

Abstract
Colorectal cancer (CRC) has an apparent hereditary component, as evidenced by the well-characterized genetic syndromes and family history associated with the increased risk of this disease. However, in a large fraction of CRC cases, no known genetic syndrome or family history can be identified, suggesting the presence of “missing heritability” in CRC etiology. The genome-wide association study (GWAS) platform has led to the identification of multiple replicable common genetic variants associated with CRC risk. These newly discovered genetic variations might account for a portion of the missing heritability. Here, we summarize the recent GWASs related to newly identified genetic variants associated with CRC risk and clinical outcome. The findings from these studies suggest that there is a lack of understanding of the mechanism of many single nucleotide polymorphisms (SNPs) that are associated with CRC. In addition, the utility of SNPs as prognostic markers of CRC in clinical settings remains to be further assessed. Finally, the currently validated SNPs explain only a small fraction of total heritability in complex-trait diseases like CRC. Thus, the “missing heritability” still needs to be explored further. Future epidemiological and functional investigations of these variants will add to our understanding of CRC pathogenesis, and may ultimately lead to individualized strategies for prevention and treatment of CRC.
Keywords: Colorectal cancer, Genome-wide association study, Single nucleotide polymorphism, Signal transduction pathways, Cell cycle control, Gene desert, Genome instability
Core tip: This review covers the recent advances in genome-wide association studies (GWASs) that have identified genetic variants associated with an altered risk of colorectal cancer (CRC). In this review, we summarize single nucleotide polymorphisms (SNPs) located in or near genes that play crucial roles in signal transduction pathways, genome stability, cell cycle control, and gene expression and regulation. SNPs that are found in gene desert regions are also discussed. The relationship between genetic variations and clinical outcomes in CRC is presented from epidemiological studies that have identified SNPs with methods other than GWASs.
INTRODUCTION
It is estimated that 35% of colorectal cancer (CRC) risk may be explained by heritable factors[1]. Heritable factors include well-characterized genetic syndromes inherited in a straightforward Mendelian manner, such as familial adenomatous polyposis and hereditary non-polyposis colorectal cancer, also known as Lynch Syndrome[2]. It is estimated that cumulatively, these and other well-characterized genetic syndromes with Mendelian mode of inheritance account for up to 10% of all CRC cases. In an estimated further 25% of cases, family history contributes to CRC risk in the absence of one of these identifiable genetic syndromes. The important role of family history in CRC risk is reflected in the guidelines published by the American College of Gastroenterology and the American Cancer Society, which recommend starting screening colonoscopies at an age cutoff that is a function of family history[3].
The combined effect of genetic syndromes and family history may explain up to 30% of CRC susceptibility, whereas the remaining genetic risk of CRC may be accounted for by a combination of high-prevalence and low-penetrance of common genetic variants. Recent advances using genome-wide association study (GWAS) have enabled the identification of multiple CRC-related single nucleotide polymorphisms (SNPs)[4-10]. These genetic variants can be broadly classified into two categories: those that affect the risk of developing CRC, and those that influence the clinical course of CRC once established. In this review, we summarize these GWAS-identified genetic variants - including functional characterizations and implications for clinical applications - and discuss some of the limitations and challenges of these studies.
GENETIC VARIANTS AND CRC RISK
By comparing the distributions of millions of tagged SNPs between CRC patients (cases) and cancer-free populations (controls), a large number of common genetic variants have been identified under the “common disease-common variant” premise. To date, more than 40 chromosome regions harboring common variants conferring altered CRC risk have been identified by the GWAS approach. These variants are dispersed amongst almost every human chromosome and the vast majority of them exhibit a small effect size (Table 1). Most of these loci confer a modest increase in CRC risk, typically with an OR of less than 1.20. Among the 48 SNPs listed in Table 1, eight had an OR of more than 1.20, of which only three exhibited an OR of more than 1.30. A higher effect size (OR = 2.64) was reported for rs6038071 located upstream of the CSNK2A1 gene and validated in familial CRC populations, although only under a recessive genetic model with least statistical power[11]. The majority of GWAS-identified CRC risk variants are involved in known biological pathways; however, a few highly significant ones reside in gene desert regions, and the mechanism by which these variants contribute to colorectal carcinogenesis remains unclear. Here, we summarize these variants in relation to their implications in pathways of signal transduction, genome instability, cell cycle control, and gene expression and regulation (Table 1). These pathways and related significant SNPs identified by GWASs are also depicted in Figure 1. This figure was produced by combining pathways from various studies[12-15].
Table 1.
Genome-wide association study-identified common genetic variants associated with colorectal cancer risk
| SNP | Loci | Gene | Full name of gene | OR | P value | Pathway/function | Ref. | Method |
| Common biological pathway-related | ||||||||
| rs12701937 | 7p14.1 | GLI3 and INHBA | GLI family zinc finger 3 and inhibin, beta A | 1.36 | 3.50E-05 | MAPK signaling pathways | [11] | G |
| rs11014993 | 10p12.1 | MYO3A | Myosin IIIA | 1.22 | 2.00E-03 | MAPK signaling pathways | [11] | G |
| rs59336 | 12q24.21 | TBX3 | T-box3 | 1.09 | 2.46E-06 | Wnt pathway | [10] | G + M |
| rs4444235 | 14q22.2 | BMP4 | Bone morphogenetic protein 4 | 1.09 | 1.95E-11 | BMP pathway | [21] | G |
| rs1957636 | 14q22.2 | BMP4 | Bone morphogenetic protein 4 | 1.08 | 1.36E-09 | BMP pathway | [21] | G |
| rs16969681 | 15q13.3 | GREM1 | DAN family BMP antagonist | - | 5.33E-08 | BMP pathway | [21] | G |
| rs4779584 | 15q13.3 | GREM1 | DAN family BMP antagonist | - | 5.27E-03 | BMP pathway | [21] | G |
| rs11632715 | 15q13.3 | GREM1 | DAN family BMP antagonist | - | 2.30E-10 | BMP pathway | [21] | G |
| rs4939827 | 18q21 | SMAD7 | SMAD family member 7 | 1.2 | 7.80E-28 | TGF-β1 pathway, cell arrest, cell proliferation | [5,6,19] | G |
| rs12953717 | 18q21 | SMAD7 | SMAD family member 7 | 1.17 | 9.10E-12 | TGF-β and Wnt signaling | [5] | G |
| rs4464148 | 18q21 | SMAD7 | SMAD family member 7 | 1.15 | 6.66E-08 | TGF-β and Wnt signaling | [5] | G |
| rs961235 | 20p12.3 | BMP2 | Bone morphogenetic protein 2 | 1.12 | 4.45E-16 | BMP pathway | [21] | G |
| rs4813802 | 20p12.3 | BMP2 | Bone morphogenetic protein 2 | 1.09 | 7.52E-11 | BMP pathway | [21] | G |
| rs6038071 | 20p13 | CSNK2A1 | Casein kinase 2, alpha 1 polypeptide | 2.64 | 3.00E-04 | MAPK signaling pathways | [11] | G |
| Genome instability-related | ||||||||
| rs11903757 | 2q32.3 | NABP1 | Nucleic acid binding protein 1 | 1.16 | 9.50E-08 | DNA repair, genomic stability | [10] | G + M |
| rs647161 | 5q31.1 | PITX1 | Paired-like homeodomain transcription factor 1 | 1.11 | 1.22E-10 | RAS pathway; activate TP53; telomerase activity | [29] | G + M |
| rs1321311 | 6p21 | CDKN1A | Cyclin-dependent kinase inhibitor 1A | 1.1 | 1.14E-10 | Microsatellite instability, DNA repair, genomic instability | [26] | G + M |
| rs3824999 | 11q13.4 | POLD3 | Polymerase DNA- directed δ3 | 1.08 | 3.65E-10 | DNA mismatch and base-excision repair | [26] | G + M |
| rs78378222 | 17p13 | TP53 | Promotor region of TP53 gene | 1.39 | 1.60E-04 | TP53 | [63] | G |
| Cell cycle control-related | ||||||||
| rs10911251 | 1q25.3 | LAMC1 | Laminin gamma 1 | 1.09 | 5.90E-08 | Gene transcription | [10] | G + M |
| rs6691170 | 1q41 | DUSP10 | Dual-specificity phosphatase | 1.06 | 9.55E-10 | Inactivates p38 and SAPK | [9] | M |
| rs6687758 | 1q41 | DUSP10 | Dual-specificity phosphatase | 1.09 | 2.27E-09 | Inactivates p38 and SAPK | [9] | M |
| rs886774 | 7q31 | LAMB1 | Laminin β1 | 1.17 | 3.00E-08 | Anchoring the single-layered epithelium, ulcerative colitis | [33] | G |
| rs3802842 | 11q23 | POU2AF1 | POU class 2 associating factor 1 | 1.1 | 5.80E-10 | Growth of multiple myeloma cells | [6] | G |
| rs10774214 | 12p13.32 | CCND2 | Cyclin D2 | 1.09 | 3.06E-08 | Cell-cycle transition | [29] | G + M |
| rs3217810 | 12p13.32 | CCND2 | Cyclin D2 | 1.2 | 3.70E-07 | Cell-cycle transition | [10] | G + M |
| rs3217901 | 12p13.32 | CCND2 | Cyclin D2 | 1.1 | < 5.0E-7 | Cell-cycle transition | [10] | G + M |
| rs11169552 | 12q13.13 | DIP2 | Disco-interacting protein 2B | 1.09 | 1.89E-10 | Cell morphogenesis | [9] | M |
| rs1728785 | 16q22 | CDH1 | E-cadherin, | 1.17 | 2.80E-08 | Epithelial restitution, repair following mucosal damage, active colitis | [33] | G |
| rs10411210 | 19q13.33 | RHPN2 | Rho GTPase binding protein 2 | 1.15 | 5.00E-09 | Actin cytoskeleton | [20] | G + M |
| rs4925386 | 20q13.33 | LAMA5 | Large laminin A5 | 1.08 | 1.89E-10 | BMP pathway | [9] | M |
| rs5934683 | Xp22.2 | SHR00M2 | Shroom family member 2 | 1.07 | 7.30E-10 | Cell morphogenesis | [26] | G + M |
| Gene expression and regulation-related | ||||||||
| rs16892766 | 8q23.3 | EIF3H | Eukaryotic translation initiation factor 3, subunit H | 1.25 | 3.30E-18 | Translation initiation | [8] | G |
| rs7014348 | 8q24 | POU5FIP1 | POU class 5 homeobox 1B | 1.19 | 8.60E-26 | Weak transcriptional activator | [6] | G |
| rs7136702 | 12q13.13 | ATF1 | Activating transcription factor 1 | 1.06 | 4.02E-08 | Transcription | [9] | M |
| rs6017342 | 20q13.12 | HNF4A | Transcription factor hepatocyte nuclear factor 4α | 1.11 | 3.20E-17 | Transcription | [33] | G |
| Gene desert and others | ||||||||
| rs7524102 | 1p36.12 | - | - | 1.1 | 3.10E-07 | - | [33] | G |
| rs16823149 | 1q31 | Clorf21 | - | - | 5.50E-08 | - | [11] | G |
| rs4574118 | 2q12 | PLGLA | Plasminogen-like A, non-coding RNA | - | 1.80E-07 | - | [11] | G |
| rs10936599 | 3q26.2 | MYNN | Myoneurn gene | 1.08 | 3.39E-08 | Unknown | [9] | M |
| rs4140904 | 4p15.3 | NCAPC | Non-SMC condensing I complex, subunit G | - | 1.40E-07 | - | [11] | G |
| rs7758229 | 6q26-27 | SLC22A3 | organic cation transporter | 1.28 | 7.92E-09 | Transport of cationic drugs, toxins, and endogenous metabolism | [19] | G |
| rs6983267 | 8q24 | - | - | 1.18 | 1.51E-08 | - | [19] | G |
| rs7837328 | 8q24 | - | - | 1.17 | 7.44E-08 | - | [19] | G |
| rs2209907 | 9q21.3 | TLE4 | Transducin-like enhancer of spit 4 | - | 3.40E-08 | - | [11] | G |
| rs10795668 | 10p14 | - | - | 1.12 | 2.50E-13 | - | [8] | G |
| rs9548988 | 13q13.3 | - | - | 1.1 | 2.70E-07 | - | [33] | G |
| rs2423279 | 20p12.3 | PLCB1 | Phospholipase C-beta 1 | 1.1 | 6.64E-09 | Unknown | [29] | G + M |
SNP: Single nucleotide polymorphisms; G: Genome-wide association study; M: Meta-analysis; G + M: Combination of GWAS and meta-analysis; BMP: Bone morphogenetic protein; MAPK: Mitogen-activated protein kinase; TGF-β: Transforming growth factor; GWAS: Genome-wide association study.
Figure 1.
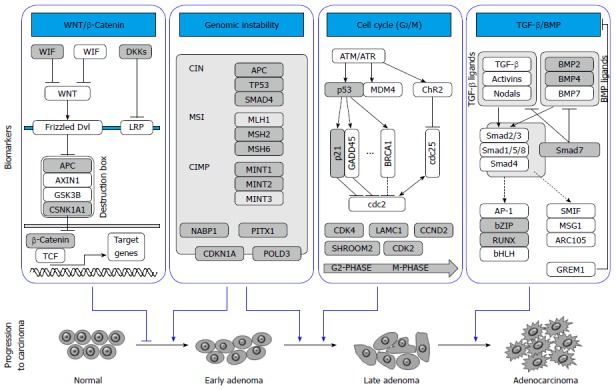
Major pathways with significant genetic variants implicated in the development of colorectal cancer. Several pathways and related genes involved in the progression of colorectal cancer are illustrated. Genes with significant single nucleotide polymorphisms that are associated with colorectal cancer risk are represented with gray color. TGF-β: Transforming growth factor-β; BMP: Bone morphogenetic protein; CIMP: CpG island methylator phenotype; MSI: Microsatellite instability; CIN: Chromosomal instability.
Genetic variants in signal transduction pathways
CRC GWASs have identified significant variants in signal transduction pathways such as those mediated by WNT/β-catenin, transforming growth factor (TGF)-β/bone morphogenetic protein (BMP), and mitogen-activated protein kinase (MAPK). Somatic mutations in the WNT/β-catenin signaling pathway were discovered in more than 95% of CRC patient tissues[16], suggesting abnormalities of genes in this pathway may play an important role in colorectal carcinogenesis. The risk allele rs59336 located in the intron of TBX3 gene, a downstream target of WNT/β-catenin pathway, has been associated with a significantly higher risk of developing CRC[8]. Changes in β-catenin and SMAD7 expression can influence WNT/β-catenin pathway signaling[17]. Moreover, perturbation of SMAD7 expression has been documented to affect CRC progression[18]. Three genetic variants of SMAD7 in chromosome 8q21 - rs4939827, rs12953717 and rs4464148 - confer an increased CRC risk[5]. These findings and other WNT/β-catenin variants were further independently identified and validated[6,19]. BMPs are closely related to signal transductions mediated by TGF-β. Two independent GWASs[9,20] identified 14 CRC risk loci, of which three were adjacent to genes involved in BMP-mediated signaling transduction, including rs4444235 on BMP4, rs961253 on BMP2, and rs4779584 on DNA family BMP antagonist GREM1. BMP-related variants were further confirmed in another independent CRC population[21]. The MAPK-mediated signaling pathway is known to be crucial for several cellular mechanisms such as cell proliferation, survival, and resistance to apoptosis. A GWAS using German familial CRC patients[9] observed that CRC risk increases significantly with an increase in the number of risk alleles in seven genes involved in MAPK signaling. The molecular basis of these observed associations remains undetermined.
Genetic variants related to genome instability
Genome instability is known to be both a contributor to, as well as a consequence of, colorectal carcinogenesis. There are several major genomic instability-related mechanisms in colorectal carcinogenesis, such as chromosomal instability, microsatellite instability, and CpG island methylator phenotypes[22]. Several loci involving these mechanisms were identified recently by GWASs. For example, Peters et al[10] identified rs11903757, a significant SNP in an intergenic locus on chromosome 2q32.3, proximal to NABP1, which encodes human single-stranded DNA binding protein 2 and plays a role in a diverse array of cellular processes such as DNA replication, recombination, transcription, and maintenance of genomic stability[23-25]. Another variant, rs1321311, is in linkage disequilibrium with a region that encompasses the CDKN1A gene[26], which encodes the p21 protein that mediates p53-dependent growth arrest, and affects multiple tumor suppressor pathways. The p21 protein also interferes with the activity of proliferating cell nuclear antigen (PCNA)-dependent DNA polymerase, thereby regulating DNA replication and repair. It has been demonstrated that down-regulation of p21 inversely correlates with microsatellite instability status[27,28]. Two additional CRC risk variants - rs248999 and rs647161 - could also potentially interact with p21[26,29]. Other genome instability related SNPs include rs248999, located in an intron of the POLD3 gene which encodes a component of the DNA polymerase-δ complex of PCNA, and rs647161 in a putative tumor suppressor homeodomain 1 gene PITX1, which has been reported to encode a protein that activates p53 protein and maintains genome stability[30,31].
Genetic variants related to cell cycle control
Genetic pathways mediating cell-cycle control are commonly implicated in colorectal carcinogenesis. Polymorphisms of several cell cycle-related genes have been reported to be associated with CRC risk in recent GWASs, including two independent SNPs (rs3217810 and rs3217901) located in the introns of CCND2. Jia et al[29] identified another SNP, rs10774214, located in 12q13.32, proximal to CCND2 in Asian populations. CCND2 encodes cyclin D2, a member of the D-type cyclin family which plays a critical role in cell cycle control, specifically at the G1/S boundary by activating cyclin-dependent kinases (CDKs), primarily CDK4 and CDK6[32]. Two significant SNPs, rs7136702 and rs11169552, lie about 275 kb apart within a large poorly-defined haplotype block covering the DIP2 gene, which encodes a protein with putative role in epithelial cell fate determination[9]. Another SNP, rs10911251, is proximal to the promoter of encoding laminin gamma 1 (LAMC1) and confers a significantly increased CRC risk by virtue of influencing LAMC1 gene expression[10]. SNPs in two additional laminin genes (laminin beta1 in 7q31 and laminin alpha 5 in 20q13) were also identified in recent CRC GWASs[9,21,33]. Laminins are known to be involved in a variety of cellular mechanisms such as regulation of cell adhesion, differentiation, and migration[34,35]. Another important cell cycle-related SNP was reported by Dunlop et al[26] using five GWAS datasets. This SNP, rs5934683, is on chromosome Xp22.2 and proximal to encoding shroom family member 2, a human homolog of the Xenopus laevis APX gene that is known to have broad functions in cell morphogenesis during endothelial and epithelial tissue development[36]. Missense mutations in this gene have been detected in a large-scale screening for recurrent mutations in cancer cell lines[37]. The relationship between Xp22.2 and CRC risk represents the first evidence for the role of X-chromosome variation in the predisposition to a non-sex-specific cancer.
Genetic variants related to gene expression and regulation
Thousands of transcription factors, cofactors, and chromatin regulators establish gene expression patterns and maintain specific cell stages in humans. Barrett et al[33] identified a significant association between CRC risk and SNP rs6017342 which maps to a recombination hot spot on chromosome 20q13 containing the 3’-untranslated region of the HNF4A gene. HNF4A encodes the transcription factor hepatocyte nuclear factor 4α, which regulates the expression of multiple organ development-related genes. In addition, HNF4A has also shown to interact with β-catenin to regulate cell-cell adhesion and gene transcription[38]. Another significant SNP, rs11169552, is close to activating transcription factor 1 (ATF1)[9], which belongs to the ATF subfamily and basic-region leucine zipper family. The protein product of ATF1 influences cellular processes by regulating the expression of many downstream target genes involved in cellular growth and survival. Previous studies have demonstrated that ATF1 protein interacts with EWSR1 to form a unique chimeric fusion protein complex which is important in the development of clear cell sarcoma[39,40]; however, its role in colorectal carcinogenesis remains to be established. Moreover, ATF1 may also form cyclin-dependent kinase 3-mdiated activating transcription factor 1 complex that is critical in cellular proliferation and malignant transformation[41].
Genetic variants in the gene desert regions and others
Although the majority CRC risk variants are related to well-established biological pathways, the functions of some reported loci remain elusive. Various independent studies have reported that multiple SNPs in chromosome region 8q24 are associated with altered risk of several solid tumor malignancies, including CRC. Three SNPs in this region, namely rs7014348, rs6983267, and rs7837328, have been significantly associated with CRC risk in recent GWASs[4,6,19,42]. In addition, variants in the 8q24 region have also been associated with cancers of breast, prostate, ovarian, bladder, pancreas, and brain[6,42-49]. Nonetheless, majority of the significant SNPs identified in this region are not located in, or close to, any well annotated genes because the 8q24 region is largely a gene desert. Therefore, details of the molecular mechanisms underlying the observed effect of these SNPs remain largely unknown. It has been speculated that these SNPs may function through their long-range linkage with causal variants within other oncogenes or tumor suppressor genes. Others have conjectured that some SNPs may influence gene expression through long-range cis-regulatory elements. Wasserman et al[50] used an in vivo bacterial artificial chromosome enhancer-trapping strategy to scan the 8q24 gene desert region and found that a highly significant CRC risk variant, rs6983267, resides within an in vivo prostate enhancer whose expression mimics that of the nearby MYC oncogene[51]. Another discovery illustrated a gene encoding a novel non-coding RNA, CCAT2, also mapped to the 8q24 gene desert region. Encompassing the rs6983267 SNP, this long non-coding RNA transcript is highly overexpressed in microsatellite-stable CRC, promoting tumor growth, metastasis, and chromosomal instability[52]. Another 8q24 locus, rs7014346, in high linkage disequilibrium with rs6983267, resides within 3 kb upstream of POU5F1P1, a pseudogene of POU5F1 that encodes an important stem cell-related protein regulating cellular pluripotency and self-renewal[53]. However, no functional implication of this SNP has been reported and it remains to be assessed whether it influences the development of CRC stem cells, a suspected small portion of cancer cells that are responsible for tumor progression and drug resistance[54]. In all, the identification of the large number of bona-fide risk variants in gene desert regions indicates that candidate-gene and pathway-based strategies may not be adequate to capture and understand the complete spectrum of common risk variants of CRC. Unbiased genome-wide interrogation in adequately powered studies, combined with meta-analysis and functional characterization is more likely to help us understand how common genetic variations play a role in CRC carcinogenesis.
GENETIC VARIANTS AND CRC CLINICAL OUTCOME
There have been reports of genetic variants associated with the clinical outcome of CRC patients which can be used to categorize patients with different survival patterns or responses to specific treatments. However, the majority of reported outcome-related SNPs are generated from candidate gene or pathway-based studies. As of yet, no GWAS has been reported to examine a direct relationship between genetic variations and CRC clinical outcome. The findings of some of recently published studies are summarized in Table 2.
Table 2.
Common genetic variants associated with colorectal cancer clinical outcome
| Genes/loci | SNP1 | Patient population | Clinical outcome | HR (95%CI) | P value | Ref. |
| MTHFR | glu429ala | Mixed colorectal cancer (CRC) patients | OS | 1.71 (1.18-2.49) | 0.005 | [64] |
| ESR2 | rs2987983 | Postmenopausal women with CRC | OS | 0.77 (0.60-0.99) | 0.002 | [65] |
| SCN1A | rs3812718 | Stage II/III patients with adjuvant 5-fuorouracil (5-FU) based chemotherapy | TTR | 2.26 (0.89-5.70) | 0.039 | [66] |
| SMAD7 | rs4939827 | Mixed CRC patients | OS | 1.16 (1.06-1.27) | 0.002 | [55] |
| mir608 | rs4919510 | Stage III patients with 5-FU based chemotherapy | RE | 1.65 (1.13-2.41) | 0.01 | [67] |
| rs4919510 | OS | 1.96 (1.19-3.21) | 0.008 | |||
| 15q13.3 | rs10318 | Stage II patients with 5-FU based adjuvant chemotherapy | ER | 2.98 (1.27-6.99) | 0.012 | [56] |
| 11q23.1 | rs10749971 | Stage III patients with 5-FU based adjuvant chemotherapy | ER | 0.46 (0.27-0.80) | 0.006 | |
| 20p12.3 | rs961253 | ER | 0.46 (0.22-0.96) | 0.038 | ||
| OS | 0.24 (0.09-0.68) | 0.007 | ||||
| 20p12.3 | rs355527 | ER | 0.48 (0.23-0.99) | 0.048 | ||
| OS | 0.29 (0.10-0.81) | 0.019 | ||||
| 18q21.1 | rs4464148 | OS | 4.34 (1.46-12.89) | 0.008 | ||
| 8q24.21 | rs6983267 | OS | 4.20 (1.13-15.64) | 0.032 | ||
| rs10505477 | OS | 4.20 (1.13-15.64) | 0.032 | |||
| 15q13 | rs4779584 | Chinese CRC patients | OS | 0.33 (0.15-0.72) | 0.007 | [57] |
| 10p14 | rs10795668 | RE | 0.55 (0.30-1.00) | 0.05 | ||
| pre-mi-423 | rs6505162 | Mixed CRC patients | OS | 2.12 (1.34-3.34) | 0.001 | [68] |
| rs6505162 | RFS | 1.59 (1.08-2.36) | 0.019 | |||
| pre-mi-608 | rs4919510 | RFS | 0.61 (0.41-0.92) | 0.017 | ||
| CLOCK | rs3749474 | Resected CRC patients | OS | 0.55 (0.37-0.81) | 0.003 | [69] |
| rs1801260 | OS | 0.31 (0.11-0.88) | 0.03 | |||
| SCD | rs7849 | Stage II patients with 5-FU based adjuvant chemotherapy | RE | 2.89 (1.54-5.41) | 0.001 | [70] |
| VEGF | -2578 | Stage II | TTR | 2.01 (1.13-3.56) | 0.02 | [71] |
| -460 | TTR | 0.50 (0.29-0.89) | 0.02 | |||
| KDR | rs10013228 | Resected CRC patients | RE | 0.53 (0.30-0.95) | 0.032 | [72] |
| CD44 | rs8193 | Stage III and high risk stage II patients with 5-FU based chemotherapy | TTR | 0.51 (0.35-0.93) | 0.022 | [73] |
| ALCAM | rs1157 | TTR | 0.56 (0.33-0.94) | 0.024 | ||
| LGR5 | rs17109924 | TTR | 0.33 (0.12-0.90) | 0.023 |
Only the most significant single nucleotide polymorphism (SNP) was shown. OS: Overall survival; RE: Recurrence; RFS: Recurrence free survival; TTR: Time to recurrence.
Three recent studies have examined the relationship between GWAS-identified CRC risk variants and the clinical outcome of the disease[55-57]. Based on the data from five GWAS populations of 2611 CRC patients, Phipps et al[55] assessed 16 SNPs and found rs4939827, a SNP in the SMAD7 gene, to be significantly associated with reduced overall survival of patients (HR = 1.16, P = 0.002) and disease-specific survival (HR = 1.17, P = 0.005). Dai et al[56] used a Caucasian population of 285 stage II or III CRC patients receiving fluorouracil-based chemotherapy to evaluate 26 CRC risk variants derived from 10 GWAS-identified chromosome loci. Although no SNP was found to be associated with the survival of all patients, they found that different SNPs might be associated with the clinical outcome of patients in specific stages. In another study of 380 Chinese CRC patients, Xing et al[57] reported two GWAS-identified CRC risk variants - rs4779584 on chromosome 15q13 and rs10795668 on 10p14 - were associated with reduced risk of both death and recurrence. Moreover, stratified analysis indicated that the beneficial effect of chemotherapy in this patient cohort was evident only in patients with the variant rs10795668, but not in those with the wild-type genotype of this SNP. This indicates that rs10795668 may potentially be useful in selecting patients for chemotherapy treatments. Taken together, these findings suggest that genetic variants associated with CRC risk may also predict the clinical outcome of CRC patients. However, these studies are limited by small sample size and heterogeneous patient population and treatments. Therefore, their findings need to be interpreted with caution and warrant further validations.
In addition to those GWAS-identified CRC risk loci, other epidemiological studies have also identified genetic variations associated with clinical outcome of CRC (Table 2). All of these studies are based on candidate gene or pathway-based approaches instead of GWAS. This is largely because compared to case-control studies, clinical outcome studies are generally based on cancer patients with highly heterogeneous characteristics and treatments that confound the very modest effect of genetic variants on patient outcomes. This issue could be partly resolved by the use of clinical trial patients that have more homogeneous characteristics and treatments, or consortium studies with much larger number of patients.
CONCLUSION
Findings from the first wave of GWASs seem to promise greater understanding of the genetic component of CRC pathogenesis on a molecular level. However, there are several major limitations in current GWAS approaches which may also pose challenges for future studies. First, the vast majority of currently identified SNPs lack known functional significance. Thus, whether they are causal variants or just surrogates that are in linkage disequilibrium with the functional loci remains largely unknown. Therefore, a major task ahead is to conduct fine-mapping in the immediate regions surrounding these loci, and narrow down the regions of association to pinpoint the causal variants[58]. Second, although the statistical significance of most GWAS-identified SNPs is high, the utility of these bona-fide variants in a clinical setting to predict the risk of developing cancer remains to be assessed. This is largely due to the modest effect size associated with most of the specific individual variants. Wacholder et al[59] reported a very modest increase in the power to predict breast cancer risk by adding 10 highly significant GWAS-identified breast cancer risk variants to the commonly recognized self-reported risk factors. Moreover, they found that the level of predicted breast cancer risk among most women barely changes by the addition of the GWAS-generated genetic information. Similarly, Park et al[60] reported that the combined use of all current genetic information derived from GWASs only has modest discriminative power (about 63.5% area under curve) in breast, prostate, and colorectal cancers. Therefore, further identification of additional low-penetrance common variants, especially the causal variants, is necessary to improve the clinical utility of GWAS-generated genetic information. Third, it is estimated that the currently validated SNPs in aggregate still explain only a small fraction of total heritability in most complex-trait diseases[61]. Several possible reasons may further account for this “missing heritability”. These include unidentified common variants, the unexplored territory of rare genetic variants that have high-penetrance but low-prevalence, and the largely un-assessed gene-gene and gene-environment interactions[21,62]. All of these issues could be partially addressed by increasing population size and thus statistical power. Thus, meta-analysis and combined analysis of multiple study populations are effective means to tackle these issues in the near future. In addition, using novel technologies such as the next generation sequencing to identify rare causal variants may also help address the missing heritability.
Despite these limitations, the identification of a host of specific genetic variants associated with elevated CRC risk through the GWAS approach does suggest the possibility of tailoring colorectal screening strategies such as age at first colonoscopy, and interval between surveillance colonoscopies. By better appreciating the mechanism by which these genetic variants alter CRC risk, morbidity and mortality could be reduced in higher risk sub-groups by more aggressive surveillance and cost could be reduced in low-risk groups requiring less intensive testing.
Footnotes
Supported by A start-up grant from Thomas Jefferson University; and National Cancer Institute Grant, CA162201
P- Reviewers: Camacho J, Tong WD, Yu B S- Editor: Gou SX L- Editor: A E- Editor: Ma S
References
- 1.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 2.Gala M, Chung DC. Hereditary colon cancer syndromes. Semin Oncol. 2011;38:490–499. doi: 10.1053/j.seminoncol.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected] Am J Gastroenterol. 2009;104:739–750. doi: 10.1038/ajg.2009.104. [DOI] [PubMed] [Google Scholar]
- 4.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–988. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 5.Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, Rowan A, Lubbe S, Spain S, Sullivan K, Fielding S, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 6.Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E, Cetnarskyj R, Cartwright N, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008;40:631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaeger E, Webb E, Howarth K, Carvajal-Carmona L, Rowan A, Broderick P, Walther A, Spain S, Pittman A, Kemp Z, et al. Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat Genet. 2008;40:26–28. doi: 10.1038/ng.2007.41. [DOI] [PubMed] [Google Scholar]
- 8.Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A, Sullivan K, et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008;40:623–630. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 9.Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, Spain SL, Broderick P, Domingo E, Farrington S, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 2010;42:973–977. doi: 10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron JA, Berndt SI, Bézieau S, Brenner H, Butterbach K, et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology. 2013;144:799–807.e24. doi: 10.1053/j.gastro.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lascorz J, Försti A, Chen B, Buch S, Steinke V, Rahner N, Holinski-Feder E, Morak M, Schackert HK, Görgens H, et al. Genome-wide association study for colorectal cancer identifies risk polymorphisms in German familial cases and implicates MAPK signalling pathways in disease susceptibility. Carcinogenesis. 2010;31:1612–1619. doi: 10.1093/carcin/bgq146. [DOI] [PubMed] [Google Scholar]
- 12.Kerr D. Clinical development of gene therapy for colorectal cancer. Nat Rev Cancer. 2003;3:615–622. doi: 10.1038/nrc1147. [DOI] [PubMed] [Google Scholar]
- 13.Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 15.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 16.Thorstensen L, Lind GE, Løvig T, Diep CB, Meling GI, Rognum TO, Lothe RA. Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability. Neoplasia. 2005;7:99–108. doi: 10.1593/neo.04448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han G, Li AG, Liang YY, Owens P, He W, Lu S, Yoshimatsu Y, Wang D, Ten Dijke P, Lin X, et al. Smad7-induced beta-catenin degradation alters epidermal appendage development. Dev Cell. 2006;11:301–312. doi: 10.1016/j.devcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 18.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Cui R, Okada Y, Jang SG, Ku JL, Park JG, Kamatani Y, Hosono N, Tsunoda T, Kumar V, Tanikawa C, et al. Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut. 2011;60:799–805. doi: 10.1136/gut.2010.215947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, Chandler I, Vijayakrishnan J, Sullivan K, Penegar S, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–1435. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomlinson IP, Carvajal-Carmona LG, Dobbins SE, Tenesa A, Jones AM, Howarth K, Palles C, Broderick P, Jaeger EE, Farrington S, et al. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 2011;7:e1002105. doi: 10.1371/journal.pgen.1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 24.Bochkarev A, Bochkareva E, Frappier L, Edwards AM. The crystal structure of the complex of replication protein A subunits RPA32 and RPA14 reveals a mechanism for single-stranded DNA binding. EMBO J. 1999;18:4498–4504. doi: 10.1093/emboj/18.16.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 26.Dunlop MG, Dobbins SE, Farrington SM, Jones AM, Palles C, Whiffin N, Tenesa A, Spain S, Broderick P, Ooi LY, et al. Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet. 2012;44:770–776. doi: 10.1038/ng.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogino S, Kawasaki T, Kirkner GJ, Ogawa A, Dorfman I, Loda M, Fuchs CS. Down-regulation of p21 (CDKN1A/CIP1) is inversely associated with microsatellite instability and CpG island methylator phenotype (CIMP) in colorectal cancer. J Pathol. 2006;210:147–154. doi: 10.1002/path.2030. [DOI] [PubMed] [Google Scholar]
- 28.Wangefjord S, Brändstedt J, Lindquist KE, Nodin B, Jirström K, Eberhard J. Associations of beta-catenin alterations and MSI screening status with expression of key cell cycle regulating proteins and survival from colorectal cancer. Diagn Pathol. 2013;8:10. doi: 10.1186/1746-1596-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia WH, Zhang B, Matsuo K, Shin A, Xiang YB, Jee SH, Kim DH, Ren Z, Cai Q, Long J, et al. Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet. 2013;45:191–196. doi: 10.1038/ng.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu DX, Lobie PE. Transcriptional activation of p53 by Pitx1. Cell Death Differ. 2007;14:1893–1907. doi: 10.1038/sj.cdd.4402209. [DOI] [PubMed] [Google Scholar]
- 31.Qi DL, Ohhira T, Fujisaki C, Inoue T, Ohta T, Osaki M, Ohshiro E, Seko T, Aoki S, Oshimura M, et al. Identification of PITX1 as a TERT suppressor gene located on human chromosome 5. Mol Cell Biol. 2011;31:1624–1636. doi: 10.1128/MCB.00470-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 33.Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, Wesley E, Parnell K, Zhang H, Drummond H, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turck N, Gross I, Gendry P, Stutzmann J, Freund JN, Kedinger M, Simon-Assmann P, Launay JF. Laminin isoforms: biological roles and effects on the intracellular distribution of nuclear proteins in intestinal epithelial cells. Exp Cell Res. 2005;303:494–503. doi: 10.1016/j.yexcr.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 35.Pouliot N, Saunders NA, Kaur P. Laminin 10/11: an alternative adhesive ligand for epidermal keratinocytes with a functional role in promoting proliferation and migration. Exp Dermatol. 2002;11:387–397. doi: 10.1034/j.1600-0625.2002.110501.x. [DOI] [PubMed] [Google Scholar]
- 36.Farber MJ, Rizaldy R, Hildebrand JD. Shroom2 regulates contractility to control endothelial morphogenesis. Mol Biol Cell. 2011;22:795–805. doi: 10.1091/mbc.E10-06-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraus C, Liehr T, Hülsken J, Behrens J, Birchmeier W, Grzeschik KH, Ballhausen WG. Localization of the human beta-catenin gene (CTNNB1) to 3p21: a region implicated in tumor development. Genomics. 1994;23:272–274. doi: 10.1006/geno.1994.1493. [DOI] [PubMed] [Google Scholar]
- 39.Yang L, Chen Y, Cui T, Knösel T, Zhang Q, Geier C, Katenkamp D, Petersen I. Identification of biomarkers to distinguish clear cell sarcoma from malignant melanoma. Hum Pathol. 2012;43:1463–1470. doi: 10.1016/j.humpath.2011.10.022. [DOI] [PubMed] [Google Scholar]
- 40.Lin WL, Chen FL, Kuo JF, Lee MY, Han CP. Cytokeratin 8/18 monoclonal antibody was dissimilar to anti-cytokeratin CAM 5.2.--a comment on: “Discovery of two novel EWSR1/ATF1 transcripts in four chimerical transcripts-expressing clear cell sarcoma and their quantitative evaluation, Experimental and Molecular Pathology 90(2): 194-200, April 2011”. Exp Mol Pathol. 2011;91:323–324. doi: 10.1016/j.yexmp.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Zheng D, Cho YY, Lau AT, Zhang J, Ma WY, Bode AM, Dong Z. Cyclin-dependent kinase 3-mediated activating transcription factor 1 phosphorylation enhances cell transformation. Cancer Res. 2008;68:7650–7660. doi: 10.1158/0008-5472.CAN-08-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007;39:989–994. doi: 10.1038/ng2089. [DOI] [PubMed] [Google Scholar]
- 43.Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, Seal S, Ghoussaini M, Hines S, Healey CS, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 45.Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, Gudmundsson J, Jakobsdottir M, Bergthorsson JT, Sigurdsson A, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet. 2008;40:1307–1312. doi: 10.1038/ng.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poynter JN, Figueiredo JC, Conti DV, Kennedy K, Gallinger S, Siegmund KD, Casey G, Thibodeau SN, Jenkins MA, Hopper JL, et al. Variants on 9p24 and 8q24 are associated with risk of colorectal cancer: results from the Colon Cancer Family Registry. Cancer Res. 2007;67:11128–11132. doi: 10.1158/0008-5472.CAN-07-3239. [DOI] [PubMed] [Google Scholar]
- 47.Park SL, Chang SC, Cai L, Cordon-Cardo C, Ding BG, Greenland S, Hussain SK, Jiang Q, Liu S, Lu ML, et al. Associations between variants of the 8q24 chromosome and nine smoking-related cancer sites. Cancer Epidemiol Biomarkers Prev. 2008;17:3193–3202. doi: 10.1158/1055-9965.EPI-08-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varghese JS, Easton DF. Genome-wide association studies in common cancers--what have we learnt? Curr Opin Genet Dev. 2010;20:201–209. doi: 10.1016/j.gde.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 49.Al Olama AA, Kote-Jarai Z, Giles GG, Guy M, Morrison J, Severi G, Leongamornlert DA, Tymrakiewicz M, Jhavar S, Saunders E, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet. 2009;41:1058–1060. doi: 10.1038/ng.452. [DOI] [PubMed] [Google Scholar]
- 50.Wasserman NF, Aneas I, Nobrega MA. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010;20:1191–1197. doi: 10.1101/gr.105361.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pomerantz MM, Beckwith CA, Regan MM, Wyman SK, Petrovics G, Chen Y, Hawksworth DJ, Schumacher FR, Mucci L, Penney KL, et al. Evaluation of the 8q24 prostate cancer risk locus and MYC expression. Cancer Res. 2009;69:5568–5574. doi: 10.1158/0008-5472.CAN-09-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ling H, Spizzo R, Atlasi Y, Nicoloso M, Shimizu M, Redis RS, Nishida N, Gafà R, Song J, Guo Z, et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013;23:1446–1461. doi: 10.1101/gr.152942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panagopoulos I, Möller E, Collin A, Mertens F. The POU5F1P1 pseudogene encodes a putative protein similar to POU5F1 isoform 1. Oncol Rep. 2008;20:1029–1033. [PubMed] [Google Scholar]
- 54.Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Colorectal cancer stem cells. Stem Cells. 2012;30:363–371. doi: 10.1002/stem.1031. [DOI] [PubMed] [Google Scholar]
- 55.Phipps AI, Newcomb PA, Garcia-Albeniz X, Hutter CM, White E, Fuchs CS, Hazra A, Ogino S, Nan H, Ma J, et al. Association between colorectal cancer susceptibility loci and survival time after diagnosis with colorectal cancer. Gastroenterology. 2012;143:51–54.e4. doi: 10.1053/j.gastro.2012.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dai J, Gu J, Huang M, Eng C, Kopetz ES, Ellis LM, Hawk E, Wu X. GWAS-identified colorectal cancer susceptibility loci associated with clinical outcomes. Carcinogenesis. 2012;33:1327–1331. doi: 10.1093/carcin/bgs147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xing J, Myers RE, He X, Qu F, Zhou F, Ma X, Hyslop T, Bao G, Wan S, Yang H, et al. GWAS-identified colorectal cancer susceptibility locus associates with disease prognosis. Eur J Cancer. 2011;47:1699–1707. doi: 10.1016/j.ejca.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Zaitlen N, Paşaniuc B, Gur T, Ziv E, Halperin E. Leveraging genetic variability across populations for the identification of causal variants. Am J Hum Genet. 2010;86:23–33. doi: 10.1016/j.ajhg.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wacholder S, Hartge P, Prentice R, Garcia-Closas M, Feigelson HS, Diver WR, Thun MJ, Cox DG, Hankinson SE, Kraft P, et al. Performance of common genetic variants in breast-cancer risk models. N Engl J Med. 2010;362:986–993. doi: 10.1056/NEJMoa0907727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–575. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci USA. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stacey SN, Sulem P, Jonasdottir A, Masson G, Gudmundsson J, Gudbjartsson DF, Magnusson OT, Gudjonsson SA, Sigurgeirsson B, Thorisdottir K, et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet. 2011;43:1098–1103. doi: 10.1038/ng.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Negandhi AA, Hyde A, Dicks E, Pollett W, Younghusband BH, Parfrey P, Green RC, Savas S. MTHFR Glu429Ala and ERCC5 His46His polymorphisms are associated with prognosis in colorectal cancer patients: analysis of two independent cohorts from Newfoundland. PLoS One. 2013;8:e61469. doi: 10.1371/journal.pone.0061469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Passarelli MN, Phipps AI, Potter JD, Makar KW, Coghill AE, Wernli KJ, White E, Chan AT, Hutter CM, Peters U, et al. Common single-nucleotide polymorphisms in the estrogen receptor β promoter are associated with colorectal cancer survival in postmenopausal women. Cancer Res. 2013;73:767–775. doi: 10.1158/0008-5472.CAN-12-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benhaim L, Gerger A, Bohanes P, Paez D, Wakatsuki T, Yang D, Labonte MJ, Ning Y, El-Khoueiry R, Loupakis F, et al. Gender-specific profiling in SCN1A polymorphisms and time-to-recurrence in patients with stage II/III colorectal cancer treated with adjuvant 5-fluoruracil chemotherapy. Pharmacogenomics J. 2014;14:135–141. doi: 10.1038/tpj.2013.21. [DOI] [PubMed] [Google Scholar]
- 67.Lin M, Gu J, Eng C, Ellis LM, Hildebrandt MA, Lin J, Huang M, Calin GA, Wang D, Dubois RN, et al. Genetic polymorphisms in MicroRNA-related genes as predictors of clinical outcomes in colorectal adenocarcinoma patients. Clin Cancer Res. 2012;18:3982–3991. doi: 10.1158/1078-0432.CCR-11-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xing J, Wan S, Zhou F, Qu F, Li B, Myers RE, Fu X, Palazzo JP, He X, Chen Z, et al. Genetic polymorphisms in pre-microRNA genes as prognostic markers of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2012;21:217–227. doi: 10.1158/1055-9965.EPI-11-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou F, He X, Liu H, Zhu Y, Jin T, Chen C, Qu F, Li Y, Bao G, Chen Z, et al. Functional polymorphisms of circadian positive feedback regulation genes and clinical outcome of Chinese patients with resected colorectal cancer. Cancer. 2012;118:937–946. doi: 10.1002/cncr.26348. [DOI] [PubMed] [Google Scholar]
- 70.Lin M, Eng C, Hawk ET, Huang M, Lin J, Gu J, Ellis LM, Wu X. Identification of polymorphisms in ultraconserved elements associated with clinical outcomes in locally advanced colorectal adenocarcinoma. Cancer. 2012;118:6188–6198. doi: 10.1002/cncr.27653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kjaer-Frifeldt S, Fredslund R, Lindebjerg J, Hansen TF, Spindler KL, Jakobsen A. Prognostic importance of VEGF-A haplotype combinations in a stage II colon cancer population. Pharmacogenomics. 2012;13:763–770. doi: 10.2217/pgs.12.38. [DOI] [PubMed] [Google Scholar]
- 72.Dong G, Guo X, Fu X, Wan S, Zhou F, Myers RE, Bao G, Burkart A, Yang H, Xing J. Potentially functional genetic variants in KDR gene as prognostic markers in patients with resected colorectal cancer. Cancer Sci. 2012;103:561–568. doi: 10.1111/j.1349-7006.2011.02194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gerger A, Zhang W, Yang D, Bohanes P, Ning Y, Winder T, LaBonte MJ, Wilson PM, Benhaim L, Paez D, et al. Common cancer stem cell gene variants predict colon cancer recurrence. Clin Cancer Res. 2011;17:6934–6943. doi: 10.1158/1078-0432.CCR-11-1180. [DOI] [PubMed] [Google Scholar]