods designed to detect association, each method has its own pros and cons. Random effects model requires knowledge of residual correlation, and misspecifying the correlation may incur inflation or power loss. Generalized estimating equations are robust to misspecification of residual correlation, but it is inflated for low-frequency variants and less powerful than random effects model in our experience. Variable reduction approaches are appealing because correlated outcomes are reduced to a single or fewer number of uncorrelated outcomes. However, in the presence of missing data in the outcomes, individuals with missing data do not contribute to the analysis, which may result in power loss. The approaches combining univariate association results are more flexible than the other methods especially when MV phenotypes consist of a mixture of continuous, discrete, and/or time-to-events data. Regression approaches have been developed to deal with such phenotypes. But they are generally complicated and few available software implements these methods. Since univariate association results are used, individuals with incomplete observations still contribute to the analysis of available phenotypes. Simulations on all continuous phenotypes indicated that the power of O’Brien’s method, one of the approaches combining univariate association results is similar to regression and variable reduction methods when the effects size are similar across multiple phenotypes [34].
All the approaches introduced here for population based approaches assume unrelated individuals. When there are related individuals in the data, not accounting for family structure can result in inflation or power loss. Extension of introduced-methods to account for family data are possible. For example, one may add a random effect in mixed effects model to account for family structure. For approaches combining univariate association results, a model that account for family structure need be used in the univariate analyses.
In terms of computational cost, mixed effects models may be most time consuming since maximization of likelihood is required.
Finally, it has been shown that traditional causal inference is useful in distinguishing true pleiotropy from other mechanisms that also result in genetic association with multiple phenotypes. A related causal inference in recent genetic literature is Mendelian randomization test [43–45]. This approach can be used to infer whether an intermediate phenotype has a causal effect on an outcome phenotype, using genetic marker(s) in association with the intermediate phenotype. Unlike a phenotype that is subject to the influence of uncontrolled environmental factors and/or reverse causation of another phenotype, genotype(s) of genetic marker(s) is(are) free of influence of environmental factors and reverse causation. For this approach, marker genotype(s) is(are) used as an instrument variable. This test requires that there is no pleiotropy effect of the genetic marker on outcome phenotype. Association of the genotype and outcome phenotype indicates that the intermediate phenotype may causally affect the outcome phenotype.
5. URLs to Software Mentioned in This Paper
SAS: http://www.sas.com/,
CUMP: http://cran.r-project.org/web/packages/CUMP/index.html,
coxme: http://cran.r-project.org/web/packages/coxme/index.html,
gee: http://cran.r-project.org/web/packages/gee/index.html,
survival: http://cran.r-project.org/web/packages/survival/index.html,
lme4: http://cran.r-project.org/web/packages/lme4/index.html,
Acknowledgments
Q. Yang’s work is supported by the National Heart, Lung, and Blood Institute’s Framingham Heart Study (Contract no. N01-HC-25195) and Grant no. R01HL093328 and R01HL093029. Y. Wang’s work is supported by NIH Grants nos. R03AG031113-01A2 and 1R01NS073671-01 1.
References
- 1.International HapMap Consortium. Frazer KA, Ballinger DG, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hindorff LA, Junkins HA, Hall PN, Mehta JP, Manolio TA. A catalog of published genome-wide association studies. National Human Genome Research Institute; 2011. http://www.genome.gov/gwastudies/ [Google Scholar]
- 4.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nature Reviews Genetics. 2010;11(6):446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–974. [PubMed] [Google Scholar]
- 7.Fitzmaurice GM, Laird NM. A likelihood-based method for analysing longitudinal binary responses. Biometrika. 1993;80(1):141–151. [Google Scholar]
- 8.Patterson HD, Thompson R. Recovery of inter-block information when block sizes are unequal. Biometrika. 1971;58:545–554. [Google Scholar]
- 9.Harville DA. Maximum likelihood approaches to variance component estimation and to related problems. Journal of the American Statistical Association. 1977;72(358):320–340. [Google Scholar]
- 10.Breslow NE, Clayton DG. Approximate inference in generalized linear mixed models. Journal of the American Statistical Association. 1993;88:9–25. [Google Scholar]
- 11.Bates DM, DebRoy S. Linear mixedmodels and penalized least squares. Journal ofMultivariate Analysis. 2004;91(1):1–17. [Google Scholar]
- 12.Kraja AT, Vaidya D, Pankow JS, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED Consortium. Diabetes. 2011;60(4):1329–1339. doi: 10.2337/db10-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Therneau TM, Grambsch PM, Pankratz VS. Penalized survival models and frailty. Journal of Computational and Graphical Statistics. 2003;12(1):156–175. [Google Scholar]
- 14.Pfeiffer RM, Hildesheim A, Gail MH, et al. Robustness of inference on measured covariates to misspecification of genetic random effects in family studies. Genetic Epidemiology. 2003;24(1):14–23. doi: 10.1002/gepi.10191. [DOI] [PubMed] [Google Scholar]
- 15.Chen MH, Liu X, Wei F, et al. A comparison of strategies for analyzing dichotomous outcomes in genome-wide association studies with general pedigrees. Genetic Epidemiology. 2011;35(7):650–657. doi: 10.1002/gepi.20614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang KY, Zeger SL. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73(1):13–22. [Google Scholar]
- 17.Cupples LA, Arruda HT, Benjamin EJ, et al. The Framingham Heart Study 100K SNP genomewide association study resource: overview of 17 phenotype working group reports. BMC Medical Genetics. 2007;8(supplement 1) doi: 10.1186/1471-2350-8-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ott J, Rabinowitz D. A principal-components approach based on heritability for combining phenotype information. Human Heredity. 1999;49(2):106–111. doi: 10.1159/000022854. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Fang Y, Jin M. A ridge penalized principal-components approach based on heritability for high-dimensional data. Human Heredity. 2007;64(3):182–191. doi: 10.1159/000102991. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Fang Y, Wang S. Clustering and principal-components approach based on heritability for mapping multiple gene expressions. BMC Proceedings. 2007;1(supplement 1):S121. doi: 10.1186/1753-6561-1-s1-s121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lange C, van Steen K, Andrew T, et al. A family-based association test for repeatedly measured quantitative traits adjusting for unknown environmental and/or polygenic effects. Statistical Applications in Genetics and Molecular Biology. 2004;3(1):1544–6115. doi: 10.2202/1544-6115.1067. [DOI] [PubMed] [Google Scholar]
- 22.Lange C, DeMeo DL, Laird NM. Power and design considerations for a general class of family-based association tests: quantitative traits. American Journal of Human Genetics. 2002;71(6):1330–1341. doi: 10.1086/344696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klei L, Luca D, Devlin B, Roeder K. Pleiotropy and principal components of heritability combine to increase power for association analysis. Genetic Epidemiology. 2008;32(1):9–19. doi: 10.1002/gepi.20257. [DOI] [PubMed] [Google Scholar]
- 24.Ferreira MAR, Purcell SM. A multivariate test of association. Bioinformatics. 2009;25(1):132–133. doi: 10.1093/bioinformatics/btn563. [DOI] [PubMed] [Google Scholar]
- 25.Lange C, Silverman EK, Xu X, Weiss ST, Laird NM. A multivariate family-based association test using generalized estimating equations: FBAT-GEE. Biostatistics. 2003;4(2):195–206. doi: 10.1093/biostatistics/4.2.195. [DOI] [PubMed] [Google Scholar]
- 26.Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177(1):577–585. doi: 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller KE, Peterson BL. Practical methods for computing power in testing the multivariate general linear hypothesis. Computational Statistics and Data Analysis. 1984;2(2):143–158. [Google Scholar]
- 28.Wu H. PhD thesis. Boston University; Boston, Mass, USA: 2009. Methods for genetic association studies using longitudinal and multivariate phenotypes in families. [Google Scholar]
- 29.Fitzmaurice GM, Laird NM. Regression models for mixed discrete and continuous responses with potentially missing values. Biometrics. 1997;53(1):110–122. [PubMed] [Google Scholar]
- 30.Liu J, Pei Y, Papasian CJ, Deng HW. Bivariate association analyses for the mixture of continuous and binary traits with the use of extended generalized estimating equations. Genetic Epidemiology. 2009;33(3):217–227. doi: 10.1002/gepi.20372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Brien PC. Procedures for comparing samples with multiple endpoints. Biometrics. 1984;40(4):1079–1087. [PubMed] [Google Scholar]
- 32.Xu X, Tian L, Wei LJ. Combining dependent tests for linkage or association across multiple phenotypic traits. Biostatistics. 2003;4(2):223–229. doi: 10.1093/biostatistics/4.2.223. [DOI] [PubMed] [Google Scholar]
- 33.Wei LJ, Johnson WE. Combining dependent tests with incomplete repeated measurements. Biometrika. 1985;72(2):359–364. [Google Scholar]
- 34.Yang Q, Wu H, Guo CY, Fox CS. Analyze multivariate phenotypes in genetic association studies by combining univariate association tests. Genetic Epidemiology. 2010;34(5):444–454. doi: 10.1002/gepi.20497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan W. Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genetic Epidemiology. 2009;33(6):497–507. doi: 10.1002/gepi.20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang JT. Approximate and asymptotic distributions of chi-squared-type mixtures with applications. Journal of the American Statistical Association. 2005;100(469):273–285. [Google Scholar]
- 37.Liu X, Yang Q. CUMP: an R package for analyzing multivariate phenotypes in genetic association studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vansteelandt S, Goetgeluk S, Lutz S, et al. On the adjustment for covariates in genetic association analysis: a novel, simple principle to infer direct causal effects. Genetic Epidemiology. 2009;33(5):394–405. doi: 10.1002/gepi.20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearl J. Causal diagrams for empirical research. Biometrika. 1995;82(4):669–688. [Google Scholar]
- 40.Robins JM. Data, design, and background knowledge in etiologic inference. Epidemiology. 2001;12(3):313–320. doi: 10.1097/00001648-200105000-00011. [DOI] [PubMed] [Google Scholar]
- 41.Lipman PJ, Liu KY, Muehlschlegel JD, Body S, Lange C. Inferring genetic causal effects on survival data with associated endo-phenotypes. Genetic Epidemiology. 2011;35(2):119–124. doi: 10.1002/gepi.20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vansteelandt S. Estimation of controlled direct effects on a dichotomous outcome using logistic structural direct effect models. Biometrika. 2010;97(4):921–934. [Google Scholar]
- 43.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? International Journal of Epidemiology. 2003;32(1):1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 44.Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Smith GD. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Statistics in Medicine. 2008;27(8):1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 45.McKeigue PM, Campbell H, Wild S, et al. Bayesian methods for instrumental variable analysis with genetic instruments (“Mendelian randomization”): example with urate transporter SLC2A9 as an instrumental variable for effect of urate levels on metabolic syndrome. International Journal of Epidemiology. 2010;39(3):907–918. doi: 10.1093/ije/dyp397. [DOI] [PMC free article] [PubMed] [Google Scholar]