

Abstract
Fragile X Syndrome is caused by loss of the FMRP translational regulator. A current hypothesis proposes that FMRP functions downstream of mGluR signaling to regulate synaptic connections. Using the Drosophila disease model, we test relationships between dFMRP and the sole Drosophila mGluR (DmGluRA) by assaying protein expression, behavior and neuron structure in brain and NMJ; in single mutants, double mutants and with an mGluR antagonist. At the protein level, dFMRP is upregulated in dmGluRA mutants, and DmGluRA is upregulated in dfmr1 mutants, demonstrating mutual negative feedback. Null dmGluRA mutants display defects in coordinated movement behavior, which are rescued by removing dFMRP expression. Null dfmr1 mutants display increased NMJ presynaptic structural complexity and elevated presynaptic vesicle pools, which are rescued by blocking mGluR signaling. Null dfmr1 brain neurons similarly display increased presynaptic architectural complexity, which is rescued by blocking mGluR signaling. These data show that DmGluRA and dFMRP convergently regulate presynaptic properties.
Keywords: Fragile X Syndrome, mental retardation, brain, translation regulator, glutamatergic signaling, axon, synapse, synaptic vesicle
Introduction
Fragile X syndrome (FXS) is a broad-spectrum neurological disease with symptoms including hyperactivity, hypersensitivity to sensory stimuli, mental retardation and autism (Turner et al. 1996; Rogers et al. 2001; Visootsak et al. 2005). The disease is caused solely by loss of expression of the fragile X mental retardation 1 (fmr1) gene, which encodes FMRP, an mRNA-binding protein that associates with polyribosomes and acts as a negative translational regulator (Garber et al. 2006). A prominent neuronal structure defect found in FXS patients and fmr1 knockout mice is denser, longer and immature appearing postsynaptic dendritic spines in the cortex, a defect also found in other mental retardation diseases (Purpura 1974; Hinton et al. 1991; Irwin et al. 2002; Grossman et al. 2006). FMRP similarly negatively regulates presynaptic growth and differentiation, with increased filipodial extensions from axon growth cones in an fmr1 mouse culture system (Antar et al., 2006) and altered presynaptic synaptogenesis in a mosaic mouse model of FXS (Hanson and Madison, 2007). FMRP also regulates synaptic functional plasticity. A prominent defect in fmr1 knockout mice is enhanced long term depression (LTD) in the hippocampus, a group I class 5 metabotropic glutamate receptor (mGluR) signaling-induced event (Huber et al. 2002). This form of LTD requires de novo protein synthesis triggered by mGluR signaling, which is sensitive to translational inhibitors and dependent on FMRP (Huber et al. 2000; Koekkoek et al. 2005; Nosyreva and Huber 2006). Based on these findings, a hypothesis has been proposed suggesting that FMRP regulates synaptic properties by regulating the level of protein synthesis downstream of mGluR signaling; “the mGluR theory of FXS” (Bear et al. 2004; Pfeiffer and Huber 2006).
Drosophila provides a powerful genetic model system to test this hypothesis. The Drosophila genome contains only one fmr1 homolog (dfmr1) and encodes a single functional mGluR (DmGluRA) (Parmentier et al. 1996; Wan et al. 2000; Zhang et al. 2001; Bogdanik et al. 2004). Null mutants of both genes are viable and therefore accessible to neurological studies throughout life. As in the mammalian system, null dfmr1 mutants display structural overgrowth and overbranching of both presynaptic and postsynaptic processes, which has been well-characterized in both the larval glutamatergic neuromuscular junction (NMJ) and the adult central brain Mushroom Body (MB) learning/memory center (Zhang et al. 2001; Lee et al. 2003; Michel et al., 2004; Pan et al. 2004; McBride et al., 2005). Loss of dFMRP also causes altered synaptic differentiation and/or function in the visual system, brain MB and NMJ (Zhang et al. 2001; Pan et al. 2004; Zhang and Broadie 2005). DmGluRA is synaptically localized in both CNS synaptic neuropil and at the NMJ (Parmentier et al. 1996; Bogdanik et al. 2004). DmGluRA is a sequence ortholog of mammalian group II/III mGluRs but, as the sole Drosophila mGluR, presumably takes on all GluR signaling functions subdivided between group I–III mGluRs in mammals. Null dmGluRA mutants display altered synaptic architecture at the NMJ and also strong defects in activity-dependent functional plasticity at the NMJ (Bogdanik et al. 2004). Roles of DmGluRA in the CNS have not yet been investigated. These data show that dFMRP and DmGluRA modulate synaptic architecture and function in the same or closely related processes.
Treatment with a group I mGluR antagonist (MPEP) can rescue two major FXS behavioral phenotypes in fmr1 knockout mice, habituation in open field tests and increased sensitivity to audiogenic seizures (Yan et al. 2005). Similarly, treating dfmr1 null mutant flies with either MPEP (Group I mGluR antagonist), or LY341495, MPPG, or MTPG (Group II/III mGluR antagonists), can effectively rescue behavioral and gross brain morphological defects, including male courtship learning/memory defects and β-lobe fusion in the Mushroom Body (McBride et al. 2005). These results have strongly supported a mechanistic relationship between DmGluRA signaling and dFMRP function. The fact that antagonists of different mammalian mGluR classes can equally rescue dfmr1 null phenotypes (McBride et al. 2005), suggests that DmGluRA does indeed mediate group 1 mGluR signaling or, alternatively, that the connection between FMRP function and mGluR signaling might be broader than is currently appreciated. The identification and elucidation of the molecular and cellular relationships between mGluR signaling and FMRP will significantly increase understanding on the mechanism of FXS, and provide insights into potential therapeutic treatments for the disease.
In this study, we examine mechanistic relationships between DmGluRA signaling and dFMRP function at genetic, molecular and cellular levels. We find dFMRP protein increased in dmGluRA null mutant CNS, and DmGluRA protein similarly increased in dfmr1 null mutants, showing a molecular feedback regulation mechanism. DmGluRA and dFMRP interact in the regulation of coordinated movement behavior, and in the regulation of synaptic architecture at the NMJ. Ultrastructure analyses show elevated synaptic vesicle pools in dfmr1 null synaptic boutons, which are largely restored to normal by also removing DmGluRA function. In the brain, blocking DmGluRA signaling rescues single-cell architectural defects in dfmr1 null neurons. Taken together, these data suggest that convergent cross-talk between DmGluRA signaling and dFMRP function controls neuron structure and presynaptic differentiation.
Results
DmGluRA and dFMRP mutual negative feedback loop
Previous work has shown that dFMRP and DmGluRA are both localized in the nervous system (Parmentier et al. 1996; Ramaekers et al. 2001; Zhang et al. 2001; Dockendorff et al. 2002), but the spatial relationship between them has not been clear. Just like mammalian FMRP, dFMRP appears to be pan-neuronally expressed with similar levels in most/all neurons, and is primarily localized in the neuronal soma cytoplasm with little/no detectable expression in the nucleus and only very faint expression in distal neuronal processes (Verheij et al., 1993; Feng et al., 1997; Zhang et al., 2001; Antar et al., 2004; Pan et al., 2004; Bagni and Greenough 2005). In the Drosophila larval CNS, dFMRP is concentrated in all neuronal soma, including midline motor neuron soma, but largely undetectable in the synaptic neuropil (Fig. 1A). In direct contrast, DmGluRA is specifically concentrated at synaptic connections (Bogdanik et al., 2004). In the Drosophila larval CNS, DmGluRA is undetectable in neuronal soma but appears throughout the synaptic neuropil (Fig. 1A). DmGluRA therefore also appears pan-neuronal, although this is impossible to say for sure in the absence of neuronal soma localization. Thus, dFMRP is primarily localized in neuronal cell bodies, whereas DmGluRA is primarily localized in synaptic neuropil (Fig. 1A). The two proteins have a largely non-overlapping expression pattern in the same neurons.
Figure 1. DmGluRA and dFMRP display mutual negative regulation.

A. Representative images of dFMRP (green) and DmGluRA (red) expression patterns in third instar larval CNS in wildtype (WT), dfmr1 null mutant, and dmGluRA null mutant. Both dFMRP and DmGluRA are highly enriched in CNS neurons, with dFMRP primary localized in neuronal cell bodies (cb), and DmGluRA primary localized in synaptic neuropil (np). Expression was assayed for each protein in the respective opposing null mutant. dFMRP level is clearly increased in dmGluRA null mutants, and DmGluRA level is also increased in dfmr1 null mutants. The scale bar is 10µm. B. Fluorescence intensity quantification of dFMRP and DmGluRA expression from (A). See Methods for detailed description of quantification method. C. Representative Western Blots showing dFMRP and DmGluRA expression compared to an α-tubulin loading control. The dFMRP level increases in the dmGluRA null, and DmGluRA expression increases in the dfmr1 null.
The mGluR theory of FXS proposes that FMRP expression is negatively regulated by mGluR signaling (Bear et al. 2004; Pfeiffer and Huber 2006). We therefore first asked whether DmGluRA signaling may regulate dFMRP by examining dFMRP protein expression in the dmGluRA112b null mutant with a companion precise excision line, 2b, as the genetic background control (Fig. 1A; green). The antibody against dFMRP does not produce any signal in dfmr1 null mutants (data not shown), proving antibody specificity. The level of dFMRP is clearly and consistently upregulated in dmGluRA null mutants (Fig. 1A). We quantified fluorescent intensity in conrtol and dmGluRA null CNS neuronal soma layers, using maximal projections of confocal Z-stacks. There is a significant (P=0.025) increase in dFMRP signal in the dmGluRA null compared to control (dmGluRA/WT=1.21±0.09, N=18 for each genotype; Fig. 1B). There is no detectable change of the expression pattern, but rather just heightened dFMRP levels in neuronal soma.
Given this evident regulation, we next assayed whether dFMRP may also regulate DmGluRA protein levels. The dfmr150M null mutant was assayed, with w1118 as the genetic background control (Fig. 1A; red). The antibody against DmGluRA does not produce any signal in dmGluRA112b (data not shown), proving antibody specificity. The synaptic neuropil expression of DmGluRA is significantly (P=0.03) increased in the dfmr1 null mutants (dfmr1/WT=1.2±0.07, N=12 for each genotype; Fig. 1B). As in the case of dFMRP, there is no detectable change in the pattern of expression, just an increase in the DmGluRA protein level in the synaptic neuropil. We also tested these two proteins expression by Western Blotting of the dissected larval CNS, and detected elevated protein levels consistent with the tissue staining results (Fig. 1C). Over-expression of dFMRP by neuronal specific elav-GAL4 did not show significantly change the DmGluRA protein level, and similarly DmGluRA over-expression did not significantly change the dFMRP protein level (data not shown).
To further examine the interaction between dFMRP and DmGluRA, we performed immunocytochemistry at an electron microscope level on ultrathin sections taken through the third instar CNS (Fig. 2). Both primary antibodies were visualized by secondary antibodies directly conjugated to gold beads (10nm diameter). In null mutant controls, the background level of adherent gold beads was virtually undetectable (data not shown), once again demonstrating antibody specificity. The dFMRP label was localized to the somal cytoplasm in neurons, often with gold particles clustered on or near large (~100nm), uniform dense-core granules resembling stress granules or P-bodies (Schneider et al., 2006; Parker and Sheth, 2007; Fig. 2A, top, arrows). In the dmGluRA112b null mutant, the density of dFMRP gold particle label was clearly and significantly (P=0.039) increased compared to control (WT=2.34±0.63 gold particles/µm2, N=8; dmGluRA=6.78±2.16 gold particles/µm 2, N=10; Fig. 2B).
Figure 2. ImmunoEM analyses of DmGluRA and dFMRP in the CNS.

A. Electon microscopy immunogold localization of dFMRP and DmGluRA in the wandering 3rd instar larval ventral nerve cord. Wildtype (WT), dfmr1 and dmGluRA null mutants immuno-labeled with 10nm gold particles against dFMRP (top) and DmGluRA (bottom). Arrows indicate representative clusters of immunogold particles in each case. Insets show higher magnification images of the gold label in each case. In dmGluRA mutants, there is a clear increase of gold particle number of dFMRP labeling. Similarly, a clear increase of DmGluRA labeling is also observed in dfmr1 null mutants. The scale is 250nm. B. Quantification of gold particle density for each genotype. See Methods for detailed description of quantification method.
The CNS neuropil is densely packed with many synaptic structures, including prominent synaptic terminals filled with mitochondria and synaptic vesicles. The synaptic arborizations are very small (<500nm mean diameter) with a packing density that exceeds by several fold the highest density found among vertebrate neurons (Strausfeld, 1998). Immunogold labeling for DmGluRA shows a moderate level of protein associated with synaptic membranes (Fig. 2A, bottom, arrows). In the dfmr150M null mutant, the density of DmGluRA gold particle label was very significantly (P=0.0007) increased compared to the genetic control (WT=0.59±0.15 gold particles/µm2, N=8; dfmr1=17.53±7.36 gold particles/µm2, N=8; Fig. 2B). Taken together, the fluorescent confocal microscopy, immunogold electron microscopy and Western Blot data all reveal that dFMRP and DmGluRA negatively regulate each other’s expression in the same neurons. This reciprocal regulation between dFMRP and DmGluRA shows a negative feedback mechanism exists between DmGluRA signaling and dFMRP function to regulate the expression of both proteins.
DmGluRA and dFMRP genetically interact in behavioral movement regulation
Behavioral tests of coordinated movement involve the integration of several sensory input modalities, coupled to sequential motor output driving the appropriate movement response. We previously devised a simple but effective test called the “roll-over” assay to measure such coordinated behavior in the Drosophila larva (Bodily et al. 2001). This assay involves placing wandering third instar larva on a smooth agar plate, turning the animal to a totally inverted position and then measuring the time the animal takes to fully right itself to the normal position. This behavior requires the integration of sensory stimuli and coordinated bilateral motor control to produce the necessary sequence of movements. We used this assay to test dfmr1 and dmGluRA single null mutants for possible defects in coordinated movement behavior, and then double null mutant combinations for modulation of behavioral responses.
Loss of DmGluRA signaling strongly impairs coordinated movement behavior, with the average response time lengthened by 48% (Fig. 3). In individual animals, the behavioral defect was clearly evident as a combination of inappropriate movement responses and defects in cooperative motor control. The dmGluRA mutant animals appear to “struggle” during a period of continuous, spastic muscle contractions that do not aid in the turning behavior, while wildtype animals always display smooth, cooperative motor control to turn over quickly. Quantitatively, dmGluRA null mutants display a very significantly (P<0.0001) slowed performance compared to the genetic wildtype (WT) control in this coordinated behavior (WT=12.4±0.7 sec, N=46; dmGluRA=18.4±1.15 sec, N=49; Fig. 3). In contrast, removal of dFMRP alone causes no defect in this coordinated behavior. We used two independent dfmr1 null mutant lines, dfmr150M and dfmr13 (Zhang et al. 2001; Dockendorff et al. 2002), and crossed them into the same genetic control (2b) background shared with dmGluRA. Both dfmr150M and dfmr13 null mutants perform comparably to the control in this coordinated movement assay (WT=12.4±0.7 sec, N=46; dfmr150M=12.9±1.04 sec, N=43, P=0.71; dfmr13=12.3±0.65, N=33; P=0.92; Fig. 3). Therefore, loss of dFMRP alone does not detectably impair the coordinated movement in the roll-over assay.
Figure 3. DmGluRA and dFMRP genetically interact in coordinated movement.

Coordinated movement behavior requires integration of sensory input and motor output. One test for this integration is the larval roll-over assay in which larvae are inverted and their righting time measured. Null dmGluRA mutants show a highly significant increase in response time, indicating grossly slowed performance. Two independent dfmr1 null alleles behave normally. However, both double mutant combinations significantly rescue the slow performance of the dmGluRA null. 0.001<P<0.05(*); P<0.0001(***).
To examine the relationship between DmGluRA signaling and dFMRP function in coordinated movement behavior, we tested the performance of two double mutant combinations; dfmr150M; dmGluRA and dfmr13; dmGluRA (Fig. 3). Both double null mutant lines displayed a remarkable rescue of the behavioral impairment caused by loss of mGluR signaling. In individual double mutant animals, the roll-over behavioral response was smooth and efficient, with a significantly shorter “struggle” time compared to the dmGluRA null. Quantitatively, the double null mutant showed comparable behavior to the wildtype control, with a very significant rescue of the impaired performance compared to the dmGluRA single mutant (dfmr150M;dmGluRA=9.0±0.62 sec, N=45, P<0.0001 to dmGluRA; dfmr13;dmGluRA=14.2±0.69 sec, N=34, P=0.007 to dmGluRA; Fig. 3). It is not clear why the dfmr150M combination actually shows improved response time compared to the control, but the average response time for the two double null mutants (11.6 seconds) is quite comparable to the wildtype performance (12.4 seconds), with no significant difference. Therefore, the coordinated movement behavior defect in the dmGluRA null mutant can be effectively rescued by removing dFMRP function.
DmGluRA and dFMRP genetically interact in regulating NMJ structure
Treating dfmr1 null flies with the mammalian group I mGluR antagonist MPEP rescues gross brain anatomical defects (McBride et al. 2005), suggesting that dFMRP and DmGluRA may be involved in the same mechanisms regulating neuronal structure. The Drosophila glutamatergic NMJ is an ideal system to test this hypothesis, since previous work has shown that both dfmr1 and dmGluRA single mutants display significant NMJ structure defects (Zhang et al., 2001; Bogdanik et al., 2004). The availability of the dfmr1; dmGluRA double mutant combinations provides the opportunity for the most rigorous genetic test of the relationship between dFMRP and DmGluRA in this synaptic architectural patterning. NMJ structure was examined in dfmr1 and dmGluRA single null mutants, dfmr1; dmGluRA double mutants, and MPEP treated dfmr1 null mutants and controls. The muscle 4 NMJ in abdominal segment A3 was used in all studies, with NMJs co-labeled for the presynaptic marker HRP (Fig. 4, red) and the postsynaptic marker DLG (Fig. 4, green). NMJ synaptic structure was quantified in three ways by measuring synaptic branch number, synaptic terminal area and synaptic bouton number (Fig. 5).
Figure 4. DmGluRA and dFMRP genetically interact in regulating NMJ structure.

Wandering 3rd instar NMJs were probed using anti-HRP (red) and anti-DLG (green) antibodies, to reveal pre- and postsynaptic domains respectively. Representative images of the NMJ on muscle 4 in abdominal segment A3 are shown. A. The wildtype (WT) 2b genetic background control. B. Null dfmr1 mutant allele dfmr150M in the 2b genetic background. C. Null dmGluRA mutant allele dmGluRA112b in the 2b genetic background. D. WT animal fed 68 µM MPEP. E. Null dfmr1 mutant fed 68 µM MPEP. F. Double mutant dfmr150M; dmGluRA112b in the 2b genetic background. Null dfmr1 mutants display increased NMJ synaptic branch number, terminal area and synaptic bouton number. MPEP-treatment and dfmr1; dmGluRA double mutant rescue the increased branch number, but not the other two defects. The scale bar is 10µm.
Figure 5. Quantification of NMJ structure in single and double mutants.
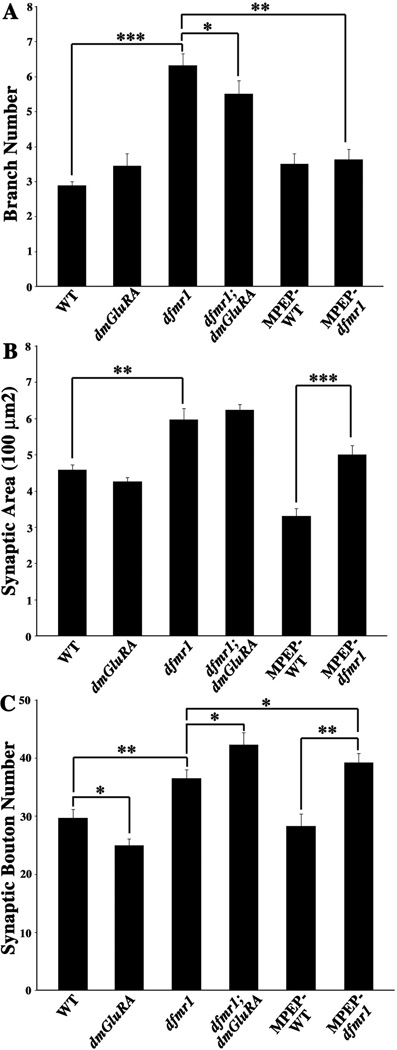
A. Quantification of NMJ synaptic arbor branch number for all genotypes. Null dfmr1 mutants display a highly significant increase of branch number, which can be rescued by blocking DmGluRA signaling genetically (double mutant) and pharmacologically (MPEP). B. Quantification of terminal synaptic area for all genotypes. Null dfmr1 mutants display a very significant increase of synaptic area, which is unaltered by blocking DmGluRA signaling in double mutant combination or with MPEP antagonist. C. Quantification of total synaptic bouton number for all genotypes. Null dfmr1 mutants display a very significant increase of bouton, whereas dmGluRA mutants display a significant decrease. Double mutants and MPEP-treated dfmr1 mutants display synergistically increased bouton number. 0.001<P<0.05(*); 0.0001<P<0.001(**); P<0.0001(***).
For synaptic branch number, dfmr1 single mutants display a very highly significant overbranching phenotype, while dmGluRA single mutants display branching comparable with the genetic control (WT=2.89±0.11, N=15; dfmr1=6.32±0.33, N=15, P<0.0001; dmGluRA=3.4±0.35, N=11, P=0.15; Fig. 4A–C, 5A). The dfmr1; dmGluRA double mutants display a tendency towards the normal level of synaptic branching with a significant rescue of the dfmr1 mutant defect (dfmr1; dmGluRA=5.5±0.38, N=11; P=0.039 to dfmr1; Fig. 4F, 5A). Consistently, treating dfmr1 null mutants with the mGluR antagonist MPEP, which blocks mGluR signaling, also effectively rescues the dfmr1 overbranching phenotype with no difference remaining with the control (MPEP-treated WT=3.5±0.3, N=11; MPEP-treated dfmr1=3.63±0.29, N=11, P=0.62 to treated WT; Fig. 4D–E, 5A). Thus, blocking DmGluRA signaling significantly rescues the synaptic overbranching phenotype of dfmr1 mutants.
For synaptic terminal area, dfmr1 single mutants display a very significant increase in total area, while dmGluRA mutants display a slight, but not quite significant decrease in synaptic area compared to the genetic control (WT=458.52±12.96µm2, N=15; dfmr1=597.45±29.61µm2, N=15, P=0.0007; dmGluRA=426.61±10.89µm2, N=11, P=0.078; Fig. 4A–C, 5B). The dfmr1; dmGluRA double mutants do not show any rescue of the increased synaptic area characteristic of dfmr1 alone, which therefore remains highly elevated compared to control (dfmr1; dmGluRA=622.88±15.77µm2, N=11; P=0.46 to dfmr1; Fig. 4F, 5B). The dfmr1 null mutants treated with the mGluR antagonist MPEP do display significantly decreased synaptic area compared to non-treated mutants (MPEP-treated dfmr1=499.95±24.88µm2, N=11, P=0.0048 to non-treated dfmr1). However, MPEP-treated control animals display a similar decrease compared to non-treated control, and therefore the difference between the MPEP-treated wildtype control and the MPEP-treated dfmr1 null mutant is still extremely significant (MPEP-treated WT=330.53±21.81µm2, N=11, P<0.0001; Fig. 4D–E, 5B). Therefore, blocking DmGluRA signaling does not significantly rescue the increased synaptic terminal area characteristic of dfmr1 mutants.
For synaptic bouton number, dfmr1 single mutants display a very highly significant increase, whereas dmGluRA mutants display a small, opposing decrease in bouton number compared to control animals (WT=29.7±1.48, N=15; dfmr1=36.5±1.5, N=15, P=0.001; dmGluRA=24.94±1.17, N=11, P=0.02 to WT; Fig. 4A–C, 5C). Surprisingly, the dfmr1; dmGluRA double mutants do not show the additive effects of the opposite phenotypes of the two single mutants, but rather display an even more severe, synergistic increase in synaptic bouton number compared to the dfmr1 single mutants (dfmr1; dmGluRA=42.3±2.09, N=11; P=0.037 to dfmr1; Fig. 4F, 5C). Consistently, treating dfmr1 null mutants with the mGluR antagonist MPEP also does not rescue the increased bouton defect of the dfmr1 mutant. Indeed, the difference between MPEP-treated control and MPEP-treated dfmr1 is more significant than the comparison of non-treated control and non-treated dfmr1 (MPEP-treated WT=28.27±2.14, N=11; MPEP-treated dfmr1=39.27±1.51, N=11, P=0.0004, Fig. 4D–E, 5C). Thus, either genetic or pharmacological block of DmGluRA signaling further exaggerates the synaptic bouton over-proliferation caused by loss of dFMRP.
Taken together, these results show a complex interaction between dFMRP and DmGluRA in regulating different aspects of presynaptic architecture. Whereas the synaptic over-branching caused by loss of dFMRP can be rescued by co-removal of DmGluRA or pharmacological block of mGluR signaling, the supernumerary boutons formed in the absence of dFMRP proliferate even more wildly when DmGluRA signaling is blocked. Thus, dFMRP and DmGluRA functions overlap in common mechanisms, but not in a simply interpretable manner.
DmGluRA and dFMRP genetically interact in regulating synaptic ultrastructure
Electron microscopy is the best means available to assay the complex assemblage occurring as a product of synaptic differentiation (Fig. 6). The presynaptic NMJ bouton is characterized by multiple, large mitochondria, dense accumulations of ~40nm diameter synaptic vesicles, an electron-dense active zone, and clustered vesicles around and docked adjacent to the T-bar presynaptic fusion sites. This bouton is deeply embedded in muscle and surrounded by the maze-like subsynaptic reticulum (SSR), which demarcates the postsynaptic domain (Fig. 6A). A detailed ultrastructural examination includes multiple parameters such as presynaptic bouton area and appearance, postsynaptic SSR area and appearance, mitochondria area and appearance, size and structure of the active zone and quantification of the different presynaptic vesicle pools (Fig. 6). At the ultrastructural level, the overall appearance of bouton and the postsynaptic SSR morphology appears normal in both dfmr1 and dmGluRA single null mutants. Quantitatively, there is no significant difference in bouton size, mitochondria size, active zone size/number or the postsynaptic SSR parameters between control and mutants (Fig. 6A and data not shown). However, there is a clear change in presynaptic vesicle density in the dfmr1 mutant, which we therefore carefully assayed in single and double mutant combinations.
Figure 6. DmGluRA and dFMRP genetically interact in presynaptic ultrastructure.

A. Representative TEM images of NMJ boutons in 2B genetic background control (WT), dfmr1 and dmGluRA single null mutants, and the dfmr1; dmGluRA double mutant. The dfmr1 mutant has a slightly larger bouton size but comparable synaptic morphology and postsynaptic SSR. Normal active zones (AZ) are visible in all panels as electron-dense synaptic membranes and T-bars. Null dfmr1 mutants display a striking increase of synaptic vesicles throughout the terminal. The scale bar is 500nm. B. High magnification images of individual synaptic active zones. In control animals (far left) the clustered area (250nm radius from T-bar center) has ~13 vesicles localized around the T-bar and ~2 docked vesicles (white arrow) contacting the presynaptic membrane adjacent to the T-bar. In dfmr1 mutants (second from left) there is an increased number of clustered vesicles and docked vesicles (arrows). The dmGluRA null mutants (second from right) display no detectable differences from control. In double mutants (far right), synaptic vesicle density and clustered synaptic vesicle number are comparable to control, but the docked synaptic vesicle number is still increased. The scale bar is 100nm. C–E) Quantitative analysis of ultrastructural phenotypes, including synaptic vesicle density (C), clustered synaptic vesicles (D; <250nm from active zone) and docked synaptic vesicles (E; <20nm from active zone). Significance indicated as 0.001<P<0.05(*); 0.0001<P<0.001(**); P<0.0001(***).
Null dfmr1 mutants display significant increases of synaptic vesicle density throughout the synaptic bouton, clustered vesicle number surrounding active zones and docked vesicle number at the T-bar membrane (Fig. 6A, B). For the overall synaptic vesicle density, single dfmr1 null mutant display a highly significant ~30% increase in overall vesicles, whereas dmGluRA mutants show vesicle density comparable to the genetic control (WT=56.36±5.6 vesicles/µm2, N=61; dfmr1=87.89±5.6 vesicles/µm2, N=40, P=0.0002; dmGluRA=59.94±4.4 vesicles/µm2, N=51, P=0.62; Fig. 6A, C). The dfmr1; dmGluRA double mutants show a very significant rescue of this elevated vesicle density phenotype (dfmr1; dmGluRA=64.22±2.8 vesicles/µm2, N=75, P=0.001 to dfmr1, P=0.14 to WT, Fig. 6A, C). Therefore, removing DmGluRA signaling can rescue the abnormal presynaptic vesicle accumulation characteristic of dfmr1 null synapses.
The synaptic vesicles clustered around the active zone form a local pool that can quickly replenish vesicles used during transmission (Fig. 6B). These vesicles are quantified by drawing a circle of 250nm radius around the electron dense T-bar and counting the number of synaptic vesicles within this radius. Null dfmr1 mutants display a highly significant ~50% increase in the pool of clustered vesicles, whereas dmGluRA mutants contain vesicle pools comparable to control (WT=13.1±0.8; dfmr1=18.8±0.7, P=0.0001; dmGluRA=14.6±0.8, P=0.22; Fig. 6B, D). The dfmr1; dmGluRA double mutants display a significant reduction in the clustered vesicle pool compared to the dfmr1 single mutant, and thus very significantly rescue the dfmr1 mutant phenotype (dfmr1; dmGluRA=16.3±0.4, P=0.005 to dfmr1; Fig. 6B, D). Therefore, removing DmGluRA signaling can also effectively rescue the increased clustered synaptic vesicle pool of the dfmr1 null mutant.
To provide for the rapidity of synaptic transmission, a subset of synaptic vesicles are maintained docked at active zones, where they form a pool that is immediately releasable upon arrival of an action potential. Electron microscopy reveals a population of synaptic vesicles morphologically adjacent to the presynaptic electron-dense membrane, which are considered to be these docked vesicles (Fig. 6B, arrows; Couteaux and Pecot-Dechavassine, 1970). Null dfmr1 mutants display a highly significant ~85% increase in the number of docked vesicles at the active zone, whereas dmGluRA mutants appear totally normal (WT=1.5±0.12; dfmr1=2.8±0.18, P=0.0001; dmGluRA= 1.5±0.14, P=0.92, Fig. 6B, E). In contrast to the above two vesicle pools, removing DmGluRA signaling does not detectably rescue the increased docked vesicle number characteristic of the dfmr1 null mutants. The dfmr1; dmGluRA double mutants, the elevation in docked vesicle number is indistinguishable from the dfmr1 single mutant alone (dfmr1; dmGluRA=2.5±0.09; P=0.19 to dfmr1, P=0.0001 to WT, Fig. 6B, E). Thus, removing DmGluRA signaling does not rescue the increased docked vesicle pool characteristic of dfmr1 mutants.
Taken together, these data also suggest that there is a complex interaction between dFMRP and DmGluRA signaling in regulating presynaptic differentiation. Loss of dFMRP results in large increases of presynaptic vesicle pools. Removing DmGluRA function alone has no effect on these vesicle pools; however removing DmGluRA in the double mutant condition can effectively rescue the enhanced overall vesicle density and increased clustered vesicle pools of the dfmr1 null mutant. In contrast, the number of docked vesicles remains elevated. These results suggest partially overlapping mechanisms between dFMRP and DmGluRA in the regulation of presynaptic differentiation at the peripheral NMJ.
DmGluRA and dFMRP interact in regulating central neuron structure
The Mushroom Body (MB) is a learning and memory center in the Drosophila brain (Heisenberg 1998; Heisenberg 2003). Our previous work has shown that dfmr1 null mutant MB neurons display increased overall architectural complexity, including axonal overgrowth and overbranching (Pan et al. 2004). The Mosaic Analysis of Repressible Cell Marker (MARCM) clonal technique provides a uniquely powerful means to examine homozygous mutant neurons in situ at a single cell level of resolution and therefore to study the cell autonomous functions for the mutant gene (Lee and Luo 1999). To test the mechanistic relationship between dFMRP and DmGluRA in central nervous system, we asked whether blocking DmGluRA signaling can rescue the architectural defects in single cell MARCM clones of dfmr1 null MB neurons. The dfmr1 gene is located on the third chromosome, while the dmGluRA gene is on fourth chromosome. It is technically not possible to do MARCM analysis for double mutants. However, our NMJ work shows that MPEP mGluR antagonist treatment totally mimics the effects of removing DmGluRA expression in all cases studied. Therefore, we used MPEP to treat MARCM clonal animals to assay the effect of blocking DmGluRA signaling on dfmr1 null MB neurons.
In MARCM clone brains, the MB axon lobes were visualized with an antibody against Fasciclin II (FasII, red), and the single-cell γ neuron MARCM clones identified by GFP (green) expression (Fig. 7A). The MB γ neuron contains a single primary axon projection in control animals (Fig. 7A; right, box) (Lee et al. 1999). The branching features of the γ neuron axon projection can be quantified both by branch number and total branch length. Null dfmr1 mutant MB neurons display obvious axon overgrowth; both the axon branch number (WT=6.11±0.7, N=9; dfmr1=8.85±0.34, N=17; P=0.0026) and the total axon branch length (WT=84.76±8.12 µm; dfmr1=106.91±8.12 µm; P=0.009) are very significantly increased in dfmr1 null neurons (Fig. 7B–D). Treating MARCM clone animals with the mGluR antagonist MPEP can effectively rescue the axon overgrowth defects of dfmr1 null mutant neurons. Blocking mGluR signaling with MPEP produces a significant rescue of the increased axon branching defect in dfmr1 null neurons (MPEP-dfmr1=6.83±0.43, N=23, P=0.018 to non-treated dfmr1=8.85±0.34). Similarly, the axon overgrowth defect is countered by MPEP treatment (MPEP-dfmr1=97.55±6.74 µm, P=0.043 to non-treated dfmr1=106.91±8.12 µm; Fig. 7B–D). Thus, MPEP-treated dfmr1 mutant neurons resemble control neurons compared to non-treated dfmr1 mutants, although the rescue provided by MPEP treatment is partial. Therefore, blocking mGluR signaling by MPEP can rescue the central neuron presynaptic over-elaboration defect of dfmr1 null mutants.
Figure 7. Blocking mGluR signaling rescues dfmr1 null neuron defects in brain.

A. Representative image of single cell MARCM clone of a Mushroom Body γ neuron. The Mushroom Body axon lobes are labeled with an antibody against Fasciclin II (FasII, red). The single γ neuron is visualized by MARCM technique-induced GFP expression (green). The boxed area indicates the axonal projection shown in the B panels. B. Representative images of axons in a non-treated WT cell, a non-treated dfmr1 null mutant cell, a MPEP-treated WT cell and a MPEP-treated dfmr1 null cell. The non-treated dfmr1 null γ neuron axon displays significant over-growth and over-branching, which is effectively rescued by MPEP treatment. Quantification of axon branch number (C) and total axon branch length (D) in both genotypes, with or without MPEP treatment. Compared to the significant increase of both axonal branch number and cumulative axon length in non-treated dfmr1 null, MPEP-treated dfmr1 mutant neurons display significant rescue of over-growth phenotypes. 0.001<P<0.05(*); 0.0001<P<0.001(**).
Discussion
Drosophila is a powerful system to test the mGluR theory of Fragile X Syndrome. There is one Drosophila homolog of the 3-member mammalian FMRP gene family (dFMRP) (Wan et al. 2000; Zhang et al. 2001) and one Drosophila homolog of the 8-member mammalian mGluR family (DmGluRA) (Parmentier et al. 1996; Bogdanik et al. 2004). Therefore, the double mutant combination of the two Drosophila null alleles provides a unique opportunity to test the relationship between all FMRP family function and all mGluR signaling throughout the nervous system. Of course this is a two-edged argument; Drosophila does not provide the means to test family member specific functions within or between these gene families, and so this sophistication of the mammalian system cannot be addressed. Nevertheless, Drosophila provides an excellent opportunity to comprehensively test interactions between mGluR signaling and FMRP functions in the in vivo context of the whole nervous system.
The mGluR theory of FXS proposes that FMRP functions downstream of mGluR signaling to regulate the synthesis of proteins critical for synaptic structural modeling and functional synaptic plasticity (Bear et al., 2004; Bagni and Greenough, 2005; Grossman et al., 2006; Pfeiffer and Huber, 2006). FMRP is a negative regulator of translation, and most known FMRP targets are constitutively upregulated in the absence of FMRP (Brown et al., 2001; Laggerbauer et al., 2001; Sung et al., 2003; Bagni and Greenough, 2005). One FMRP target is its own message, providing an interesting negative feedback loop on FMRP expression (Ashley et al., 1993; Sung et al., 2000; Schaeffer et al., 2001). In this work, we find that dFMRP and DmGluRA also mutually negatively regulate each other’s expression levels (Figure 1, 2). The protein level of dFMRP is increased in the dmGluRA null mutant, and the DmGluRA level is increased in the dfmr1 null mutant, providing bidirectional negative feedback regulation between the two proteins. Although these two proteins are co-expressed in the same neurons, consistent with their mutual co-regulation, they occupy quite distinct subcellular domains (Figure 1); dFMRP is highly enriched in the soma cytoplasm and is largely undetectable at synapses, whereas DmGluRA is highly enriched in the synaptic plasma membrane and is largely undetectable in the soma. This protein distribution suggests that the effect of mGluR signaling on dFMRP expression must be somewhat indirect, involving a long-distance second messenger mechanism.
The increase in dFMRP level in the absence of mGluR signaling is somewhat surprising in light of mammalian studies showing that increased synaptic activity increases FMRP expression and, more specifically, that mGluR activation increases FMRP expression (Weiler et al., 1997; Todd et al., 2003). The current study suggests instead that the overall role of mGluR signaling is to strongly suppress FMRP expression in the nervous system, at least in Drosophila. The increase in the DmGluRA in the absence of dFMRP indicates that the receptor is negatively regulated by dFMRP function. Some studies have suggested that mGluR transcripts may be direct targets for FMRP binding and, presumably, negative translation regulation. Such a mechanism would be consistent with the findings of this study. However, such direct regulation is not required, as there are obviously numerous possibilities of indirect regulation. In any case, the existence of this mutual negative feedback loop shows that mGluR signaling and FMRP function clearly interact to mutually repress each other in the Drosophila nervous system, providing molecular support for the theory of their functional interaction.
Both Group I and Group II/III mGluRs have been shown to play roles in the generation of locomotor activity and regulation of movement behavior (Vezina and Kim, 1999; Cauli et al., 2005; Nistri et al., 2006). In Drosophila, mGluR signaling is similarly required for effective performance in coordinated movement behavior (Figure 3). Null dmGluRA mutants display a grossly slowed response time in a relatively simple movement behavior, as revealed by the roll-over assay, indicating sluggish integration of sensory input and sequential motor output. Obviously, there are many possible causes of such a behavioral defect. In contrast, dfmr1 null mutants perform this task as well as wildtype. This is somewhat surprising given that dfmr1 mutants are more obvious impaired than dmGluRA mutants in many published cellular assays of synaptic structuring and function (Zhang et al., 2001; Bogdanik et al., 2004; Xu et al., 2004). The most important finding here, however, is that the dmGluRA defect can be rescued entirely simply by removing dFMRP in double null mutants (Figure 3). This is a rather remarkable finding as it shows that the behavioral impairment caused by the absence of mGluR signaling can be entirely compensated for by the co-absence of FMRP function. Although the nature of the dysfunction is unknown, this interaction shows that FMRP must act to enable a mechanism that is over active in the absence of mGluR signaling; a mechanism important for facilitating coordinated movement behavior. These results provide in vivo genetic evidence for a functional connection between dFMRP and DmGluRA in the sensory and motor response loop.
One obvious place for dFMRP-DmGluRA pathway regulation is the NMJ synapse. Both dfmr1 and dmGluRA single null mutants display striking defects in NMJ synaptic properties (Zhang et al. 2001; Bogdanik et al. 2004). Null dfmr1 mutants display elevated synaptic branch number, increased synaptic terminal area and supernumerary synaptic boutons (Figures 4 and 5). Blocking mGluR signaling by genetically removing DmGluRA or treating animals with the mGluR antagonist MPEP rescues the branch number defects, but has no significant effect on synaptic area defect. Moreover, although the dmGluRA single null mutant displays the opposing phenotype of decreased synaptic bouton number, both double null mutants and MPEP-treated dfmr1 animals actually display a more severe increase in synaptic bouton number. This complex interaction clearly supports a relationship between dFMRP function and DmGluRA signaling in the regulation of presynaptic architecture, but suggests that this relationship is not a simple direct upstream-downstream signaling cascade. Rather, dFMRP function is likely controlled by several converging intercellular signaling pathways, of which DmGluRA-mediated glutamatergic synaptic signaling is only one. Some of the synaptic structuring mechanisms may involve overlapping dFMRP and DmGluRA functions, while others likely involve quite independent pathways that can converge at multiple levels. In addition, note that treating dfmr1 null animals with MPEP creates a stronger rescue of synaptic over-branching than genetically removing DmGluRA, suggesting MPEP may affect other targets than just DmGluRA.
A similar conclusion derives from our ultrastructural studies of presynaptic differentiation. dFMRP clearly plays critical roles in the regulation of synaptic vesicle pools; dfmr1 null mutants display elevated overall synaptic vesicle density, an increased pool of clustered vesicles at active sites and an elevated number of docked vesicles at the presynaptic membrane (Figure 6). These defects provide a mechanistic explanation for the elevated presynaptic glutamate release that we previously characterized at this synapse (Zhang et al. 2001). In addition, many presynaptic protein transcripts have been identified as putative direct targets of FMRP binding, including MUNC-13, NAP-22, SEC-7 and RAB-5 (Brown et al. 2001; Miyashiro et al. 2003). Several of these encoded proteins are known to play important roles in vesicle cycling and trafficking, consistent with the presynaptic vesicle defects reported here in dfmr1 mutants. In contrast, DmGluRA plays no detectable function in the modulation of any of these vesicle pools (Figure 6). In mammalian system, although Group II/III mGluR signaling has been implicated in vesicular endocytosis and exocytosis cycling in the presynaptic terminal, there is similarly no evidence that loss of mGluR signaling results in defects in presynaptic vesicle pools (Hay et al., 2001; Pamidimukkala and Hay, 2001; Zakharenko et al., 2002). The most important finding here, however, is that the dfmr1 defects can be rescued simply by blocking DmGluRA signaling in double null mutants (Figure 6). Removing DmGluRA function significantly rescues the defects in both the total synaptic vesicle pool and clustered vesicle pool. These findings reinforce the conclusion that dFMRP and DmGluRA functions must intersect in the regulation of presynaptic properties, in both structuring and functional manifestations.
Fragile X Syndrome is a mental retardation disorder, and therefore it is obviously critical to move into the brain and assay functions in neuronal circuits relevant to learning and memory. In Drosophila, the relevant brain region is the Mushroom Body, a well-characterized center of learning and memory consolidation (Heisenberg 1998; Heisenberg 2003). We have shown previously that all classes of MB neurons display a cell-autonomous requirement of dFMRP in mediating correct axonal patterning (Pan et al., 2004). In the absence of dFMRP, these learning circuit neurons display improper axonal growth and branching, and strong defects in presynaptic connectivity (Figure 7). Similarly, mammalian FMRP is localized in the axon process and growth cone in mouse hippocampal neurons (Antar, Afroz et al. 2004; Antar, Li et al. 2006) and loss of FMRP results in excess axonal filopodia and altered motility of the axonal growth cone (Antar et al., 2006). Moreover, recent studies in mouse brain hippocampal slices mosaic for FMRP expression have revealed that presynaptic axons lacking FMRP form fewer functional presynaptic terminals than control axons (Hanson and Madison, 2007). Thus, mammalian FMRP and dFMRP clearly play conserved roles is establishing correct presynaptic connectivity in the brain.
We do not have similar knowledge of the roles of DmGluRA signaling in the brain. Technical limitations with the MARCM clonal technique mean that comparable studies are not possible for dmGluRA null MB neurons, so we do not know whether there are dmGluRA single mutant defects in related presynaptic mechanisms. Moreover, the MARCM technique does not allow us to pursue the same powerful double mutant analyses we performed at the NMJ. Therefore, we are limited to blocking DmGluRA signaling by treating MARCM clonal animals with the mGluR antagonist MPEP, in order to pharmacologically block mGluR signaling. The most important finding here, however, is that the dfmr1 defects can be rescued simply by blocking mGluR signaling (Figure 7). MPEP treatment significantly rescues both the increased axon branching and presynaptic axon overgrowth that would otherwise occur in dfmr1 null mutant neurons. This single cell level finding is consistent with a previous report that treating dfmr1 null animals with MPEP rescues the gross anatomical defect of MB axonal lobe fusion (McBride et al. 2005). This MPEP-treatment rescue shows that an overlapping mechanism exists between mGluR signaling and FMRP function in regulating neuronal circuit architecture in central nervous system. Thus, the results in both the peripheral and central presynaptic processes consistently support a mechanistic interaction between mGluR and FMRP in controlling presynaptic structure and differentiation.
Experimental Methods
Drosophila Genetics
All Drosophila stocks were maintained at 25°C on standard food under standard conditions. The P-element imprecise excision dmGluRA112b null mutant was used as the dmGluRA single mutant, and dmGluRA2b (hereafter called 2b), a p-element precise excision line from the same screen, was used as its genetic background control (Bogdanik, Mohrmann et al. 2004). The w1118; dfmr150M and w1118; dfmr13 null mutant strains were used as two independent dfmr1 single mutants, with w1118 as the genetic background control (Zhang, Bailey et al. 2001; Dockendorff, Su et al. 2002). For all assays involving dmGluRA and dfmr1 mutants, both dfmr1 alleles were back-crossed into the 2b genetic background, to generate a common background for all single and double mutants. The following multiply mutant strains were used: 1) the dfmr150M; dmGluRA112b and dfmr13; dmGluRA112b double null mutants, and 2) the dfmr150M; 2b and dfmr13; 2b genetic background control combinations. For drug treatment studies, MPEP, a generous gift from Fragile X Foundation, was dissolved in ddH2O, and added to standard fly food at a final concentration of 86µM as previously reported (McBride et al. 2005). In MARCM analysis, the following strains were used 1, heatshock-FLP, mouse CD8-GFP; FRT82B, tubulin P-GAL80/TM3; GAL4-OK107. 2, y, w; FRT82B/TM3. 3, FRT82B, dfmr50M/TM6 (Pan et al., 2004).
Immunocytochemistry
Wandering 3rd instar larvae were dissected in standard saline, followed by 4% paraformaldehyde fixation for 30 mins (for staining dFMRP, HRP and DLG) or Bouin’s Fixative for 30 mins (for staining DmGluRA). The monoclonal mouse antibody against dFMRP (6A15; Sigma) was used at 1:1000. The monoclonal mouse antibody against DmGluRA (7G11), a generous gift from Dr. Irmi Sinning (Universität Heidelberg), was used at 1:50. The monoclonal mouse antibody against DLG (4F3; used at 1:500) was obtained from the Developmental Studies Hybridoma Bank (University of Iowa). The Texas Red-conjugated anti-Horseradish Peroxidase (HRP; used at 1:200) was from Jackson. All primary antibodies were visualized using fluorescent dye-conjugated secondary antibodies, including Alexa Fluor 488 goat anti-mouse IgG (1:200; Molecular Probes) and cy3-conjugated goat anti-mouse IgG (1:200; Jackson). All fluorescent images were collected using a Zeiss LSM 510 meta laser scanning confocal microscope and Zeiss image-collection software. All image processing was done with Adobe Photoshop 7.0.
Fluorescent intensity quantification
For any given experiment, animals of control and mutant genotypes were simultaneously processed, together in the same tube, and imaged using identical confocal settings. All images used in fluorescent intensity quantification were 3D-projections from complete Z-stacks through the entire 3rd instar CNS. All images were analyzed using LSM 5 Confocal Image Examiner software in the “histogram” display mode. For dFMRP quantification, 8 neuronal soma regions (1000 µm2 area) were defined along the lateral boundary of CNS for each animal. For DmGluRA quantification, two large, bilateral synaptic neuropil regions (10,000 µm2 area) were defined for each animal. The software output reported fluorescence intensity for each region. The fluorescence intensity was calculated as an average of all test regions to produce each single data point. Statistical analysis was done using GraphPad InStat 3 software.
Western Blotting
The wandering 3rd instar larval CNS (2 each for dFMRP assay, 4 each for DmGluRA assay) was dissected in 1×PBS, homogenized in 20 µl of 1× Nupage LDS Sample Buffer (Invitrogen, Carlsbad, CA) with 5% β-Mercaptoethanol (Acros), and boiled for 10 mins. Extracts were loaded onto a 4–12% Bis-Tris gel and run at 200 V in 1× MES buffer (Invitrogen, Carlsbad, CA). Protein was transferred to nitrocellulose membrane in 1× transfer buffer (Invitrogen, Carlsbad, CA) plus 10% methanol at 100 V for 1 hr. The membrane was blocked for at least 1 hr in Odyssey Blocking Buffer (Li-Cor, Lincoln, NE) and probed for 12–16 hrs at 4°C with the following antibodies: dFMRP (6A15, Sigma, 1:5000); DmGluRA (7G11, gift from Dr. Irmi Sinning, 1:5); α-tubulin (Sigma, 1:400,000). Membranes were washed 3× with buffer (25 mM Tris pH8.0, 150 mM sodium chloride, 0.05% Ige-PAL-CA630). The secondary anti-mouse IgG IR680 (Invitrogen, Carlsbad, CA) was diluted 1:10,000 in Odyssey Blocking Buffer and applied for 1 hr at 25°C. The blot was washed 3× with buffer and scanned on an Odyssey Infrared Imaging System.
Immuno-Electron Microscopy
Wandering 3rd instar larvae were processed by adapting published methods (McDonald, 1999; Edelmann, 2002; Spehner et al., 2002). Dissected larvae were fixed for 1 hr in 4% paraformaldehyde plus 0.5% glutaraldehyde, rinsed in PBS for 6 mins, and passed through an ethanol series (50%, 70%, 100%; 20 mins each). 1:1 propylene oxide: araldite was used as a transition media to 100% araldite. Tissue was placed in a flat embedding mold and cured overnight in a 60°C oven. Gold thin sections were obtained from Leica UCT Ultracut microtome, using 200-mesh nickel grids on which to collect sections. Grids were blocked for 15 mins with 1% BSA in DPBS, and incubated overnight at 4°C in either DmGluRA or dFMRP primary antibody. Grids were washed for 1 hr in DPBS with Tween-20, and for 15 mins in TRIS buffer with 0.05% Tween-20, blocked for 15 mins with 1% BSA in TRIS buffer, and incubated for 1 hr at room temperature in secondary antibody conjugated to 10 or 25nm gold particles (1/50 dilution). Grids were washed for 15 mins in TRIS buffer + 0.05%, washed again for 15 mins in dH2O, and blotted dry. Sections were stained with uranyl acetate and lead citrate, and imaged as for TEM. Gold particle densities were calculated by counting the total number of gold particles inside one bouton section and then dividing by total bouton area. Significance levels were calculated by paired t-test.
Behavior Assay
Animals were cultured at 25°C in regular food to the wandering 3rd instar larval stage. All assays were done at room temperature (RT). Before every assay, larvae and test agar plates were placed at RT for 2 hrs to acclimatize. For the assay, an individual animal was placed on the agar plate, and allowed to move freely for 2 mins. Using a soft brush, the test animal was then rolled over to a completely inverted position, as defined by the ventral midline. The time that the animal spent to totally right itself was recorded. Three assays were done for each animal, and then averaged to produce one data point. Statistical analysis was done using GraphPad InStat 3 software. P-values were calculated by 2-tailed T-test to compare each group.
NMJ structure quantification
All images used in NMJ structure quantification were 3D-projections from complete Z-stacks through the entire NMJ. The lateral, longitudinal muscle 4 in abdominal segment A3 was used for all quantification. Data from the two-paired hemisegments were averaged for each animal, to produce each single data point. Synaptic boutons were defined according to HRP (presynaptic) and DLG (postsynaptic) staining. Branches originating directly from the nerve entry point were defined as primary braches, and each higher order branch was counted only when two or more boutons could be observed in a subsequent branch fork. For total synaptic area, LSM 5 Confocal Image Examiner software was used in the “histogram” display mode. Synaptic regions were user-defined with the closed free shape curve drawing tools, defined by the boundary of DLG staining. The software output reports the area for each region automatically. Statistical analysis was done using GraphPad InStat 3 software. P-value was calculated by T-test to compare each matched group.
Ultrastructural Analysis
Wandering 3rd instar larvae were dissected, fixed, sectioned and visualized in parallel using standard TEM techniques, as reported previously (Featherstone et al., 2001; Haas et al., 2007). Staged animals were dissected in 1×PBS and subsequently fixed with 2.0% glutaraldehyde in 0.05M PBS for 15 mins; replaced with fresh 2.0% glutaraldehyde for 1 hr. Preparations were washed three times in PBS, transferred to 1% OsO4 in dH2O for 2 hrs, and then washed three times in dH2O. Preparations were stained en bloc in 1% aqueous uranyl acetate for 1 hr, washed three times in dH2O, dehydrated in an EtoH series (30–100%), passed through propylene oxide, transferred to a 1:1 araldite: propylene oxide mixture, and embedded in araldite embedding media. Ultra-thin serial sections (50–60nm) were made on a Leica UCT Ultracut microtome and transferred to formvar-coated grids. Grids were examined and images collected on a Phillips CM10 TEM equipped with an AMT 2 mega pixel camera. NMJs were sectioned, and profiles for each synaptic bouton were quantified in sections containing only a single prominent electron-dense active zone (AZ) and T-bar structure. Synaptic vesicles in the “clustered” pool were defined as those within 250 nm of an AZ. Docked vesicle were defined as those <0.5 vesicles diameter (< 20 nm) from the electron-dense plasma membrane at the AZ. Measurements and quantifications were made using Image J 1.32j free software from NIH. Each profile was scored for bouton/mitochondria area, and the number of docked, clustered vesicles and total vesicle density (corrected for mitochondria area). Mean quantified parameters were statistically compared using the Mann-Whitney test, and presentation images were processed in Adobe Photoshop.
Mushroom Body MARCM analysis
The Mosaic Analysis of Repressible Cell Marker (MARCM) clonal technique was employed as first described in Lee and Luo (1999). Single neuron MARCM clones were made in the brain Mushroom Body, within the population of γ neurons. Staged embryos were collected within a 4 hr window and cultured at 25°C. Mature embryos at 20 hrs were heat-shocked at 37°C for 1 hr to induce recombination and clone formation. Animals were then cultured to maturity at 25°C. Adult brains were dissected out within 1 day following eclosion. Brains were dissected in 1×PBS, fixed in 4% paraformaldehyde for 30 mins and processed with immuno-staining. MB axon lobe was labeled by mouse anti-Drosophila Fasciclin II 1D4 (1:20, Developmental Studies Hybridoma Bank, University of Iowa), and the MARCM clone was labeled by rat anti-mouse CD8 (1:100, Caltag). Primary antibodies were visualized using Cy3-conjugated goat anti-mouse IgG (1:100, Jackson), and FITC-conjugated goat anti-rat IgG (1:100, Jackson). For γ-neuron axonal quantification, the primary axon branch was identified first as the single projection joined γ-lobe, and all other axon processes extended from this main trunk were counted as branches. The length of each branch was measured based on 3D-projections from complete Z-stacks from confocal microscopy. All branch lengths of single axon branches were added together to obtain the total cumulative axon length. Statistical analysis was done using GraphPad InStat 3 software. P-value was calculated by paired T-test to compare each matched group.
Statistics
Significance levels were calculated by paired t-test comparisons, unless otherwise noted for individual techniques. Significance is displayed in all figures as 0.001<P<0.05(*); 0.0001<P<0.001(**); P<0.0001(***). All error bars represent Standard Error of Mean (SEM), appropriate for comparison of the mean of means distribution, unless otherwise noted.
Acknowledgements
We are particularly grateful to Dr. Irmi Sinning for the generous gift of DmGluRA antibodies, and the Fragile X Foundation for the generous gift of MPEP. We thank the Iowa Developmental Studies Hybridoma Bank for providing critical antibodies, and the Bloomington Drosophila Stock Center for providing essential stocks. This work was supported by NIH grant GM54544 to K.B.
References
- Antar LN, Afroz R, et al. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar LN, Li C, et al. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32(1–2):37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Ashley CT, Jr, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6(5):376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, et al. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bodily KD, Morrison CM, et al. A novel member of the Ig superfamily, turtle, is a CNS-specific protein required for coordinated motor control. J Neurosci. 2001;21(9):3113–3125. doi: 10.1523/JNEUROSCI.21-09-03113.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanik L, Mohrmann R, et al. The Drosophila metabotropic glutamate receptor DmGluRA regulates activity-dependent synaptic facilitation and fine synaptic morphology. J Neurosci. 2004;24(41):9105–9116. doi: 10.1523/JNEUROSCI.2724-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107(4):477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Cauli O, Llansola M, Rodrigo R, El Mlili N, Errami M, Felipo V. Altered modulation of motor activity by group I metabotropic glutamate receptors in the nucleus accumbens in hyperammonemic rats. Metab Brain Dis. 2005;20:347–358. doi: 10.1007/s11011-005-7918-0. [DOI] [PubMed] [Google Scholar]
- Couteaux R, Pecot-Dechavassine M. [Synaptic vesicles and pouches at the level of "active zones" of the neuromuscular junction] C R Acad Sci Hebd Seances Acad Sci D. 1970;271:2346–2349. [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, et al. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34(6):973–984. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- Edelmann L. Freeze-dried and resin-embedded biological material is well suited for ultrastructure research. J Microsc. 2002;207:5–26. doi: 10.1046/j.1365-2818.2002.01033.x. [DOI] [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17:1539–1547. doi: 10.1523/JNEUROSCI.17-05-01539.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber K, Smith KT, et al. Transcription, translation and fragile X syndrome. Curr Opin Genet Dev. 2006;16(3):270–275. doi: 10.1016/j.gde.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Grossman AW, Aldridge GM, et al. Local protein synthesis and spine morphogenesis: Fragile X syndrome and beyond. J Neurosci. 2006;26(27):7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Madison DV. Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J Neurosci. 2007;27:4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- aas KF, Woodruff E, Iii, Broadie K. Proteasome function is required to maintain muscle cellular architecture. Biol Cell. 2007 doi: 10.1042/BC20070019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay M, Hoang CJ, Pamidimukkala J. Cellular mechanisms regulating synaptic vesicle exocytosis and endocytosis in aortic baroreceptor neurons. Ann N Y Acad Sci. 2001;940:119–131. doi: 10.1111/j.1749-6632.2001.tb03671.x. [DOI] [PubMed] [Google Scholar]
- Heisenberg M. What do the mushroom bodies do for the insect brain? an introduction. Learn Mem. 1998;5(1–2):1–10. [PMC free article] [PubMed] [Google Scholar]
- Heisenberg M. Mushroom body memoir: from maps to models. Nat Rev Neurosci. 2003;4(4):266–275. doi: 10.1038/nrn1074. [DOI] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, et al. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41(3):289–294. doi: 10.1002/ajmg.1320410306. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, et al. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51(4):441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, et al. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99(11):7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, et al. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288(5469):1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Idupulapati M, et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet. 2002;111(2):140–146. doi: 10.1002/ajmg.10500. [DOI] [PubMed] [Google Scholar]
- Koekkoek SK, Yamaguchi K, et al. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005;47(3):339–352. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Lee A, Li W, et al. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development. 2003;130(22):5543–5552. doi: 10.1242/dev.00792. [DOI] [PubMed] [Google Scholar]
- Lee T, Lee A, et al. Development of the Drosophila mushroom bodies: sequential generation of three distinct types of neurons from a neuroblast. Development. 1999;126(18):4065–4076. doi: 10.1242/dev.126.18.4065. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22(3):451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- McBride SM, Choi CH, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45(5):753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- McDonald K. High-pressure freezing for preservation of high resolution fine structure and antigenicity for immunolabeling. Methods Mol Biol. 1999;117:77–97. doi: 10.1385/1-59259-201-5:77. [DOI] [PubMed] [Google Scholar]
- Michel CI, Kraft R, Restifo LL. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J Neurosci. 2004;24:5798–5809. doi: 10.1523/JNEUROSCI.1102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, et al. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37(3):417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Nistri A, Ostroumov K, Sharifullina E, Taccola G. Tuning and playing a motor rhythm: how metabotropic glutamate receptors orchestrate generation of motor patterns in the mammalian central nervous system. J Physiol. 2006;572:323–334. doi: 10.1113/jphysiol.2005.100610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95(5):3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Pamidimukkala J, Hay M. Frequency dependence of endocytosis in aortic baroreceptor neurons and role of group III mGluRs. Am J Physiol Heart Circ Physiol. 2001;281:H387–H395. doi: 10.1152/ajpheart.2001.281.1.H387. [DOI] [PubMed] [Google Scholar]
- Pan L, Zhang YQ, et al. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14(20):1863–1870. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Parmentier ML, Pin JP, et al. Cloning and functional expression of a Drosophila metabotropic glutamate receptor expressed in the embryonic CNS. J Neurosci. 1996;16(21):6687–6694. doi: 10.1523/JNEUROSCI.16-21-06687.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. Current advances in local protein synthesis and synaptic plasticity. J Neurosci. 2006;26(27):7147–7150. doi: 10.1523/JNEUROSCI.1797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purpura DP. Dendritic spine "dysgenesis" and mental retardation. Science. 1974;186(4169):1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- Ramaekers A, Parmentier ML, et al. Distribution of metabotropic glutamate receptor DmGlu-A in Drosophila melanogaster central nervous system. J Comp Neurol. 2001;438(2):213–225. doi: 10.1002/cne.1310. [DOI] [PubMed] [Google Scholar]
- Rogers SJ, Wehner DE, et al. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22(6):409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, Moine H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. Embo J. 2001;20:4803–4813. doi: 10.1093/emboj/20.17.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MD, Najand N, Chaker S, Pare JM, Haskins J, Hughes SC, Hobman TC, Locke J, Simmonds AJ. Gawky is a component of cytoplasmic mRNA processing bodies required for early Drosophila development. J Cell Biol. 2006;174:349–358. doi: 10.1083/jcb.200512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spehner D, Drillien R, Proamer F, Hanau D, Edelmann L. Embedding in Spurr's resin is a good choice for immunolabelling after freeze drying as shown with chemically unfixed dendritic cells. J Microsc. 2002;207:1–4. doi: 10.1046/j.1365-2818.2002.01038.x. [DOI] [PubMed] [Google Scholar]
- Strausfeld NJ. Crustacean-insect relationships: the use of brain characters to derive phylogeny amongst segmented invertebrates. Brain Behav Evol. 1998;52(4–5):186–206. doi: 10.1159/000006563. [DOI] [PubMed] [Google Scholar]
- Sung YJ, Dolzhanskaya N, Nolin SL, Brown T, Currie JR, Denman RB. The fragile X mental retardation protein FMRP binds elongation factor 1A mRNA and negatively regulates its translation in vivo. J Biol Chem. 2003;278:15669–15678. doi: 10.1074/jbc.M211117200. [DOI] [PubMed] [Google Scholar]
- Turner G, Webb T, et al. Prevalence of fragile X syndrome. Am J Med Genet. 1996;64(1):196–197. doi: 10.1002/(SICI)1096-8628(19960712)64:1<196::AID-AJMG35>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT, Oostra BA. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature. 1993;363:722–724. doi: 10.1038/363722a0. [DOI] [PubMed] [Google Scholar]
- Vezina P, Kim JH. Metabotropic glutamate receptors and the generation of locomotor activity: interactions with midbrain dopamine. Neurosci Biobehav Rev. 1999;23:577–589. doi: 10.1016/s0149-7634(98)00055-4. [DOI] [PubMed] [Google Scholar]
- Visootsak J, Warren ST, et al. Fragile X syndrome: an update and review for the primary pediatrician. Clin Pediatr (Phila) 2005;44(5):371–381. doi: 10.1177/000992280504400501. [DOI] [PubMed] [Google Scholar]
- Wan L, Dockendorff TC, et al. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol. 2000;20(22):8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Bogert BA, Li W, Su K, Lee A, Gao FB. The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr Biol. 2004;14:1025–1034. doi: 10.1016/j.cub.2004.05.055. [DOI] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, et al. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49(7):1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002;35:1099–1110. doi: 10.1016/s0896-6273(02)00898-x. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107(5):591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Broadie K. Fathoming fragile X in fruit flies. Trends Genet. 2005;21(1):37–45. doi: 10.1016/j.tig.2004.11.003. [DOI] [PubMed] [Google Scholar]