

Abstract
Colorectal cancer is the most prevalent among digestive system cancers. Carcinogenesis relies on disrupted control of cellular processes, such as metabolism, proliferation, DNA damage recognition and repair, and apoptosis. Cell, tissue, organ and body physiology is characterized by periodic fluctuations driven by biological clocks operating through the clock gene machinery. Dysfunction of molecular clockworks and cellular oscillators is involved in tumorigenesis, and altered expression of clock genes has been found in cancer patients. Epidemiological studies have shown that circadian disruption, that is, alteration of bodily temporal organization, is a cancer risk factor, and an increased incidence of colorectal neoplastic disease is reported in shift workers. In this review we describe the involvement of the circadian clock circuitry in colorectal carcinogenesis and the therapeutic strategies addressing temporal deregulation in colorectal cancer.
Keywords: Colorectal cancer, Circadian rhythm, Clock gene
Core tip: The biological clock drives crucial cell processes, such as growth, proliferation, differentiation and apoptosis, controls metabolic pathways, and regulates tissue functions and behavioral cycles. Derangement of these phenomena is involved in colorectal carcinogenesis. The circadian clock circuitry is a leading actor in physiological regulation, a drawn in bystander in colorectal tumorigenesis, and a possible therapeutic target.
INTRODUCTION
Digestive system cancers account for approximation 20% of neoplastic disease incidence, and colorectal cancer (CRC) is the most prevalent, representing > 50% of all these cancers. CRC is the third most commonly diagnosed cancer, representing approximation 10% of all newly diagnosed cancers apart from skin cancers, with 19.4 new cases per 100000 of the male population and 17 new cases per 100000 of the female population. CRC is the fourth leading cause of cancer deaths, accounting for 8.1% of all cancer-related deaths worldwide. Epidemiological data report a 5-year CRC prevalence of 66.3 cancer survivors per 100000 population worldwide, and a cumulative CRC risk in persons aged < 75 years of 1.96% worldwide; 2.35% in men and 1.62% in women[1,2]. At present, the first-choice treatment is surgical excision with or without adjuvant chemotherapy, and the identification of novel biomarkers for prognosis and molecular targets for therapeutic intervention is urgently needed. CRC onset and progression are related to mutational pathways, such as chromosomal instability and microsatellite instability (MSI). MSI is distinguished as H (high frequency or probability), L (low frequency or probability) and S (stable); is determined by defects in the normal DNA damage response and mismatch repair process, leading to the loss or gain of repeated units on the daughter strand and resulting in length variation[3,4]; and influences cancer-specific survival and time to recurrence in CRC patients[5,6]. An early step in colorectal carcinogenesis is represented by mutations in the gene adenomatous polyposis coli (APC), causing large deletions in the C terminus of the proteins that are found in > 80% of human colon cancers. This region is involved in the binding of β-catenin and axin, and its deletion leads to post-translational stabilization and accumulation of β-catenin. In turn, this transcriptional factor translocates from the cytoplasm to the nucleus, where it relieves T-cell factor (TCF)/lymphocyte enhancer factor (LEF)1-mediated transcriptional repression, to activate several oncogenes and other gene targets, such as c-MYC and CCND1, which stimulate target-gene expression and eventually tumor phenotype development[7,8].
Circadian clock circuitry and the bowel
Colorectal carcinogenesis is related to the progressive loss of homeostatic control of cell proliferation, differentiation and apoptosis. Cellular processes and functions in living organisms show time-related variations[9-11]. The patterns of oscillation may be rhythmic with a period of approximation 24 h and are called circadian[12]. The circadian timing system responsible for the generation of these rhythmic variations is composed of central and peripheral oscillators[13]. The central pacemaker is located in the suprachiasmatic nuclei of the brain, which are entrained to the environmental light-dark cycle by photic inputs conveyed by the retino-hypothalamic tract, and synchronize self-sustained oscillators in peripheral tissues through autonomic nervous system fibers and hormone output; mainly represented by cortisol and melatonin[14,15]. The molecular clock responsible for the generation of circadian rhythmicity consists of a set of interlocking transcription-translation feedback loops that complete one cycle each day and are driven by the core clock genes encoding the circadian proteins brain and muscle aryl-hydrocarbon receptor nuclear translocator-like (ARNT)l (BMAL1/2 also called ARNTL/2), circadian locomotor output cycles kaput (CLOCK) or its paralog neuronal PAS domain protein (NPAS)2, PERIOD (PER) 1-2, CRYPTOCHROME (CRY) 1-2[16]. Other proteins that in some way are related to the clock gene machinery are represented by PER3, TIMELESS, timeless-interacting protein (TIPIN), and protein kinases. The genes PER1-2 and CRY1-2 are transcriptionally activated by the basic helix-loop-helix/PAS (period, aryl-hydrocarbon-receptor, single minded) transcription factors CLOCK and BMAL1, which heterodimerize and bind to E-box enhancer elements in the promoters of these genes[16]. In turn, PER and CRY proteins form a repression complex that translocates back into the nucleus and interacts with CLOCK and BMAL1, hindering their activity. PER3 is believed to be the product of one of the output genes, more than a core clock gene, considering that Per3 knockout mice do not show any circadian phenotype, whereas PER1 and PER2 play an essential role in the molecular clockwork[17]. TIMELESS is the homolog of a core circadian gene of Drosophila melanogaster and is maintained in mammals, but its role in the function of the mammalian molecular clock is still unclear. TIMELESS and its partner TIPIN interact with components of the DNA replication system to regulate DNA replication processes under both normal and stress conditions and are essential for ataxia telangiectasia and Rad3-related (ATR)-checkpoint kinase (Chk)1 and ataxia telangiectasia mutated (ATM)-Chk 2-mediated signaling and S-phase arrest[18,19]. CLOCK-BMAL1 heterodimer activates an assisting loop that promotes expression of the nuclear receptors reverse transcript of erythroblastosis gene (REV-ERB) α/β (encoded by NR1D1 and NR1D2, respectively), and retinoic acid-related (RAR) orphan receptor (ROR)α/γ, which in turn compete for ROR-responsive elements (ROREs) of the BMAL1 promoter, and control negatively and positively the rhythmic transcription of BMAL1, as the REV-ERBs repress BMAL1 transcription, while RORs activate it. This stabilizing negative loop is important for precise control of the circadian pacemaker, and these nuclear receptors regulate a number of physiological functions, including circadian rhythmicity, lipid metabolism, and cellular differentiation[20-23]. The correct functioning of the clock gene machinery relies on post-translational modifications of circadian proteins, represented by phosphorylation, O-GlcNAcylation, SUMOylation, acetylation, and deacetylation. Phosphorylation is operated by protein kinases, such as casein kinase (CK)1-ε (encoded by CSNK1Ε), which targets the proteins BMAL1, PER1, PER2, and CRY1; CK2, which targets BMAL1 and PER2; AMP-activated kinase (AMPK), which targets the CRY proteins; and glycogen synthase kinase (GSK)-3β, which targets the proteins CLOCK, BMAL1, PER2 and CRY2[24]. In the absence of GSK-3β-mediated phosphorylation BMAL1 becomes stabilized, decreasing the dependent circadian gene expression[25]. In turn, GSK-3β regulates the activity of O-GlcNAc transferase, which works as a metabolic sensor gauging the nutrient flux into the hexosamine biosynthesis pathway, and fine-tunes the regulation of the circadian clock through reversible change of structure and transcriptional activity of the circadian proteins CLOCK and PER, as well as stabilization of BMAL1 and CLOCK as a result of inhibition of their ubiquitination[26,27]. Furthermore, BMAL1 is SUMOylated in vivo on Lys259; CLOCK is necessary to stimulate this post-translational modification; and BMAL1 SUMOylation and activation oscillate with circadian rhythmicity in the mouse liver[28,29]. Acetylation is operated by CLOCK, whereas deacetylation is operated by SIRT1, a type III NAD+-dependent histone/protein deacetylase that is required for high-magnitude circadian transcription of several proteins encoded by core clock genes, including BMAL1, PER2, and CRY1, which counterbalances the histone acetyltransferase activity of CLOCK, and promotes the deacetylation and degradation of PER2. The interaction of SIRT1 with CLOCK:BMAL1 is not time dependent, whereas the NAD+-dependent SIRT1 activity changes in a circadian manner, and the circadian regulation of SIRT1 activity depends on CLOCK:BMAL1-mediated regulation of expression of nicotinamide phosphoribosyltransferase (NAMPT); the rate-limiting enzyme involved in NAD+ synthesis[30-34]. The clock gene machinery controls the expression of hundreds of genes (clock-controlled genes) that drive the expression of proline-and acidic amino acid-rich domain basic leucine zipper (PAR bZIP), including DBP (albumin D-site binding protein), TEF (thyrotroph embryonic factor), HLF (hepatic leukemia factor), and the nuclear factor, interleukin-3-regulated protein (NFIL3, also known as E4BP4), whose promoter contains a RORE and is transcriptionally regulated by REV-ERBs[35]. Many physiological processes in the gastrointestinal tract, such as motility of gut sections, activity of mucosal enzymes, function of mucosal transporters, and proliferation rate of different cell types, exhibit diurnal rhythms[36,37]. Studies in animal models and observations in humans have demonstrated that a circadian biological clock operates in the gastrointestinal tract, and the core clock genes drive the circadian expression of clock-controlled genes and tissue-specific output genes[38,39], coding for proteins involved in gut functions, such as NHE3, encoding an electroneutral Na+/H+ exchanger, and ATP1A1, ENaCg, DRA, AE1 and NHERF1, involved in colonic NaCl absorption[40]. In addition, the clock gene machinery drives the circadian rhythmicity of gut physiology and motility[41,42], suggesting possible links between disruptions of circadian biology and diseases of the gastrointestinal tract, such as motility disorders, inflammation, and cancer[43,44].
The biological clock in colorectal carcinogenesis
The clock genes drive the circadian rhythmicity of expression of so-called clock-controlled genes, which represent 5%-20% of the genome, comprising key cell-cycle regulators, tumor suppressor genes and oncogenes, and their expression regulates timing of basic cell functions, such as metabolism, xenobiotic detoxification, DNA damage repair, and autophagy[45,46]. Cell-cycle progression is tightly regulated and the circadian system is involved in the control of cell proliferation and apoptosis, driving the transcription and/or post-translational modification of key proteins that are essential for DNA replication such as thymidylate synthase, and molecules that gate cell division in mitosis such as WEE-1[47-49]. Disruption of the circadian clock may lead to deregulated cell proliferation and deregulation of the circadian clock has been implicated in CRC[49-52]. The alteration of bodily temporal organization, defined as circadian disruption, is considered a cancer risk factor, and an increased incidence of CRC has been reported in shift workers by epidemiological studies[53-55].
The role of the disruption of the molecular clockwork in colorectal carcinogenesis and CRC progression is hinted by experimental data showing distortion of circadian regulation in colonic neoplastic tissue and gene-specific disruption in the matched nontumorous tissues. Overexpression of PER1 in human cancer cell lines results in reduced colony formation and clonogenic expansion, in sensitization to radiation-induced apoptosis, and in altered expression of transcriptional target genes such as c-MYC and p21. In contrast, Per2-null mice show an increase in hyperplasia and neoplasia in response to γ radiation, and Per2 mutation has been shown to accelerate intestinal polyp formation in ApcMin/+ mice[56-58]. PER1 and PER2 are involved in ATM-Chk1/Chk2 DNA damage response pathways and modulate β-catenin, encoded by the clock-controlled gene CTNNB1, whose promoter shows BMAL1 occupancy, indicating direct circadian regulation by this transcription factor[59,60], and that can function as an oncogene, influencing cell proliferation in colon cancer cells. In turn, intestinal tumorigenesis may alter clock function as a result of increased β-catenin destabilization of PER2 protein and impairing circadian clock gene expression in intestinal mucosa of ApcMin/+ mice[61].
In mice, mutation in the Period genes leads to altered temporal expression of genes involved in cell-cycle regulation and tumor suppression, such as c-Myc, Cyclin D1, Cyclin A, Mdm-2 and GADD45Α, deregulation of DNA-damage response, accelerated intestinal polyp formation in ApcMin/+ mice, and increased neoplastic growth[62]. Moreover, the BMAL1 gene is a fundamental hinge in the clock gene machinery and plays a key role in the regulation of tumor cell apoptosis, cell-cycle progression, and DNA damage response. In in vitro and in vivo experiments the knockdown of BMAL1 by RNA interference in murine colon cancer cells (C26) reduced the expression of Per1, Per2, Per3, Wee1 and Tp53; decreased apoptosis induced by etoposide; reduced the distribution in the G2/M phases of cells treated by docetaxel; and decreased DNA damage induced by cisplatin, accelerating cell proliferation in vitro and promoting tumor growth in mice[63]. A role in the early stages of colorectal carcinogenesis is played also by CK1ε, as demonstrated by in vitro and in vivo studies, and knock down of CSNK1E or use of a kinase inhibitor specific to CK1ε induced tumor-cell-selective cytotoxicity, because tumor cells depend more on the kinase activity of CK1ε than normal cells do[64,65].
The function of the circadian clock during neoplastic transformation was evaluated in a mouse model of chemically induced primary CRC, and the daily profiles of the core clock genes Per1, Per2, Rev-Erbα and Bmal1, the clock-controlled gene Dbp and the clock-controlled cell cycle genes Wee1, c-Myc and p21 were assessed in the tumor, matched nontumorous tissue, and the liver[66]. The circadian rhythmicity of Per1, Per2, Rev-Erbα and Dbp was significantly decreased in CRC compared with nontumorous tissue, and the expression of Bmal1 was not rhythmic. Besides, the circadian expression of Per1, Per2, Rev-Erbα and Dbp was present in the nontumorous matched colonic tissue, but the expression of Bmal1 did not show a circadian rhythm[66]. The expression patterns of Wee1, c-Myc and p21 did not show circadian rhythmicity in tumors or the colon of healthy animals. A phase shift was evidenced for the rhythmic expression of the clock genes in the liver of tumor-bearing mice[66].
Clock genes in human colorectal cancer
There has been much interest in the alteration of expression of clock genes and clock-controlled genes in humans affected by CRC[67,68]. CLOCK is reported to be mutated in cancer, and may be involved in carcinogenesis through changed response to DNA damage. Microarray gene expression data combined with public gene sequence information have identified CLOCK as a potential target of somatic mutations in microsatellite unstable CRCs, and CLOCK mutations occurred in 53% of MSI CRCs[69]. Restoring CLOCK expression in a human colon adenocarcinoma cell line derived from a primary colon cancer lacking wild-type CLOCK, and testing the effects of UV-induced apoptosis and radiation by DNA content analysis using flow cytometry, demonstrated protection against UV-induced apoptosis and decreased G2/M arrest in response to ionizing radiation. Novel CLOCK-binding elements were identified near DNA damage genes p21, NBR1, BRCA1 and RAD50 by means of chromatin immunoprecipitation with parallel DNA sequencing[69]. Furthermore, changes in the expression of PER2 were demonstrated by immunohistochemical staining and real-time polymerase chain reaction in tumor tissue of CRC patients, with heterogeneous, and negative or weak staining patterns in cancerous cells, with higher PER2 expression in well-differentiated cancer cells when compared to poorly differentiated ones, and associations of decreased PER2 levels with patient age, histological grade, TNM stage and expression of nuclear proliferation-related antigen Ki67[70]. Besides, downregulation of PER3 was seen in tumor tissues of CRC patients, associated with various clinicopathological factors, comprising tumor location, differentiation, and stage, and to poorer survival, suggesting an important role in CRC progression[71]. Among the clock-controlled genes that drive intestinal physiology, an important role is played by PPARG, which codes for the transcription factor peroxisome proliferator-activated receptor (PPAR)γ, which is involved in several physiological processes, and an important interplay has been demonstrated between PPARγ and β-catenin[72]. β-Catenin is crucial in the WNT signaling pathway, and plays an essential role in the regulation of gene expression interacting with several molecular partners, such as APC and the TCF/LEF1 family transcription factors, and in cell adhesion, bridging between cadherins and the actin cytoskeleton at the cell-cell adherens junctions. Cytoplasmic but not membrane-bound β-catenin forms a complex with APC, the associated protein axin/conductin, CK2 and GSK-3β, which phosphorylate β-catenin and other elements of the molecular proteolytic complex. Activation of WNT signaling involves the inhibition of GSK-3β through mechanisms that may involve axin binding to the proteins Dishevelled or low-density lipoprotein receptor-related protein (LRP)-5, determining nuclear β-catenin accumulation, binding to TCF/LEF1 transcription factors, and expression of target genes including c-MYC and CCND1[73]. The protein kinase CK1ε is involved in cell proliferation by stabilizing β-catenin, and CK1ε overexpression mimics the effect of WNT signalling, resulting in cytoplasmic accumulation of β-catenin and its subsequent nuclear localization, to control transcription and support tumorigenesis[74-76]. The role of CK1ε in cell proliferation and in circadian clock function in the context of colorectal carcinogenesis might be not crucial. In the absence of WNT signaling, β-catenin is destabilized by GSK-3β, which plays a role in mammalian circadian clock function, counteracts the ability of CK1ε to promote β-catenin stability, and is involved in circadian control of cell-cycle progression[77].
PPARγ is a ligand-activated transcription factor belonging to the large superfamily of nuclear receptors, regulates cellular proliferation/differentiation, and plays a role in colorectal carcinogenesis[78]. In the normal mucosa of the colon and rectum there is high PPARγ expression, and in mouse small intestine and colon, decreased intestinal PPARγ levels are associated with enhanced tumorigenicity[79]. In CRC patients, PPARγ expression levels are not uniformly changed, suggesting that PPARG deregulation plays different and context-dependent roles in CRC onset and progression, and that this nuclear receptor inhibits tumor growth only in the presence of functional APC[80].
The functioning of the clock gene machinery is severely altered in patients affected by CRC, with downregulation of BMAL1, PER1, PER2, PER3 and CRY2, upregulation of BMAL2 and TIMELESS (in particular in MSI-H and MSI-L tumors), and significant differences in survival related to differential expression of clock genes[44,81]. The altered expression of clock genes influences also phenotypic characteristics of colon cancer cells and disease outcome, and a correlation between high expression of the BMAL1 gene and low expression of the PER1 gene and liver metastasis, and between high expression of the PER2 gene and significantly better outcomes of CRC have been observed[82]. The upregulation of BMAL2 is accompanied by high expression of SERPINE1 in CRC patients[81]. Invasion by cancer cells requires proteases to degrade the cellular matrix. Cathepsins, which are cysteine proteases, play a crucial role in this process through the destruction of the cell-surrounding extracellular matrix[83]. Plasmin is formed from its zymogen, plasminogen; a reaction catalyzed by the serine protease urokinase-type plasminogen activator (uPA) and partially regulated by plasminogen activator inhibitors, such as plasminogen activator inhibitor (PAI)-1[84]. A high concentration of PAI1 in tumor biopsy specimens, the major physiological regulator of the pericellular plasmin-generating cascade, is associated with poor prognosis, and high preoperative plasma concentrations of PAI-1 are associated with shorter survival in CRC patients[85]. Both CLOCK:BMAL1 and CLOCK:BMAL2 heterodimers powerfully activate the promoter of the gene encoding PAI-1, officially called SERPINE1 and located on the seventh chromosome (7q21.3-q22), underlying the circadian variation in circulating PAI-1[86,87]. CLOCK:BMAL2, binding two E-box enhancers in the promoter, is more potent than CLOCK:BMAL1 in its ability to activate SERPINE1[88-90].
Colorectal tumorigenesis is influenced also by SIRT1 that may behave as a tumor suppressor or as an oncogene in different in vitro and in vivo conditions. The involvement of SIRT1 in onset and progression of CRC is corroborated by the observation that transgenic mice overexpressing SIRT1 in the intestine show reduced development of tumors caused by ApcMin/+ mutation, and that SIRT1–/– mice show increased tumor incidence when crossed to a p53–/– background. In CRC patients, the expression levels are unevenly modified, reflecting the different settings that characterize the experimental conditions, and different colon cancer cell lines show dissimilar levels of SIRT1 expression and variable changes in clock gene expression after ectopic upregulation[91]. In normal colon, significant levels of nuclear SIRT1 are expressed in proliferating colon epithelial cells at the base of the crypt, and expression gradually decreases in cells migrating toward the lumen. In contrast, in samples of benign adenomas (polyps), SIRT1 is detected in all cells with an adenomatous morphology of benign adenomas (polyps), but not in the adjacent normal mucosa. Colonic adenocarcinoma shows a heterogeneous SIRT1 expression profile, suggesting that in normal colon epithelium, SIRT1 levels correlate with active cell proliferation. SIRT1 is expressed at high levels in normal colon and benign lesions, and high SIRT1 expression is an intrinsic response to cell proliferation in untransformed mucosa and premalignant adenoma. In CRC tissue, SIRT1 expression is downregulated in a subset of tumors to facilitate tumor growth, and in particular in high grade tumors, where the growth-inhibitory activity of SIRT1 becomes a rate-limiting factor for tumor progression, whereas some low- grade tumors may overexpress SIRT1 to benefit from its antiapoptotic effects[92].
Circadian rhythmicity and CRC treatment
The body systems involved in xenobiotic detoxification show circadian variations of function and activity driven by the clock gene machinery, determining time-dependent toxicity of xenobiotics and drugs[93-95]. The product of the multidrug resistance (mdr1a) gene, P-glycoprotein, works as a xenobiotic transporter contributing to the intestinal barrier, and intestinal expression of the mdr1a gene and its efflux pump function show oscillation with 24-h periodicity driven by the circadian clock through the opposing action of HLF and E4BP4, connecting the circadian timekeeping system to xenobiotic detoxification[96]. The time-of-day-dependent variations in cancer cell proliferation, as well as drug metabolism, toxicity and effectiveness represent the rationale for the timing of drug administration over a 24-h period[97-100], and they form the basis for chronomodulated chemotherapy of advanced-stage CRC, characterized by an opposite phase of delivery of oxaliplatin in the afternoon, with respect to 5-fluorouracil and leucovorin that are delivered late at night (chronoFLO4)[101]. Chronomodulated delivery of chemotherapy has shown better tolerability and antitumor activity compared with conventional chemotherapy, with less myelosuppression and more gastrointestinal toxicity for the chronoschedule, but with a favorable difference between treatments in median survival time only in male patients. Indeed, sex dependency in the effects of this schedule has been demonstrated with shorter survival and greater toxicity reported in female CRC patients in the European Organisation for Research and Treatment of Cancer (EORTC) Chronotherapy Group trial[102]. From these studies, sex emerges as the single predictor of survival, conditioning outcome of chronoFLO4 and determining genetic variation of metabolic responses that influence time-related variables and hinder administration of maximal effective dosing. A different genotypic profile between men and women could characterize CRC patients, and the higher burden of toxicity reported in women treated with 5-fluorouracil-based chemotherapy may be also due to the lower expression of dihydropyrimidine dehydrogenase in the tumors in female patients and/or to sex dependency of circadian pharmacology[68]. Xenobiotic detoxification and metabolic pathways differ between male and female mammals including humans. In double mutant Cry1–/–Cry2–/– male mice, sex dimorphism in hepatic drug metabolism is altered. Upon inactivation of the Cryptochrome genes, the levels of sex-specific liver products, including several cytochrome P450 enzymes, expressed by male mice are similar to those expressed by female mice[103]. In humans, the decreased expression level of Cryptochrome genes observed in female patients affected by CRC[44] might explain the different median survival and increased toxicity observed after chronomodulated chemotherapy administration in the EORTC Chronotherapy Group trial[104]. High Cryptochrome gene expression and protein levels in tumor tissue are correlated with tumor progression and poorer overall and disease-free survival in CRC patients[104].
Many cytotoxic anticancer drugs damage DNA and may activate DNA checkpoints permitting attempted DNA repair. This process is essential for cell survival, but may reduce the cytotoxicity of anticancer drugs. TIMELESS is required for ATM-dependent Chk2-mediated signaling of doxorubicin-induced DNA double-strand breaks, and downregulation of TIMELESS by siRNA significantly attenuates doxorubicin-induced G2/M cell cycle arrest and sensitizes cancer cells to doxorubicin-induced cytotoxicity. Therefore, upregulation of TIMELESS in CRC may predict altered drug sensitivity, and TIMELESS inhibition is a potential novel anticancer drug target to enhance the cytotoxic effectiveness of chemotherapeutic drugs known to activate DNA response pathways within cancer cells[105]. TIMELESS expression is also significantly associated with MSI status in CRC patients, and higher expression is found in patients with MSI-H and MSI-L[44]. Approximately 15% of CRC is characterized by defects in the DNA mismatch repair system, leading to MSI that generates a large number of substitution, as well as insertion and deletion mutations. These mutations typically target microsatellite sequences and lead to shifts in the reading frame, resulting in truncation or other alterations of the protein product. The presence of premature stop codons decreases the expression of mRNAs with such frameshift mutations and results in degradation of some of the mutant mRNA through the nonsense-mediated decay pathway. Response to adjuvant chemotherapy may be influenced by MSI status in CRC patients, because MSI-H tumors show better outcome with irinotecan-containing than 5-fluorouracil-containing regimens[106]. The increased expression of TIMELESS in MSI-H and MSI-L CRC may be related to the process of tumorigenesis and might cause reduced response to possible adiuvant chemotherapy. The circadian CLOCK-BMAL1 transactivation complex may be directly involved in the response to stress at the individual cell level, because Clock mutant and Bmal1 knockout mice are highly sensitive to cyclophosphamide treatment in a time-of-day-independent manner, suggesting that CLOCK and BMAL1 directly regulate the expression of cell-cycle- and apoptosis-related genes, and that time-related and allelic-dependent variations in response to chemotherapy correlate with the functional status of the CLOCK-BMAL1 heterodimer[93-95,107]. The frequencies of the 311T>C CLOCK gene CC genotype and C allele were significantly higher among CRC patients compared to controls, increasing the risk of CRC by 2.78- and 1.78-fold, respectively[108]. Furthermore, higher levels of CLOCK gene and protein expression were observed in human CRC tissues, particularly in poorly differentiated, advanced Dukes’ stage tumors and in cases with lymph node involvement, and a strong positive linear correlation was found with ARNT, hypoxia-inducible factor-1α and vascular endothelial growth factor expression in tumor tissue[109,110]. BMAL1 protein suppresses the proto-oncogene c-MYC, and stimulates the tumor suppressor WEE1, confirming the important role played by the altered expression of this core clock gene in the process of carcinogenesis[50]. In addition, a tight interplay has been demonstrated between aryl hydrocarbon receptor (AHR)/ARNT system and circadian pathways, and this interconnection might be critical to influence onset, promotion, and progression of colorectal malignant neoplasms[46]. An important role is played in carcinogenesis by the transcription factors of the AHR/ARNT system, which bind to a xenobiotic response element and control the expression of genes encoding enzymes involved in the metabolism and detoxification of exogenous and endogenous compounds, but they are antagonized by the AHR repressor[111] (Figure 1). Upon ligand-mediated activation, the AHR/ARNT system modulates metabolic pathways and cell processes, such as survival, cell cycle, proliferation, apoptosis, differentiation, epithelial to mesenchymal transition, angiogenesis, inflammation, cell contact inhibition, cell-matrix interaction, and extracellular matrix remodeling, which are crucial in the initiation and evolution of neoplastic disease[112-114].
Figure 1.
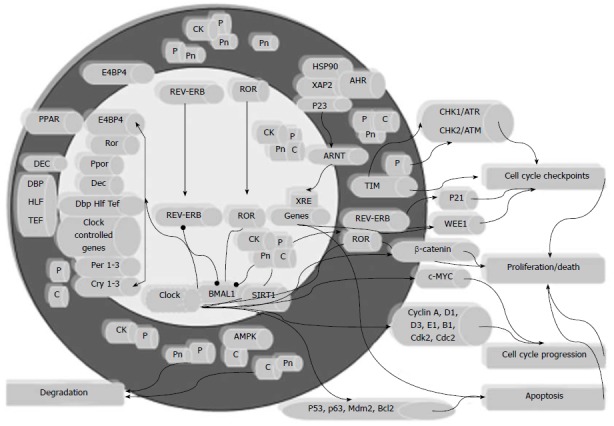
Biological clock and the cell processes involved in colorectal carcinogenesis. The scheme renders the transcriptional/translational feedback loop through which the molecular clockwork operates, and depicts its interplay with the aryl hydrocarbon receptor (AHR)/ARNT system in the control of the cell cycle and apoptosis, and ultimately in the regulation of the processes of cell proliferation and death, whose deregulation brings about colorectal cancer (CRC) onset and progression. CK: Casein kinase; AHR: Aryl hydrocarbon receptor; PPAR: Peroxisome proliferator-activated receptor; CHK: Checkpoint kinase; ATR: Ataxia telangiectasia and rad3-related; ATM: Ataxia telangiectasia mutated; ARNT: Aryl hydrocarbon receptor nuclear translocator; HLF: Hepatic leukaemia factor; TEF: Thyrotroph embryonic factor.
CONCLUSION
The biological clock plays an important role in bowel physiology, and alteration of the molecular clockwork is involved in colorectal carcinogenesis. The circadian genes and proteins are variably altered in colorectal malignant neoplasm and influence phenotypic characteristics of colon cancer cells, disease progression, patient survival, and response to chemotherapy. A better understanding of the mechanisms controlled by the clock gene machinery in normal conditions and in time- and biological-clock-related derangements in colorectal carcinogenesis will allow further advances in the comprehension of the pathophysiological mechanism operating in intestinal cancer, and will provide more effective therapeutic strategies for patients suffering from colorectal malignant tumors.
Footnotes
Supported by The “5x1000” voluntary contribution and by a grant to GM from the Italian Ministry of Health through Department of Medical Sciences, Division of Internal Medicine and Chronobiology Unit, IRCCS Scientific Institute and Regional General Hospital “Casa Sollievo della Sofferenza”, Opera di Padre Pio da Pietrelcina, San Giovanni Rotondo (FG), Italy, Nos. RC1203ME46 and RC1302ME31; by a grant to AP from the Italian Ministry of Health through Department of Medical Sciences, Division of Gastroenterology and Research Laboratory, Nos. RC1203GA55 and RC1203GA56; and by a grant to MV from AIRC, No. MFAG-AIRC2012-13419
P- Reviewers: Hirayama J, Shi Q, Xiong XJ S- Editor: Ma YJ L- Editor: Kerr C E- Editor: Liu XM
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Parkin DM, Curado MP, Bray F, Edwards B, Shin HR, Forman D. Cancer Incidence in Five Continents, Volumes I to IX: IARC CancerBase No. 9 [Internet] Lyon, France: International Agency for Research on Cancer; 2010. Available from: http://ci5.iarc.fr. [Google Scholar]
- 3.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jensen SA, Vainer B, Kruhøffer M, Sørensen JB. Microsatellite instability in colorectal cancer and association with thymidylate synthase and dihydropyrimidine dehydrogenase expression. BMC Cancer. 2009;9:25. doi: 10.1186/1471-2407-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright CM, Dent OF, Newland RC, Barker M, Chapuis PH, Bokey EL, Young JP, Leggett BA, Jass JR, Macdonald GA. Low level microsatellite instability may be associated with reduced cancer specific survival in sporadic stage C colorectal carcinoma. Gut. 2005;54:103–108. doi: 10.1136/gut.2003.034579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 7.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 8.Kobayashi M, Honma T, Matsuda Y, Suzuki Y, Narisawa R, Ajioka Y, Asakura H. Nuclear translocation of beta-catenin in colorectal cancer. Br J Cancer. 2000;82:1689–1693. doi: 10.1054/bjoc.1999.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. 2012;74:246–260. doi: 10.1016/j.neuron.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Lemmer B. Discoveries of rhythms in human biological functions: a historical review. Chronobiol Int. 2009;26:1019–1068. doi: 10.3109/07420520903237984. [DOI] [PubMed] [Google Scholar]
- 11.Bonny O, Vinciguerra M, Gumz ML, Mazzoccoli G. Molecular bases of circadian rhythmicity in renal physiology and pathology. Nephrol Dial Transplant. 2013;28:2421–2431. doi: 10.1093/ndt/gft319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duguay D, Cermakian N. The crosstalk between physiology and circadian clock proteins. Chronobiol Int. 2009;26:1479–1513. doi: 10.3109/07420520903497575. [DOI] [PubMed] [Google Scholar]
- 13.Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Adv Genet. 2011;74:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004;119:693–705. doi: 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 17.Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun ZS, Eichele G, Bradley A, et al. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell. 2001;105:683–694. doi: 10.1016/s0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- 18.Smith KD, Fu MA, Brown EJ. Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. J Cell Biol. 2009;187:15–23. doi: 10.1083/jcb.200905006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kemp MG, Akan Z, Yilmaz S, Grillo M, Smith-Roe SL, Kang TH, Cordeiro-Stone M, Kaufmann WK, Abraham RT, Sancar A, et al. Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J Biol Chem. 2010;285:16562–16571. doi: 10.1074/jbc.M110.110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burris TP. Nuclear hormone receptors for heme: REV-ERBalpha and REV-ERBbeta are ligand-regulated components of the mammalian clock. Mol Endocrinol. 2008;22:1509–1520. doi: 10.1210/me.2007-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ripperger JA, Albrecht U. REV-ERB-erating nuclear receptor functions in circadian metabolism and physiology. Cell Res. 2012;22:1319–1321. doi: 10.1038/cr.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin L, Wu N, Lazar MA. Nuclear receptor Rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl Recept Signal. 2010;8:e001. doi: 10.1621/nrs.08001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature. 2012;485:123–127. doi: 10.1038/nature11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchida Y, Hirayama J, Nishina H. A common origin: signaling similarities in the regulation of the circadian clock and DNA damage responses. Biol Pharm Bull. 2010;33:535–544. doi: 10.1248/bpb.33.535. [DOI] [PubMed] [Google Scholar]
- 25.Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS One. 2010;5:e8561. doi: 10.1371/journal.pone.0008561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaasik K, Kivimäe S, Allen JJ, Chalkley RJ, Huang Y, Baer K, Kissel H, Burlingame AL, Shokat KM, Ptáček LJ, et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013;17:291–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li MD, Ruan HB, Hughes ME, Lee JS, Singh JP, Jones SP, Nitabach MN, Yang X. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab. 2013;17:303–310. doi: 10.1016/j.cmet.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardone L, Hirayama J, Giordano F, Tamaru T, Palvimo JJ, Sassone-Corsi P. Circadian clock control by SUMOylation of BMAL1. Science. 2005;309:1390–1394. doi: 10.1126/science.1110689. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Lee Y, Lee MJ, Park E, Kang SH, Chung CH, Lee KH, Kim K. Dual modification of BMAL1 by SUMO2/3 and ubiquitin promotes circadian activation of the CLOCK/BMAL1 complex. Mol Cell Biol. 2008;28:6056–6065. doi: 10.1128/MCB.00583-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 32.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153:1448–1460. doi: 10.1016/j.cell.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bozek K, Relógio A, Kielbasa SM, Heine M, Dame C, Kramer A, Herzel H. Regulation of clock-controlled genes in mammals. PLoS One. 2009;4:e4882. doi: 10.1371/journal.pone.0004882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoogerwerf WA. Role of biological rhythms in gastrointestinal health and disease. Rev Endocr Metab Disord. 2009;10:293–300. doi: 10.1007/s11154-009-9119-3. [DOI] [PubMed] [Google Scholar]
- 37.Polidarová L, Soták M, Sládek M, Pacha J, Sumová A. Temporal gradient in the clock gene and cell-cycle checkpoint kinase Wee1 expression along the gut. Chronobiol Int. 2009;26:607–620. doi: 10.1080/07420520902924889. [DOI] [PubMed] [Google Scholar]
- 38.Polidarová L, Sládek M, Soták M, Pácha J, Sumová A. Hepatic, duodenal, and colonic circadian clocks differ in their persistence under conditions of constant light and in their entrainment by restricted feeding. Chronobiol Int. 2011;28:204–215. doi: 10.3109/07420528.2010.548615. [DOI] [PubMed] [Google Scholar]
- 39.Mazzoccoli G, Francavilla M, Pazienza V, Benegiamo G, Piepoli A, Vinciguerra M, Giuliani F, Yamamoto T, Takumi T. Differential patterns in the periodicity and dynamics of clock gene expression in mouse liver and stomach. Chronobiol Int. 2012;29:1300–1311. doi: 10.3109/07420528.2012.728662. [DOI] [PubMed] [Google Scholar]
- 40.Soták M, Polidarová L, Musílková J, Hock M, Sumová A, Pácha J. Circadian regulation of electrolyte absorption in the rat colon. Am J Physiol Gastrointest Liver Physiol. 2011;301:G1066–G1074. doi: 10.1152/ajpgi.00256.2011. [DOI] [PubMed] [Google Scholar]
- 41.Hoogerwerf WA, Shahinian VB, Cornélissen G, Halberg F, Bostwick J, Timm J, Bartell PA, Cassone VM. Rhythmic changes in colonic motility are regulated by period genes. Am J Physiol Gastrointest Liver Physiol. 2010;298:G143–G150. doi: 10.1152/ajpgi.00402.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoogerwerf WA. Role of clock genes in gastrointestinal motility. Am J Physiol Gastrointest Liver Physiol. 2010;299:G549–G555. doi: 10.1152/ajpgi.00147.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazzoccoli G, Palmieri O, Corritore G, Latiano T, Bossa F, Scimeca D, Biscaglia G, Valvano MR, D’Incà R, Cucchiara S, et al. Association study of a polymorphism in clock gene PERIOD3 and risk of inflammatory bowel disease. Chronobiol Int. 2012;29:994–1003. doi: 10.3109/07420528.2012.705935. [DOI] [PubMed] [Google Scholar]
- 44.Mazzoccoli G, Panza A, Valvano MR, Palumbo O, Carella M, Pazienza V, Biscaglia G, Tavano F, Di Sebastiano P, Andriulli A, et al. Clock gene expression levels and relationship with clinical and pathological features in colorectal cancer patients. Chronobiol Int. 2011;28:841–851. doi: 10.3109/07420528.2011.615182. [DOI] [PubMed] [Google Scholar]
- 45.Mazzoccoli G, Pazienza V, Vinciguerra M. Clock genes and clock-controlled genes in the regulation of metabolic rhythms. Chronobiol Int. 2012;29:227–251. doi: 10.3109/07420528.2012.658127. [DOI] [PubMed] [Google Scholar]
- 46.Anderson G, Beischlag TV, Vinciguerra M, Mazzoccoli G. The circadian clock circuitry and the AHR signaling pathway in physiology and pathology. Biochem Pharmacol. 2013;85:1405–1416. doi: 10.1016/j.bcp.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 47.Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–259. doi: 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- 48.Hunt T, Sassone-Corsi P. Riding tandem: circadian clocks and the cell cycle. Cell. 2007;129:461–464. doi: 10.1016/j.cell.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 49.Filipski E, King VM, Li X, Granda TG, Mormont MC, Liu X, Claustrat B, Hastings MH, Lévi F. Host circadian clock as a control point in tumor progression. J Natl Cancer Inst. 2002;94:690–697. doi: 10.1093/jnci/94.9.690. [DOI] [PubMed] [Google Scholar]
- 50.Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- 51.Fu L, Lee CC. The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer. 2003;3:350–361. doi: 10.1038/nrc1072. [DOI] [PubMed] [Google Scholar]
- 52.Lee CC. Tumor suppression by the mammalian Period genes. Cancer Causes Control. 2006;17:525–530. doi: 10.1007/s10552-005-9003-8. [DOI] [PubMed] [Google Scholar]
- 53.Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I, Fuchs CS, Colditz GA. Night-shift work and risk of colorectal cancer in the nurses’ health study. J Natl Cancer Inst. 2003;95:825–828. doi: 10.1093/jnci/95.11.825. [DOI] [PubMed] [Google Scholar]
- 54.Kloog I, Haim A, Stevens RG, Portnov BA. Global co-distribution of light at night (LAN) and cancers of prostate, colon, and lung in men. Chronobiol Int. 2009;26:108–125. doi: 10.1080/07420520802694020. [DOI] [PubMed] [Google Scholar]
- 55.Parent MÉ, El-Zein M, Rousseau MC, Pintos J, Siemiatycki J. Night work and the risk of cancer among men. Am J Epidemiol. 2012;176:751–759. doi: 10.1093/aje/kws318. [DOI] [PubMed] [Google Scholar]
- 56.Yang X, Wood PA, Ansell C, Hrushesky WJ. Circadian time-dependent tumor suppressor function of period genes. Integr Cancer Ther. 2009;8:309–316. doi: 10.1177/1534735409352083. [DOI] [PubMed] [Google Scholar]
- 57.Wood PA, Yang X, Taber A, Oh EY, Ansell C, Ayers SE, Al-Assaad Z, Carnevale K, Berger FG, Peña MM, et al. Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol Cancer Res. 2008;6:1786–1793. doi: 10.1158/1541-7786.MCR-08-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wood PA, Yang X, Hrushesky WJ. Clock genes and cancer. Integr Cancer Ther. 2009;8:303–308. doi: 10.1177/1534735409355292. [DOI] [PubMed] [Google Scholar]
- 59.Guo B, Chatterjee S, Li L, Kim JM, Lee J, Yechoor VK, Minze LJ, Hsueh W, Ma K. The clock gene, brain and muscle Arnt-like 1, regulates adipogenesis via Wnt signaling pathway. FASEB J. 2012;26:3453–3463. doi: 10.1096/fj.12-205781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin F, Chen Y, Li X, Zhao Q, Tan Z. Over-expression of circadian clock gene Bmal1 affects proliferation and the canonical Wnt pathway in NIH-3T3 cells. Cell Biochem Funct. 2013;31:166–172. doi: 10.1002/cbf.2871. [DOI] [PubMed] [Google Scholar]
- 61.Yang X, Wood PA, Ansell CM, Ohmori M, Oh EY, Xiong Y, Berger FG, Peña MM, Hrushesky WJ. Beta-catenin induces beta-TrCP-mediated PER2 degradation altering circadian clock gene expression in intestinal mucosa of ApcMin/+ mice. J Biochem. 2009;145:289–297. doi: 10.1093/jb/mvn167. [DOI] [PubMed] [Google Scholar]
- 62.Yang X, Wood PA, Ansell CM, Quiton DF, Oh EY, Du-Quiton J, Hrushesky WJ. The circadian clock gene Per1 suppresses cancer cell proliferation and tumor growth at specific times of day. Chronobiol Int. 2009;26:1323–1339. doi: 10.3109/07420520903431301. [DOI] [PubMed] [Google Scholar]
- 63.Zeng ZL, Wu MW, Sun J, Sun YL, Cai YC, Huang YJ, Xian LJ. Effects of the biological clock gene Bmal1 on tumour growth and anti-cancer drug activity. J Biochem. 2010;148:319–326. doi: 10.1093/jb/mvq069. [DOI] [PubMed] [Google Scholar]
- 64.Modak C, Chai J. Potential of casein kinase I in digestive cancer screening. World J Gastrointest Oncol. 2009;1:26–33. doi: 10.4251/wjgo.v1.i1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang WS, Stockwell BR. Inhibition of casein kinase 1-epsilon induces cancer-cell-selective, PERIOD2-dependent growth arrest. Genome Biol. 2008;9:R92. doi: 10.1186/gb-2008-9-6-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soták M, Polidarová L, Ergang P, Sumová A, Pácha J. An association between clock genes and clock-controlled cell cycle genes in murine colorectal tumors. Int J Cancer. 2013;132:1032–1041. doi: 10.1002/ijc.27760. [DOI] [PubMed] [Google Scholar]
- 67.Mostafaie N, Kállay E, Sauerzapf E, Bonner E, Kriwanek S, Cross HS, Huber KR, Krugluger W. Correlated downregulation of estrogen receptor beta and the circadian clock gene Per1 in human colorectal cancer. Mol Carcinog. 2009;48:642–647. doi: 10.1002/mc.20510. [DOI] [PubMed] [Google Scholar]
- 68.Krugluger W, Brandstaetter A, Kállay E, Schueller J, Krexner E, Kriwanek S, Bonner E, Cross HS. Regulation of genes of the circadian clock in human colon cancer: reduced period-1 and dihydropyrimidine dehydrogenase transcription correlates in high-grade tumors. Cancer Res. 2007;67:7917–7922. doi: 10.1158/0008-5472.CAN-07-0133. [DOI] [PubMed] [Google Scholar]
- 69.Alhopuro P, Björklund M, Sammalkorpi H, Turunen M, Tuupanen S, Biström M, Niittymäki I, Lehtonen HJ, Kivioja T, Launonen V, et al. Mutations in the circadian gene CLOCK in colorectal cancer. Mol Cancer Res. 2010;8:952–960. doi: 10.1158/1541-7786.MCR-10-0086. [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, Hua L, Lu C, Chen Z. Expression of circadian clock gene human Period2 (hPer2) in human colorectal carcinoma. World J Surg Oncol. 2011;9:166. doi: 10.1186/1477-7819-9-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, Yan D, Teng M, Fan J, Zhou C, Li D, Qiu G, Sun X, Li T, Xing T, et al. Reduced expression of PER3 is associated with incidence and development of colon cancer. Ann Surg Oncol. 2012;19:3081–3088. doi: 10.1245/s10434-012-2279-5. [DOI] [PubMed] [Google Scholar]
- 72.Panza A, Pazienza V, Ripoli M, Benegiamo G, Gentile A, Valvano MR, Augello B, Merla G, Prattichizzo C, Tavano F, et al. Interplay between SOX9, β-catenin and PPARγ activation in colorectal cancer. Biochim Biophys Acta. 2013;1833:1853–1865. doi: 10.1016/j.bbamcr.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 73.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 74.Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl Acad Sci USA. 2002;99:1182–1187. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee E, Salic A, Kirschner MW. Physiological regulation of [beta]-catenin stability by Tcf3 and CK1epsilon. J Cell Biol. 2001;154:983–993. doi: 10.1083/jcb.200102074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwarz-Romond T, Asbrand C, Bakkers J, Kühl M, Schaeffer HJ, Huelsken J, Behrens J, Hammerschmidt M, Birchmeier W. The ankyrin repeat protein Diversin recruits Casein kinase Iepsilon to the beta-catenin degradation complex and acts in both canonical Wnt and Wnt/JNK signaling. Genes Dev. 2002;16:2073–2084. doi: 10.1101/gad.230402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van de Wetering M, de Lau W, Clevers H. WNT signaling and lymphocyte development. Cell. 2002;109 Suppl:S13–S19. doi: 10.1016/s0092-8674(02)00709-2. [DOI] [PubMed] [Google Scholar]
- 78.Pancione M, Sabatino L, Fucci A, Carafa V, Nebbioso A, Forte N, Febbraro A, Parente D, Ambrosino C, Normanno N, et al. Epigenetic silencing of peroxisome proliferator-activated receptor γ is a biomarker for colorectal cancer progression and adverse patients’ outcome. PLoS One. 2010;5:e14229. doi: 10.1371/journal.pone.0014229. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79.Dai Y, Qiao L, Chan KW, Yang M, Ye J, Ma J, Zou B, Gu Q, Wang J, Pang R, et al. Peroxisome proliferator-activated receptor-gamma contributes to the inhibitory effects of Embelin on colon carcinogenesis. Cancer Res. 2009;69:4776–4783. doi: 10.1158/0008-5472.CAN-08-4754. [DOI] [PubMed] [Google Scholar]
- 80.Pazienza V, Vinciguerra M, Mazzoccoli G. PPARs Signaling and Cancer in the Gastrointestinal System. PPAR Res. 2012;2012:560846. doi: 10.1155/2012/560846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mazzoccoli G, Pazienza V, Panza A, Valvano MR, Benegiamo G, Vinciguerra M, Andriulli A, Piepoli A. ARNTL2 and SERPINE1: potential biomarkers for tumor aggressiveness in colorectal cancer. J Cancer Res Clin Oncol. 2012;138:501–511. doi: 10.1007/s00432-011-1126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oshima T, Takenoshita S, Akaike M, Kunisaki C, Fujii S, Nozaki A, Numata K, Shiozawa M, Rino Y, Tanaka K, et al. Expression of circadian genes correlates with liver metastasis and outcomes in colorectal cancer. Oncol Rep. 2011;25:1439–1446. doi: 10.3892/or.2011.1207. [DOI] [PubMed] [Google Scholar]
- 83.Talieri M, Papadopoulou S, Scorilas A, Xynopoulos D, Arnogianaki N, Plataniotis G, Yotis J, Agnanti N. Cathepsin B and cathepsin D expression in the progression of colorectal adenoma to carcinoma. Cancer Lett. 2004;205:97–106. doi: 10.1016/j.canlet.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 84.Troy AM, Sheahan K, Mulcahy HE, Duffy MJ, Hyland JM, O’Donoghue DP. Expression of Cathepsin B and L antigen and activity is associated with early colorectal cancer progression. Eur J Cancer. 2004;40:1610–1616. doi: 10.1016/j.ejca.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 85.Sakakibara T, Hibi K, Koike M, Fujiwara M, Kodera Y, Ito K, Nakao A. Plasminogen activator inhibitor-1 as a potential marker for the malignancy of colorectal cancer. Br J Cancer. 2005;93:799–803. doi: 10.1038/sj.bjc.6602743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oishi K. Plasminogen activator inhibitor-1 and the circadian clock in metabolic disorders. Clin Exp Hypertens. 2009;31:208–219. doi: 10.1080/10641960902822468. [DOI] [PubMed] [Google Scholar]
- 87.Oishi K, Miyazaki K, Uchida D, Ohkura N, Wakabayashi M, Doi R, Matsuda J, Ishida N. PERIOD2 is a circadian negative regulator of PAI-1 gene expression in mice. J Mol Cell Cardiol. 2009;46:545–552. doi: 10.1016/j.yjmcc.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 88.Schoenhard JA, Eren M, Johnson CH, Vaughan DE. Alternative splicing yields novel BMAL2 variants: tissue distribution and functional characterization. Am J Physiol Cell Physiol. 2002;283:C103–C114. doi: 10.1152/ajpcell.00541.2001. [DOI] [PubMed] [Google Scholar]
- 89.Schoenhard JA, Smith LH, Painter CA, Eren M, Johnson CH, Vaughan DE. Regulation of the PAI-1 promoter by circadian clock components: differential activation by BMAL1 and BMAL2. J Mol Cell Cardiol. 2003;35:473–481. doi: 10.1016/s0022-2828(03)00051-8. [DOI] [PubMed] [Google Scholar]
- 90.Sasaki M, Yoshitane H, Du NH, Okano T, Fukada Y. Preferential inhibition of BMAL2-CLOCK activity by PER2 reemphasizes its negative role and a positive role of BMAL2 in the circadian transcription. J Biol Chem. 2009;284:25149–25159. doi: 10.1074/jbc.M109.040758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pazienza V, Piepoli A, Panza A, Valvano MR, Benegiamo G, Vinciguerra M, Andriulli A, Mazzoccoli G. SIRT1 and the clock gene machinery in colorectal cancer. Cancer Invest. 2012;30:98–105. doi: 10.3109/07357907.2011.640650. [DOI] [PubMed] [Google Scholar]
- 92.Kabra N, Li Z, Chen L, Li B, Zhang X, Wang C, Yeatman T, Coppola D, Chen J. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J Biol Chem. 2009;284:18210–18217. doi: 10.1074/jbc.M109.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Antoch MP, Kondratov RV, Takahashi JS. Circadian clock genes as modulators of sensitivity to genotoxic stress. Cell Cycle. 2005;4:901–907. doi: 10.4161/cc.4.7.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kondratov RV, Antoch MP. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 2007;17:311–317. doi: 10.1016/j.tcb.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 95.Antoch MP, Kondratov RV. Circadian proteins and genotoxic stress response. Circ Res. 2010;106:68–78. doi: 10.1161/CIRCRESAHA.109.207076. [DOI] [PubMed] [Google Scholar]
- 96.Murakami Y, Higashi Y, Matsunaga N, Koyanagi S, Ohdo S. Circadian clock-controlled intestinal expression of the multidrug-resistance gene mdr1a in mice. Gastroenterology. 2008;135:1636–1644.e3. doi: 10.1053/j.gastro.2008.07.073. [DOI] [PubMed] [Google Scholar]
- 97.Brandi G, Calabrese C, Pantaleo MA, Morselli Labate A, Di Febo G, Hakim R, De Vivo A, Di Marco MC, Biasco G. Circadian variations of rectal cell proliferation in patients affected by advanced colorectal cancer. Cancer Lett. 2004;208:193–196. doi: 10.1016/j.canlet.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 98.Lévi F, Focan C, Karaboué A, de la Valette V, Focan-Henrard D, Baron B, Kreutz F, Giacchetti S. Implications of circadian clocks for the rhythmic delivery of cancer therapeutics. Adv Drug Deliv Rev. 2007;59:1015–1035. doi: 10.1016/j.addr.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 99.Hrushesky WJ, Grutsch J, Wood P, Yang X, Oh EY, Ansell C, Kidder S, Ferrans C, Quiton DF, Reynolds J, et al. Circadian clock manipulation for cancer prevention and control and the relief of cancer symptoms. Integr Cancer Ther. 2009;8:387–397. doi: 10.1177/1534735409352086. [DOI] [PubMed] [Google Scholar]
- 100.Antoch MP, Kondratov RV. Pharmacological modulators of the circadian clock as potential therapeutic drugs: focus on genotoxic/anticancer therapy. Handb Exp Pharmacol. 2013;(217):289–309. doi: 10.1007/978-3-642-25950-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628. doi: 10.1146/annurev.pharmtox.47.120505.105208. [DOI] [PubMed] [Google Scholar]
- 102.Giacchetti S, Bjarnason G, Garufi C, Genet D, Iacobelli S, Tampellini M, Smaaland R, Focan C, Coudert B, Humblet Y, et al. Phase III trial comparing 4-day chronomodulated therapy versus 2-day conventional delivery of fluorouracil, leucovorin, and oxaliplatin as first-line chemotherapy of metastatic colorectal cancer: the European Organisation for Research and Treatment of Cancer Chronotherapy Group. J Clin Oncol. 2006;24:3562–3569. doi: 10.1200/JCO.2006.06.1440. [DOI] [PubMed] [Google Scholar]
- 103.Bur IM, Cohen-Solal AM, Carmignac D, Abecassis PY, Chauvet N, Martin AO, van der Horst GT, Robinson IC, Maurel P, Mollard P, et al. The circadian clock components CRY1 and CRY2 are necessary to sustain sex dimorphism in mouse liver metabolism. J Biol Chem. 2009;284:9066–9073. doi: 10.1074/jbc.M808360200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu H, Meng X, Wu J, Pan C, Ying X, Zhou Y, Liu R, Huang W. Cryptochrome 1 overexpression correlates with tumor progression and poor prognosis in patients with colorectal cancer. PLoS One. 2013;8:e61679. doi: 10.1371/journal.pone.0061679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang X, Wood PA, Hrushesky WJ. Mammalian TIMELESS is required for ATM-dependent CHK2 activation and G2/M checkpoint control. J Biol Chem. 2010;285:3030–3034. doi: 10.1074/jbc.M109.050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bertagnolli MM, Niedzwiecki D, Compton CC, Hahn HP, Hall M, Damas B, Jewell SD, Mayer RJ, Goldberg RM, Saltz LB, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol. 2009;27:1814–1821. doi: 10.1200/JCO.2008.18.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gorbacheva VY, Kondratov RV, Zhang R, Cherukuri S, Gudkov AV, Takahashi JS, Antoch MP. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc Natl Acad Sci USA. 2005;102:3407–3412. doi: 10.1073/pnas.0409897102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Karantanos T, Theodoropoulos G, Gazouli M, Vaiopoulou A, Karantanou C, Stravopodis DJ, Bramis K, Lymperi M, Pektasidis D. Association of the clock genes polymorphisms with colorectal cancer susceptibility. J Surg Oncol. 2013;108:563–567. doi: 10.1002/jso.23434. [DOI] [PubMed] [Google Scholar]
- 109.Karantanos T, Theodoropoulos G, Gazouli M, Vaiopoulou A, Karantanou C, Lymberi M, Pektasides D. Expression of clock genes in patients with colorectal cancer. Int J Biol Markers. 2013;28:280–285. doi: 10.5301/jbm.5000033. [DOI] [PubMed] [Google Scholar]
- 110.Wang L, Chen B, Wang Y, Sun N, Lu C, Qian R, Hua L. hClock gene expression in human colorectal carcinoma. Mol Med Rep. 2013;8:1017–1022. doi: 10.3892/mmr.2013.1643. [DOI] [PubMed] [Google Scholar]
- 111.Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, Martínez A, Narayan G, Kirsch I, Franklin W, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118:640–650. doi: 10.1172/JCI30024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feng S, Cao Z, Wang X. Role of aryl hydrocarbon receptor in cancer. Biochim Biophys Acta. 2013;1836:197–210. doi: 10.1016/j.bbcan.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 113.Niestroy J, Barbara A, Herbst K, Rode S, van Liempt M, Roos PH. Single and concerted effects of benzo[a]pyrene and flavonoids on the AhR and Nrf2-pathway in the human colon carcinoma cell line Caco-2. Toxicol In Vitro. 2011;25:671–683. doi: 10.1016/j.tiv.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 114.Xie G, Peng Z, Raufman JP. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1006–G1015. doi: 10.1152/ajpgi.00427.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]